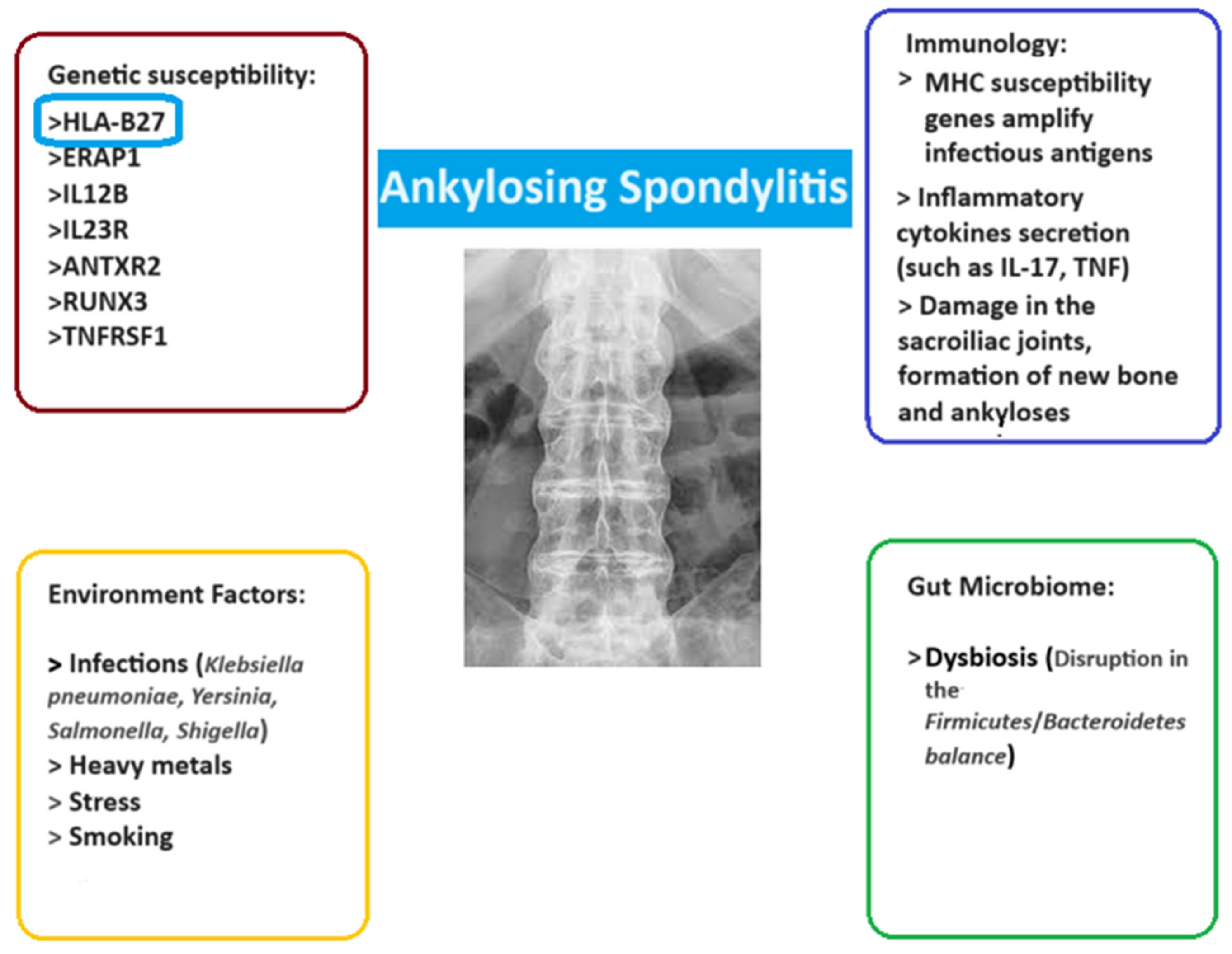
3.1. The Genomic Grammar of Ankylosing Spondylitis
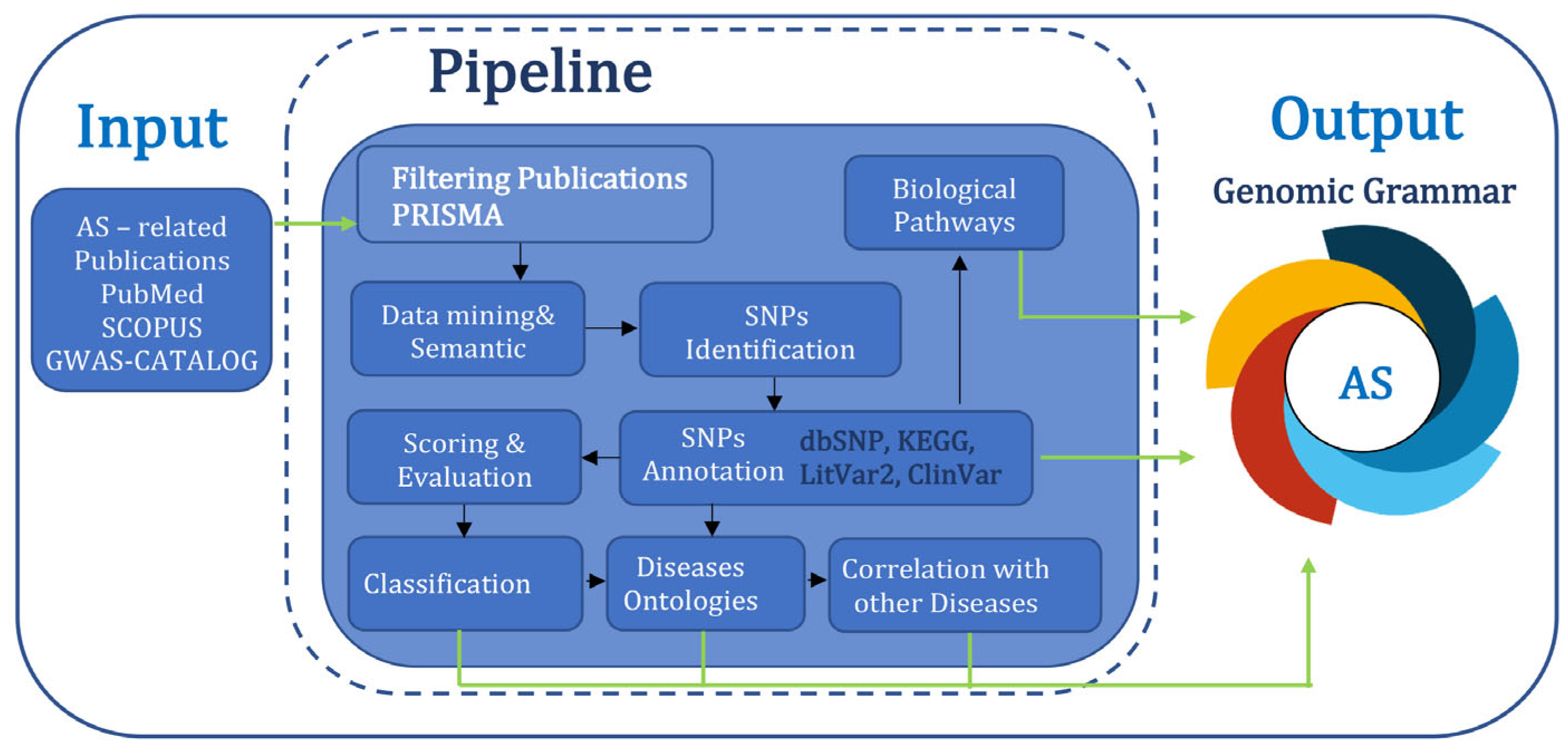
A total number of 57,909 publications containing or associated with the term “Ankylosing Spondylitis’” were retrieved from PubMed and SCOPUS, including information dated between 1940 and 2025. Most of them were registered in the last 20 years, indicating an increased awareness from the scientific community. After a filtering process based on the PRISMA flowchart, 539 publications were selected (
Supplementary Table S1), from which 793 SNPs have been identified. The dataset created was then annotated, evaluated, and classified based on the information found in other biological databases such as NCBI dbSNP, LitVar2, ClinVar, and KEGG Pathway. The final dataset, corresponding to the disease genomic grammar of AS, contains 793 genetic variants, of which 32 SNPs are strongly associated (Class A) with AS tand 44 are highly associated (Class B), while a vast majority, 717 of them, received a lower score (Class C), suggesting either a weak association or that more research is needed to assess their importance (
Table 1,
Supplementary Table S2). For the purposes of this study, further analysis focuses on SNPs of Classes A and B (
Supplementary Table S2). The number of significant SNPs rises to a total of 76 and affects 43 different genetic loci, from which, 376 are genes, 3 are pseudogenes, and 4 are ncRNA (
Figure 3). Most of these variants lie on non-coding regions, acting as regulators of genes, 50 of which are intronic; four lie on three prime UTR regions; and five are 2KB upstream variants (
Figure 3). A smaller portion of variants lie on coding regions, 16 of which cause a missense mutation, ultimately altering the protein’s ability to interact, and 1 is synonymous, altering gene expression levels (
Figure 3).
Most of the SNPs reported affect ERAP1, an aminopeptidase involved in trimming HLA class I-binding precursors, which has been widely studied as a risk factor for AS. This gene’s variants are thought to confer risk, acting through an epistatic interaction with
HLA-B27 as well as other MHC proteins [
52]. Additionally, a great number of SNPs were found to affect IL23R, a receptor involved in
IL23A signaling that is known to be related to AS as well as some autoimmune diseases, and has been previously studied as a possible therapeutic target (
Figure 3) [
22,
53]. Also, at the chromosome level, chromosomes 1, 2, and 5 accumulated the most SNPs (
Figure 3). Interestingly, although most of the SNPs (84%) affected genes, 6.8% affected pseudogenes, and 9% affected ncRNAs. It is important to note that recent studies suggest that SNPs in intronic regions and pseudogenes, though their functions remain poorly understood, may influence gene regulation through mechanisms such as alternative splicing and enhancer activity [
54]. This could possibly explain a part of the ‘missing’ AS heritability; however, it is good to keep in mind that while GWASs can identify genetic associations, functional validation is essential to establish causal relationships, and that there are some inherent limitations of GWASs in multifactorial diseases. Further analysis of protein interactions and related pathways showed a strong association between the involved genes and immunological pathways, especially cytokine and interleukin signaling, in which 12 genes were included, namely
IL6R,
IL33,
IL12B,
IL37,
IL1R1,
IL1A,
TNFRSF1A,
TNF,
IL23R,
IL1RN,
IL1F10,
LTBR, and
SH2B3. The involvement of interleukins in signaling in AS is well studied, especially for the
IL-23/
IL-17 axis, which plays a role in the Th17 immune response and has been previously associated with many immune-mediated inflammatory diseases, including RA, PsO, IBD, and MS. It has been shown that exposure of Th17 cells to
IL-23 induces pathogenesis and leads to the expression of their master regulator,
RORγt, as well as to the production of pro-inflammatory cytokines [
55]. Also, the
IL-23/IL-17 axis has attracted attention as a potential therapeutic target for AS. While NSAIDs, medicines which target the cyclooxygenase (COX) enzymes, relieving pain and reducing inflammation, are considered as the first-line treatment for AS, there are cases in which they might not be successful in suppressing AS symptoms, or side effects may occur due to their long-term use. In these cases, treatment with biologics, such as the inhibitors anti-TNF and anti-IL17, seems very promising for AS patients [
2,
10,
55].
Besides immunological pathways, the analysis also showed association with pathways that involve signal transduction, metabolism, programmed cell death, the transport of small molecules, and gene expression. Furthermore, gene ontology analysis showed that the encoded proteins are involved in biological processes that pertain to the immune system, such as cytokine signaling, the inflammatory response, the positive regulation of IFN-γ production, the positive regulation of JAK-STAT, and the cellular response to LPS. It also showed that most of the proteins are located at the external side of the plasma membrane, as well as in the extracellular space and at receptor complexes, while their molecular functions involved cytokine binding, especially of IL-1, and metallo-aminopeptidase activity.
Similar findings have been previously reported in studies that investigate the possible role of the immune system and bacterial factors in AS pathogenesis. It has been suggested that
HLA-B27-positive individuals are more susceptible to AS after microbial infection (especially by
K. pneumoniae), which leads to increased LPS levels and, therefore, triggers the activation of innate immune cells. These, in response, promote inflammation, a process driven by cytokine production (including
TNF,
IL-23, and
IL-17), while hematopoietic stem cells invoke
NF-κB,
RANKL, and
M-CSF, leading to abnormal bone formation [
56]. In addition, it has been previously suggested that the
IL-1 gene cluster plays a role in AS, due to the observation that there is an aberrant expression of this cytokine in AS patients (among others, including
TNF and
IL-17), and this cytokine has also been used as a potential therapeutic target; however, the causal relationship between this gene and AS pathogenesis needs further investigation [
57]. Also, metalloaminopeptidase activity has been previously studied in association with AS through the role of endoplasmic reticulum aminopeptidase 1 (
ERAP1) and
ERAP2 enzymes, which work closely with MHC-I molecules (such as the
HLA-B27) for antigen presentation [
52].
Furthermore, these SNPs were examined for possible overlapping associations with autoimmune diseases, and it was found that many of the AS-related genetic variants are also associated, primarily, with PsO as well as RA and secondly with IMD, such as CD and UC (
Table 2). All of these diseases have been previously studied in association with AS; however, cases reported for comorbidity are quite rare due to the rarity of AS patients alone [
58,
59,
60]. In such cases, where clinical reports are scarce due to disease rarity or a difficulty in diagnosing patients, genetic research can prove to be very useful for detecting possible associations. However, these associations do not prove causality and should be carefully examined [
61].
Overall, many of the AS-related SNPs were found to be associated with autoimmune diseases; however, 16 SNPs were found to be uniquely associated with AS, with only a few studies trying to associate them with other diseases (
Supplementary Figures S2 and S3). These are the rs7711564 and rs27038 located at
ERAP1, the rs6692977 located at
FCRL5, the rs27037 located at the
CAST gene, the rs4333130 located at
ANTXR2, the rs2242944 located at RPL23AP12, the rs116488202 located between
MICA, the rs26307 located at
OTULIN, the rs4648889 located at
RUNX3, the rs1894399 and rs2856836 located at
IL1A, the rs2192752 located at
IL1R1, the rs27356 located at
ANKH, the rs3750996 located at
STIM1,the rs4349859 located at
MICA-AS1, and, lastly, the rs1929992 located at
IL33 and LOC107987046. Especially in the case of rs116488202, this SNP has also been greatly associated with acute anterior uveitis, a very common extra-articular manifestation of AS, which has been noticed in 25,8% of AS cases [
62].
In this study, we chose to focus on the genetic variants that AS shares with RA and PsO, two chronic autoimmune diseases that have more established genetic overlap and are more commonly compared in the literature. These have been previously strongly associated with MHC molecules (
HLA-DRB1 shared epitope alleles and
HLA-C*0602,
respectively), but altogether hold some etiological and clinical differences (
Figure 4) [
63,
64]. Moreover, we examined the genetic overlap between AS and autoimmune diseases that are more distantly related or less genetically overlapping with AS. These include IBD, systemic lupus erythematosus (SLE), uveitis, and MS, and the SNPs that appeared in at least three of these diseases were further discussed (
Figure 4). The aim was to investigate possible underlying molecular mechanisms that these autoimmune diseases may share with AS. Therefore, the shared genetic variants were collected and analyzed regarding their molecular mechanisms as well as the gene ontology and the related pathways of the encoded proteins that they affected (
Figure 3).
3.2. Genetic Overlap Between AS and Stongly Associated Autoimmune Diseases—RA and PsO
In this study, nine SNPs were found to be common in the diseases AS, RA, and PsO, located in six distinct genetic loci, namely
IL23R,
IL6R,
CDC37,
IL12B,
TNF, and
PTPN22, most of which pertain to immunological processes (
Figure 4,
Table 3). More specifically,
IL23R binds
IL-23, promoting Th17 cell differentiation; IL6R binds IL-6 and initiates inflammatory responses through
JAK/
STAT3 signaling;
CDC37 acts as a co-chaperone that stabilizes protein kinases via
HSP90, including immune-related kinases, and indirectly can affect signaling pathways through kinase stabilization (
JAKs,
IKKs);
IL12B encodes a p40 subunit shared by
IL-12 and
IL-23, which activate Th1 and Th17 cells, respectively;
TNF is a pro-inflammatory cytokine that promotes immune cell activation, apoptosis, and inflammation; and, lastly,
PTPN22 negatively regulates T cell receptor (TCR) signaling.
Within the locus of
IL23R, four SNPs (rs11209026, rs2201841, rs1343151, and rs10489629) were found to be associated with the three diseases, with one being exonic (rs11209026) and causing a missense variation, while the other three were intronic. More specifically, the missense SNP leads to an Arg381Gln mutation that affects the cytoplasmic tail of the receptor of
IL23. This domain is responsible for signal transduction by interacting with its associated
JAK2 kinase; therefore, this missense SNP may functionally affect the
IL-23R transducing pathway [
65]. It has also been shown that this SNP reduces
IL-17A and
IL-22 serum levels [
66]. In some cases, protective haplotypes, including R381Q, may influence mRNA splicing by modifying the SF2 splicing enhancer binding site, leading to increased production of a soluble
IL-23R isoform (
IL23RΔ9), which binds
IL-23 and blocks its pro-inflammatory signaling pathway. Thus, a therapeutic strategy using antisense oligonucleotides to induce this splicing shift offers a promising approach with which to reduce
IL-23 signaling in autoimmune conditions [
67]. Interestingly, while it has been reported that this SNP is protective for AS, in a meta-analysis it has been suggested that it confers risk to RA and it has also been associated with PsO through GWASs [
68,
69]. On the other hand, the intronic rs2201841 was found to be associated with AS susceptibility, and the G allele was also associated with an increased risk for PsO, as shown in GWASs [
70,
71,
72,
73]. However, the latter has been shown to be protective in the case of RA, as shown in a meta-analysis [
69]. In the case of the intronic SNPs (rs1343151 and rs10489629), the A and C allele, respectively, seemed to confer risk to both AS and PsO, as shown in GWASs [
70,
71,
72,
73]. Lastly, in the case of RA, these SNPs were associated with increased risk, and even though previous studies showed controversial results, another study suggests that the susceptibility character of these SNPs might be explained by studying the surrounding haplotype, rather than the SNPs alone [
74].
Another SNP, rs4129267, located at intron 8 of
IL6R, might regulate gene expression, as it has been shown that patients who carried the T allele in homozygosity had 73% higher concentrations of serum
IL6R [
22]. It could also predict a worse ASAS-20 (Assessment of Spondyloarthritis international Society) response in a Taiwanese population. This SNP has been associated with both AS and RA susceptibility [
75,
76], but was shown to be associated with reduced PsO activity in GWASs [
77]. Also, the intronic rs35164067 located in
CDC37 has been associated with AS susceptibility in GWASs and has also been used in an RA association study, investigating a possible connection between SNPs and anti-citrullinated protein antibody (ACPA)-positive RA genetic risk score [
22,
78]. On the contrary, it was found to be protective of PsO [
22,
79]. rs3212227, located at the 3′ UTR of the
IL12B region, has been suggested to be protective against AS [
80]. However, another study showed that this allele may amplify the genetic risk for AS, as well as maybe contributing to elevated
IL-23 and
IL-12p40 serum levels if individuals that carry it also carry the rs17860508 allele 2, suggesting a leading role for rs17860508 in genetic susceptibility imposed by
IL12B polymorphisms [
81]. Although its relationship with AS needs further research, in the case of RA this SNP has been strongly associated with RA susceptibility by multiple studies [
82,
83,
84,
85]. Another study in a Chinese population showed that patients with PsO who carry the minor allele might have a better response in acitretin therapy [
72,
80,
86].
In the case of rs361525, located 2KB upstream from the
TNF locus, the A allele was found to be protective against AS and it was associated with a later age of onset as well as a lower erythrocyte sedimentation rate (ESR). Serum TNF-α levels were not significantly different and anti-TNF treatment was not influenced. However, another study suggested that patients carrying the G allele could predict a better response in etanercept therapy (an inhibitor that targets TNF) [
87,
88]. In the case of RA, the G allele was shown to confer risk, while patients carrying this allele also showed elevated TNF serum levels, but also had a better response to anti-TNF treatment for that reason [
89]. Also, the minor allele is strongly associated with PsO type I (characterized by the early onset of symptoms) but seems to be protective of AS. Specifically, patients carrying the GA/AA genotype developed symptoms at a later age and they had a lower erythrocyte sedimentation rate (ESR); however, they gave no insight regarding serum TNF-α levels and responses to anti-TNF [
88,
90].
Lastly, the intronic rs1217414, located at intron 1 of the
PTPN22 locus, might affect splicing, leading to abnormal
PTPN22 expression levels. The TT genotype of this SNP has been suggested to confer risk to AS, as shown in a meta-analysis; however, another study conducted in a Han Chinese population suggested that this SNP might be protective [
91,
92]. This SNP has also been suggested to be protective in RA Han Chinese patients [
93]. The rs1217414 in the
PTPN22 locus seems to cause splicing dysregulation by retaining additional introns after splicing (which can lead to the expression of a dysfunctional protein or the alteration of expression levels), and the T allele has been suggested to confer risk for AS. The encoded protein of this gene is thought to suppress the TCR signal transduction of T cells, and polymorphisms in this gene have been associated with many autoimmune diseases [
91]. This SNP was also significantly associated with type I psoriasis (early onset), as shown in another study [
94].
Beyond the genetic variants shared across AS, RA, and PsO, another set of SNPs appears to be specifically shared between AS and RA, suggesting a partially overlapping genetic architecture distinct from that of PsO, as discussed in the next section.
3.2.1. The Genetic Basis Unique to Ankylosing Spondylitis and Rheumatoid Arthritis
RA and AS are considered to be two of the most common rheumatic (joint) conditions; however, there are some etiological and clinical differences between these two. The most distinct clinical difference, which was found in 1974, is that RA patients often show elevated levels of rheumatoid factors in the blood, whereas AS patients are seronegative. This has helped clinicians to better categorize AS as part of seronegative spondyloarthropathy (SpA), and to make a more accurate diagnosis, since AS was often misdiagnosed as RA. Another difference is that RA usually affects the smaller joints, such as in the hands and feet, whereas in AS the spine and especially the lower vertebrae are primarily affected [
95,
96]. In the case of RA many types of immune cells, such as macrophages, T and B cells, fibroblasts, chondrocytes, and dendritic cells seem to play a role, while pathologic activation of the osteoclasts, a process carried out by fibroblast-like synoviocytes (FLSs), leads to damage of the cartilage, bones, and tendons [
97].
Possible SNPs that are associated with both RA and AS were gathered. In total, 22 polymorphisms were found, 4 of which are located at an exonic region, causing a missense variation, while the rest are intronic, regulating gene expression (
Figure 4,
Table 4). Most of the genes reported play a role in immunological processes, such as interleukin signaling, complement activation, and T-lymphocyte activation, as well as in other biological processes, including cell proliferation, differentiation, and apoptosis. Previous studies have also supported the idea that there is an interplay between both complement activation and regulation by T cells, which activates and maintains the process of inflammation in RA, confirming these findings [
98]. Also, interleukin signaling has previously been studied in association with RA, as will be discussed below. Further analysis using DAVID tools and cross-validation by Reactome showed that most of the encoded proteins are located at the extracellular space and more specifically in receptor complexes, such as the
IL-23 receptor complex. Also, it was shown that most of the proteins play a role in the cytokine-mediated signaling pathway, the positive regulation of
IFN-γ production, the cellular response to lipopolysaccharide, the positive regulation of gene expression, and the positive regulation of the Th-17-type immune response, while these are involved in molecular functions that include protein binding and, more specifically,
IL-1 and
IL-12 receptor binding.
As mentioned earlier, the
IL23/
IL17 axis has been widely studied as it is known to be associated closely with AS and it has been used as a potential therapeutic target, like in the case of the anti-IL17 drug ‘Secukinumab’, which has been approved in the USA and by several countries in the EU [
99]. Even though RA and AS include chronic inflammation mediated by the immune system and the
IL23/
IL17 axis is thought to play a critical role in both [
100], there are some differences in the therapeutic approaches used for each. In the case of RA, there are still no anti-IL17 approved drugs; however, there are novel approaches that are being used in the clinic, targeting cytokines such as
TNF,
IL-1,
IL-1R,
IL6, and
IL6R, which have been proven to be useful [
101]. Also, the
IL-12 family has been previously reported to play a role in RA by promoting T cell differentiation to Th-1, which subsequently leads to IFN-γ production, while
IL-1 is known to play a role in osteoclast differentiation, which leads to bone resorption [
102,
103].
In this study, many of the AS and RA common variants were found to affect interleukins. More specifically, two SNPs located at the
IL23R locus were found, which are in non-coding regions (rs10889677 and rs1004819). In the case of rs10889677, a 3′ UTR variant was found to be associated with both AS and RA susceptibility in the general population, especially in Caucasians [
104]. Furthermore, another study performed in RA Iranian patients indicated that this SNP might be associated with the overexpression of
IL23R [
105]. In the case of the intronic rs1004819, the A allele has been associated with AS susceptibility in the overall population; however, in the subgroup analysis, no significant association was shown with an Asian population [
106]. It has also been associated with an increased risk of RA in Turkish and Iranian populations; however, another meta-analysis suggests otherwise, and further research is therefore needed [
107,
108,
109].
Other SNPs located in coding regions of interleukin genes are rs3811047, rs3811058, and rs17561, affecting the genes
IL37,
IL1F10, and
IL-1A, respectively. rs3811047 is located at a coding region of the
IL37 locus, causing a missense variance and is associated with AS susceptibility in the Han Chinese; however, a meta-analysis showed otherwise, meaning that more research is needed to assess its susceptibility character [
110,
111]. It has also been suggested to be strongly associated with more severe RA disease activity [
112]. rs3811058 located at exon 3 of
IL1F10, causing a missense variance, has been shown to be involved in AS risk; however, it was restricted to a non-AS phenotype [
113]. Also, it has been strongly associated with RA and with slight differences in CRP levels [
114]. The rs17561, located at exon 5 of
IL-1A, causing a missense variance (Ser114Ala), has been shown to confer risk to both AS and RA. This SNP has been shown to influence
IL-1 expression and to be more resistant to calpain cleavage, without which
IL-1A cannot be activated [
115,
116].
Other SNPs affecting genes that pertain to immunological processes include rs1800629, located 2 KB upstream of the TNF locus. Individuals carrying rs1800629 in a Bulgarian population had a lower risk of developing AS. Also, as was shown in a later study, RA and AS patients carrying the GA genotype instead of the GG genotype did not respond to anti-TNF treatment [
81,
117]. However, these results may vary depending on ethnicity [
118]. Other SNPs also include rs3091244, a 2KB upstream variant located at the
CRP locus, which has been associated with both AS and RA susceptibility. More specifically, it has been shown that AS patients carrying the A allele had higher
CRP levels and that they responded better to etanercept treatment, as was shown by their ASAS20 and ASAS40 scores. For that reason, the study suggested that this SNP should be taken into consideration when assessing disease score in AS patients [
119]. In the case of RA patients, the A allele contributed to higher CRP levels; however, this finding was secondary when assessing the disease score in patients whose disease had progressed [
120,
121]. Another intronic variant, rs11065898, located at the
SH2B3 locus, which has been positively associated with CD4+ lymphocyte counts, was suggested to be a risk factor for AS in GWASs, while only a marginal association was found between this SNP and RA in a Taiwanese population analysis [
22,
122].
Other SNPs that affect genes involved in other biological processes as well as other genetic loci, such as pseudogenes, include rs2283790, an intronic variant located at the
UBE2L3 gene. This SNP was associated with AS in European populations in GWASs and this was confirmed by a second analysis on a Chinese population, where a marginal association was found [
22,
87]. Additionally, it reached the genome-wide significance threshold in an RA study; however, there are no studies reporting the possible functional consequence that it may have [
123]. Furthermore, rs13202464, which is located upstream of the
FGFR3P1 pseudogene, is a tag SNP, which strongly represents the risk effect of HLA-B*27 and has been previously shown to be a risk factor for AS in both European and Chinese populations [
124]. However, it should be taken into account that this screening marker may not be suitable for every population due to differences in genomics and metagenomics [
125]. This SNP has also been shown to be associated with RA in a genome-wide study [
123]. Lastly, three SNPs, namely rs6759298, rs12186979, and rs2836883, which affect
RN7SL51P,
PTGER4, and
RPL23AP12, respectively, have been associated with AS susceptibility in GWASs, while they were also used in an RA association study, investigating a possible connection between these SNPs and ACPA+ RA genetic risk score [
22,
78]. More specifically, rs6759298, an intronic variant located in a gene desert at chromosome 2p15, has been significantly associated with AS in both European and Chinese populations, as shown in GWASs [
22,
126]. It is suggested that this SNP might impact non-coding RNA sequence or transcription effects [
127]. The variant rs12186979, located in an intronic region at
PTGER4, as well as rs2836883 and an intergenic SNP located nearby the pseudogene
RPL23AP12 were shown to be associated with AS in European populations in GWASs, but further research is needed to assess their association as well as their functional consequences [
22].
3.2.2. The Genetic Basis Unique to Ankylosing Spondylitis and Psoriasis
In this study, the number of common genetic associations between AS and PsO was higher than that of AS and RA, an autoimmune disease widely thought to be closely related to AS. In the case of PsO, an excessive proliferation of epidermal cells with a simultaneous inflammation at the dermis (the middle layer of skin) leads to the creation of patches of red thick skin which causes itchiness to the patient and the characteristic lesions of PsO. It is a complex disease with no clearly defined causes, but it has been hypothesized that the amplification of this disease is T-cell-mediated, while in the pathophysiology of the disease multiple types of cells, including dendritic cells, T cells, and keratinocytes, are involved in its initiation and maintenance [
128].
Based on the results, 25 variations were found to be overlapping between the two diseases, 7 of which are located at an exonic region, causing a missense variation, while the rest are intronic, regulating gene expression, modifying regulatory nucleic acid binding reactions, or dysregulating splicing (
Figure 4,
Table 5). Most of the genes reported play a role in immunological processes, such as trimming HLA class I-binding precursors, promulgating cytokine signals, and regulating T cell receptor signaling. This is not surprising, since it has been previously supported that professional antigen-presenting cells (APCs) play a central role in PsO pathogenesis, while activated T cells promote inflammation via cytokine secretion [
129]. Other genes were also found to be involved in biological processes such as gene expression, signal transduction, amino acid metabolism, coagulation, cell differentiation, and apoptosis. Further analysis using DAVID tools and cross-validation by Reactome showed that most of the encoded proteins are located at the external side of the plasma membrane (mostly receptors), while one-fifth of them are located at the ER lumen. Additionally, it was shown that most of the proteins play a role in the tyrosine phosphorylation of the STAT protein, as well as in activation of JAK, the production of IFN-γ, and the regulation of osteoclast differentiation. Indeed, a strong association has been previously observed between psoriatic inflammation and the differentiation of osteoclasts, which can lead to PsA, a type of SpA that is similar to RA but has been linked to PsO, yet the underlying pathogenic mechanisms are not completely understood [
130]. Lastly, the analysis showed that the affected proteins are involved in molecular functions of interleukin signaling and, more specifically, of
IL-6 and
IL-12.
Both
IL-6 and
IL-12 have been previously reported to play a role in PsO. More specifically, it has been suggested that in the process of PsO pathogenesis, an initial trigger causes stress to keratinocytes, which, in response, activate plasmacytoid dendritic cells that secrete
IFN,
TNF-a,
IL-1β, and
IL-6. Subsequently, myeloid dendritic cells are activated and secrete, among others things,
IL-12, inducing the Th-1 response, which contributes to the recurring cycle of psoriatic plaque creation [
128]. Also, it has been shown that treatment that blocks the
IL-12/
IL-23 signaling pathway seems very promising for PsO patients [
131].
In this study, one SNP located in the
IL23R locus was found at a non-coding region (rs11209032). More specifically, the minor allele seems to confer risk for both AS and PsO, as shown in GWASs [
70,
71,
72,
73]. The intergenic rs11209032 is located within an enhancer region and seems to be involved in elevated Th1 cells [
72]. On the other hand, the exonic rs11209032 is a well-studied SNP that is thought to be protective of several immune-mediated diseases, among which are AS and PsO [
68]. It involves a change in the negatively charged Arg to a neutral Gln in position 381, in the intra-cytoplasmic tail of the receptor, perhaps affecting interaction with
JAK2 and, therefore, reducing the signal transduction of
IL23 [
65]. One other intronic variant was found within the
IL12B locus (rs6556416), which was associated with AS and PsO in GWASs [
73]. Another intronic variant located within
IL1R1 and
IL1R2 loci (rs4851529) is associated with many chronic inflammatory diseases, including AS and PsO, and seems to modulate the
IL-1 response [
22,
132].
Besides the involvement of interleukins in AS susceptibility, there have been many previous reports discussing the interplay between
ERAP1 and
HLA-B27 in AS susceptibility, highlighting a possible epistatic interaction, since
ERAP1 polymorphisms only affect AS risk in
HLA-B27-positive individuals [
133].
ERAP1 is responsible for shaping the
HLA-B27 peptidome (the trimmed precursors that will bind to MHC-I molecules such as
HLA-B27), while
HLA-B27 is responsible for presenting these peptides to T cells, determining the immunological reaction [
134]. In this study, six SNPs in the
ERAP1 locus were revealed to confer risk for both AS and PsO, located at exonic regions (rs30187, rs27044, rs10050860, rs17482078, rs26653, and rs2287987) and affecting enzymatic activity (which alters trimming and therefore the
HLA-B27 peptidome) [
134,
135,
136,
137]. The variants rs30187 (K528R) and rs27044 (Q730E) seem to be protective of AS and patients who carry them express significantly lower levels of HLA class I FHCs [
138]. However, two GWASs associated them with PsO susceptibility [
139,
140]. Likewise, the minor alleles of variants rs10050860 (D575N), rs17482078 (R725Q), rs26653 (R127P), and rs2287987 (M349V) seem to alter ERAP1 trimming and confer protection from AS and PsO. For example, the R725Q substitution of rs17482078 is thought to lead to the loss of two hydrogen bonds between R725 and D766, affecting the stability of the C-terminus of ERAP1, leading to impaired ERAP enzymatical activity [
137]. Also, the variants rs26653 and rs2287987 showed the same pattern as rs30187, according to a meta-analysis between trans-ethnic populations [
136]. Altogether, previous studies have confirmed that each
ERAP1 SNP contributes to AS susceptibility individually rather than by the haplotypes of SNPs [
141]. Two other intronic variants were found in the
ERAP2 locus (rs2910686 and rs2248374), from which rs2910686 seems to confer risk to both AS and PsO, while the minor allele of rs2248374 seems to be protective to AS, due to leading to the complete absence of
ERAP2 expression, while it was shown to confer risk in PsO [
22,
132,
142,
143]. Lastly, it is worth noting that the association of
ERAP2 with AS appears independent of the presence of
HLA-B27, in contrast to
ERAP1 [
142].
Other intronic variants affecting proteins involved in immunological processes are the primary associated SNP at
TNFRSF1A, rs1860545, which was found to be associated with both AS and PsO in two GWASs [
22,
132]. Another SNP is rs10865331, found in an intergenic region on 2p15. This region has been strongly associated with AS in Europeans in previous GWASs [
23,
144] and has also been found to be associated with PsO in another GWAS [
145]. It has also been associated with the
BASFI (an index used for the estimation of the disease activity of AS), and in the case of AA/AG genotypes it has been associated with a higher ESR in AS patients of a Taiwanese population [
146]. Some studies suggest that this region may also affect the promoter of
COMMD1, a gene involved in
NF-κB signaling and located over approximately 435 kb upstream [
147]. The intronic rs35164067 located in
CDC37, which encodes a protein that promulgates cytokine signals, was also genotyped in GWASs and it was found to be protective of PsO, while on the contrary was found to confer risk to AS [
22,
79].
Intronic variants that affect proteins of other biological processes include rs6600247, which has been associated with both AS and PsO in GWASs, and is located in a 15 kb LD block upstream of the
RUNX3 promoter and is associated with lower CD8+ T cell counts. It has been suggested that it may disrupt a binding site of the c-MYC transcription factor, affecting the regulation of
RUNX3, a transcription factor that also functions as a tumor suppressor [
132,
148]. Lastly, two significantly associated SNPs located in intergenic lncRNAs (possibly acting as transcriptional regulators) were extracted from GWASs. These are rs11624293, which is found in LINC01147, and rs11190133, found between LINC01475 and
GOT1-DT, two ncRNAs [
22,
132].
3.3. Genetic Overlap Between AS and More Distantly Related Autoimmune Diseases—IBD, SLE, Uveitis, and MS
In addition to RA and PsO, the genetic overlap between AS and autoimmune diseases with lower association scores—namely IBD (CD and UC), SLE, uveitis, and MS—was examined and the analysis was performed in sets of three-disease combinations (
Table 6,
Figure 4). In all of these diseases persistent inflammation is central, each affecting a different type of tissue. IBD, encompassing CD, UC, and unclassified IBD (when patients have features that overlap CD and UC), involves chronic intestinal inflammation triggered by genetic, microbial, environmental, and immune factors. Genome-wide studies have identified over 200 susceptibility loci, from which 137 are shared between CD and UC and most are non-coding, affecting gene regulation. Both the innate and adaptive immune systems are implicated in IBD pathogenesis, with CD being associated with
Th1/
Th17 responses, producing cytokines like
IL-17,
IFN-γ, and
TNF-α, whereas ulcerative colitis features a Th2 response, with
IL-5 and
IL-13 activating B cells and NK T cells [
149,
150]. Although IBD primarily targets the gastrointestinal tract, it frequently leads to extraintestinal manifestations. These involve (but are not limited to) the musculoskeletal, dermatologic, hepatobiliary, and ocular systems [
151]. Epidemiological studies show that clinically apparent IBD occurs in approximately 6–14% of AS patients, while microscopic gut inflammation is present in up to 60% [
152]. Conversely, AS develops in up to 3% of IBD patients, particularly those with CD. Mendelian randomization and transcriptome analyses further support a causal role of IBD in promoting AS development and suggest that disease activity in IBD may influence AS progression through shared immune pathways and risk gene expression profiles [
153]. Moreover, insights from genetic associations suggest the implication of the gut–joint axis, involving T cell migration and dysregulated microbiota, as an underlying factor for this bidirectional interaction [
152].
SLE is a chronic autoimmune disease marked by systemic inflammation and multi-organ involvement, with a strong female predominance and distinct immunopathology compared to AS [
154]. Although both conditions are associated with immune dysregulation, their co-occurrence is rare, and genetic overlap remains limited. However, recent GWASs have identified shared susceptibility loci in genes regulating immune responses, such as
TNFAIP3 and
IL23R, suggesting that subtle common pathways may exist in immune regulation [
53,
155]. The immunopathogenesis of SLE involves the hyperactivation of autoreactive B cells and the production of pathogenic autoantibodies, particularly anti-dsDNA and anti-Sm, in addition to dysregulated T helper cell subsets. Key roles are played by plasmacytoid dendritic cells (pDCs), which produce large amounts of type I interferons, as well as by CD4+ T cells that provide B cell help and perpetuate inflammation. Elevated levels of cytokines such as
IFN-α,
IL-6, and
BAFF (B cell activating factor) contribute to chronic immune activation and tissue damage [
156]. Despite the distinct clinical presentations and immune profiles of SLE and AS, emerging evidence indicates potential overlap in cytokine-mediated signaling pathways that may support the development of shared therapeutic strategies [
157].
On the other hand, autoimmune (non-infectious) uveitis is a major ocular manifestation of systemic rheumatic diseases such as RA, AS, juvenile idiopathic arthritis, and PsA, and can occur independently or alongside these conditions. Uveitis refers to inflammation primarily affecting the uveal tract with multiple clinical manifestations (acute anterior uveitis—AAU, intermediate uveitis—IU, Vogt–Koyanagi–Harada syndrome—VKH, and others), classified by type (granulomatous, non-granulomatous), anatomical location (anterior, posterior, intermediate, and panuveitis), time course, and relapse. It is thought that the abnormal immune response is mediated from T cells. Several forms of uveitis are strongly linked to specific
HLA genes, and a well-known example is the strong link between AAU and the
HLA-B27 genes, which also explains its frequent association with AS [
158,
159]. Other genes, including
IL23R, which is implicated in multiple forms of uveitis (including AAU, Behçet’s disease, and VKH), also plays a role in Th17 pathway activation, and
ERAP1/
ERAP2, which are involved in antigen processing, are strongly correlated with AAU and VKH, as well as the CFB, which is part of the alternative complement pathway and
STAT4, which is associated mostly with Behçet’s disease, and serves as a crucial transcription factor in the Th1 immune response [
160].
Lastly, MS is also a chronic inflammatory disease; however, it is thought to be unrelated to AS, while cases for comorbidity are rare. In this disease, activated immune cells penetrate the central nervous system and cause inflammation, which leads to myelin destruction and as a result partial, or even total, loss of cognitive and physical function. In the pathogenesis mechanism, it has been suggested that an interplay between APCs and Th cells (CD4+) initiates the disease, while
IL-4,
IL12, and
IL23 induce Th-1 differentiation, which, subsequently, promotes inflammation. Furthermore, TNF-α and FAS ligand (a transmembrane protein produced by lymphocytes) bind to TNF receptors on oligodendrocyte cells of the neuron, initiating apoptosis [
161].
In this study, four key SNPs were identified to be overlapping between these diseases, all of which are in intronic or intergenic regions (
Table 5), namely rs11465804 located at
IL23R, rs1860545 located at
TNFRSF1A, rs1250550 located at
ZMIZ1, and rs1495965 located between
IL12RB2 and
DNAJB6P4. The variants are located in genes that pertain to immunological processes, immune cell signaling, cell apoptosis, and transcription regulation (among others). More specifically,
IL23R encodes a subunit of the receptor of IL23, which is a cytokine critical for Th17 cell development and plays a key role in multiple chronic inflammatory diseases [
162,
163,
164,
165]. Also,
ZMIZ1 acts as a modulatory regulator (can either enhance or repress transcription) of STAT signaling. The
TNFRSF1A plays a key role in immune cell signaling, particularly in the TNF-α pathway, where it mediates inflammation, apoptosis, immune regulation, and, lastly,
IL12RB2.
The intronic rs11465804 located at
IL23R, with an unclear functional impact, has been previously associated with susceptibility to AS, but a reduced risk for IBD as well as a lower response to infliximab in contrast to the CC genotype for IBD [
166,
167]. However, in another study, it has been suggested that this SNP may confer risk to IBD in Swedish, Finnish, Hungarian, and Italian populations [
168]. Moreover, this SNP has been associated with uveitis in sarcoidosis in European populations; however, this was not the case with Japanese or Czech cohorts, with the variant being monomorphic in the Japanese population [
169]. On the other hand, the intronic rs1860545 in the
TNFRSF1A gene has been associated with multiple immune-mediated diseases, including AS, CD, UC, and MS through GWASs, but without well-studied functional consequences [
22,
132]. This SNP is in strong linkage disequilibrium with rs1800696, which causes alternative splicing that leads to the loss of exon 6 of the protein (a transmembrane domain), which normally anchors the TNFR1 protein to the cell. As a result, a soluble form of the TNFR1 receptor is produced that acts as a soluble receptor for TNF—similarly to TNF inhibitor drugs like etanercept—and inhibits
TNF signaling. While this may reduce inflammation and be beneficial in AS, it could have the opposite effect in MS, where
TNF inhibition is known to worsen disease, explaining the opposite direction of genetic association [
22]. The intronic rs1250550 is found in the
ZMIZ1 locus and has been previously associated with both AS and MS in GWASs. In AS the G allele is the risk variant, whereas in MS the A allele is the risk factor [
22]. On the contrary, the T allele was associated with a decreased risk of developing pediatric-onset CD and UC, as well as adult-onset CD, as was confirmed by GWASs, with no subsequent studies published since 2016 [
170,
171]. In another study, a significant overlap of this SNP was found within active and weak promoters as well as enhancers of regions that are functionally active in B cells, suggesting that genetic regions associated with MS may induce disease via the dysregulation of B cells [
172]. Lastly, the intergenic rs1495965 has been suggested as a risk factor for AS by multiple studies [
70,
106,
173], and has also been suggested to play a role in CD, as shown in a meta-analysis [
174], as well as Bechet’s uveitis; however, its functional consequences are not understood [
175].
In all diseases, IL23R played a role in disease susceptibility—as has been extensively discussed—and most of the involved variants lie in genomic regions that pertain to immune biological processes. Notably, rs1860545 seemed to play a role in multiple immune-mediated diseases, namely PsO, CD, UC, MS, and sclerosing cholangitis, suggesting a common underlying molecular mechanism. It is important to note, however, that these findings highlight only some of the most frequently referenced genetic variants and do not impose a direct association with the disease, whereas there is still a large portion of genetic associations that remain to be studied. Therefore, more research is needed to fully understand the underlying mechanisms of these diseases.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}