
Cardiovascular Toxicity of Antineoplastic Treatments in Hematological Diseases: Focus on Molecular Mechanisms to Improve Therapeutic Management

Abstract
1. Introduction

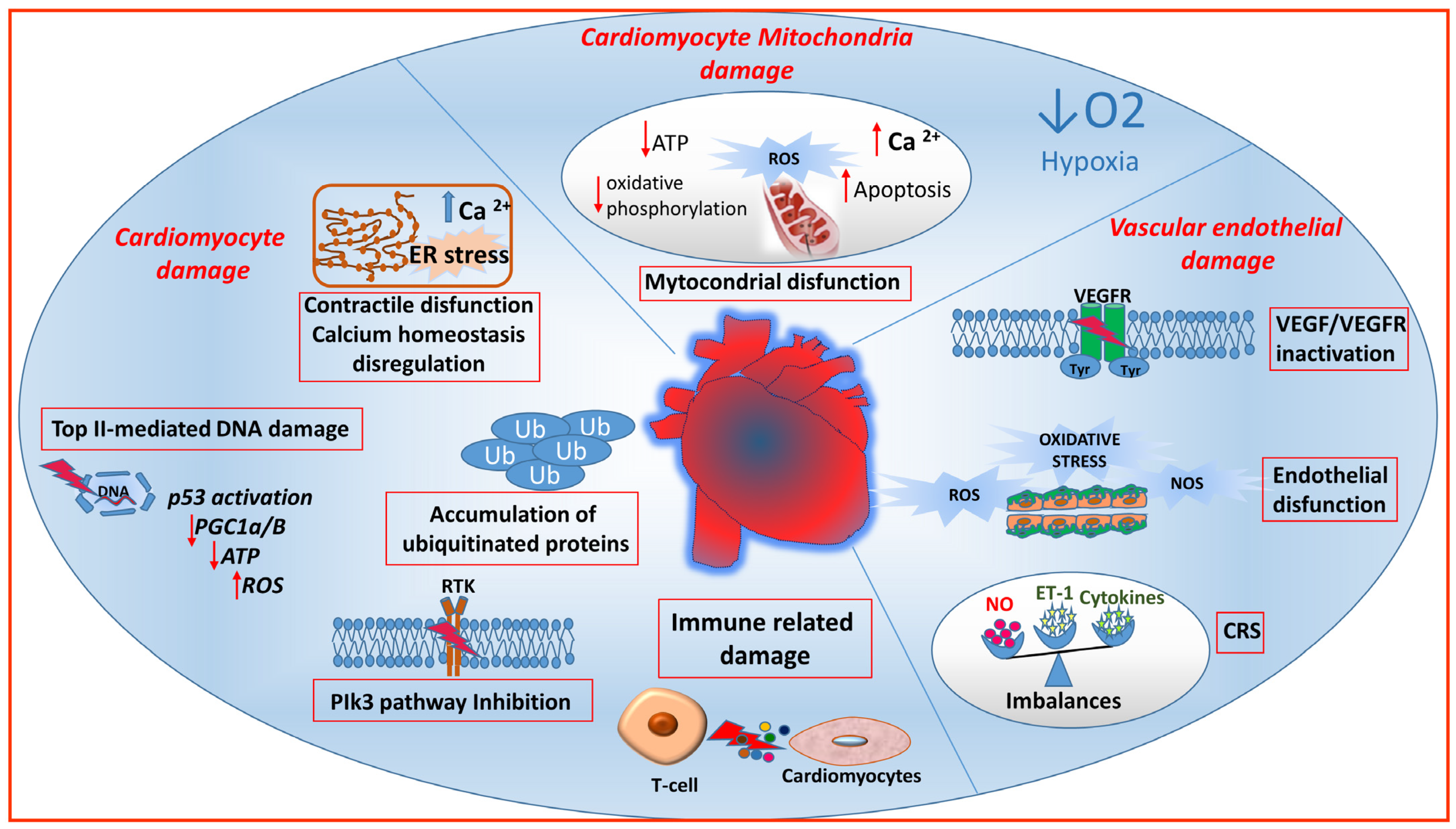
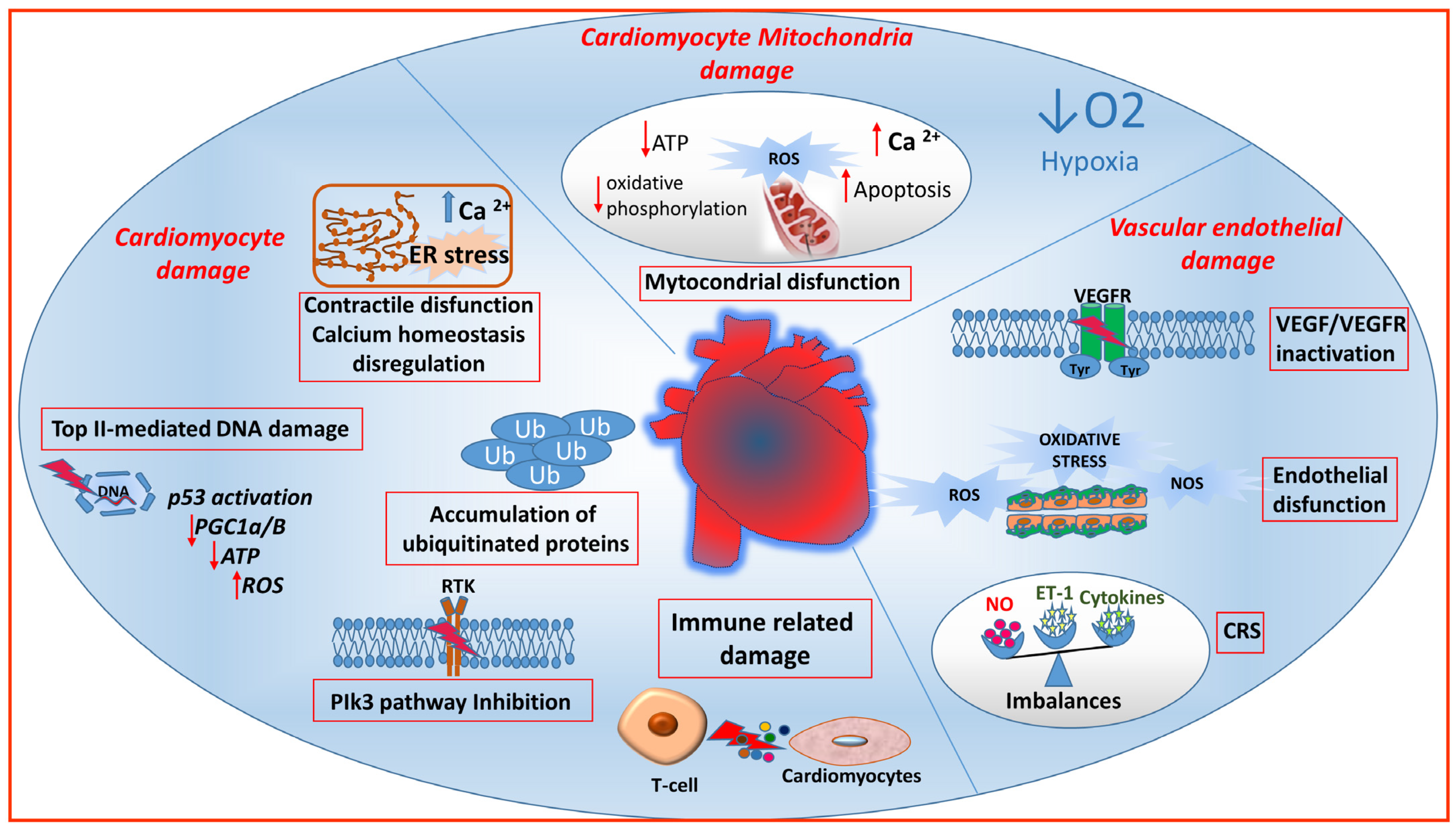{kind=link}
| Class of Drugs | Drug | Mechanism of Action | Adverse Event | Reference |
|---|---|---|---|---|
| Liposomal anthracyclines | Myocet (liposomal doxorubicin) | DNA intercalating agent, topoisomerase 2 inhibitor | Impairment of LVEF, HF | [13,14] |
| Vyxeos (liposomal cytarabine plus daunorubicin) | DNA intercalating agent | QT interval prolongation and left ventricular dysfunction | [15] | |
| BTK inhibitors | Ibrutinib | BTK | AF, ventricular tachycardia, ventricular fibrillation, sudden cardiac death | [16,17,18] |
| Acalabrutinib (ACP-196) | BTK | Ischemic vascular events, AF, hypertension, ventricular arrhythmia, HF | [19] | |
| Zanubrutinib | BTK | AF, hypertension, ventricular arrhythmia | [16,18] | |
| Pirtobrutinib | BTK | AF | [20] | |
| BCR-ABL inhibitors | Imatinib | BCR-ABL, PDGFR-α, and c-KIT | Cerebrovascular accidents | [21,22] |
| Nilotinib | BCR-ABL, PDGFR-α, and c-KIT | Hypertension, ischemic vascular events, prolongation QT, hyperlipidemia, and hyperglycemia | [23,24] | |
| Bosutinib | BCR-ABL, FGFR 2, VEGFR 2, PDGFRβ, SRC | Ischemic vascular events, prolongation QT, hypertension, AF | [21,22] | |
| Ponatinib | ABL, FGFR 1,2 and 3, VEGFR 1, 2, and 3, PDGFR-α, PDGFR-β, c-KIT, SRC, TIE2 | Hypertension, Ischemic vascular events, HF, pulmonary embolism | [25,26,27] | |
| JAK inhibitors | Ruxolitinib | JAK/STAT | AF, dysregulation of lipolysis | [28,29,30] |
| PI3K inhibitors | Copanlisib | PI3K-α/PI3K-δ | Hypertension | [31,32,33,34] |
| HDAC inhibitors | Romidepsin | HDAC enzymes | Prolongation QT | [35,36,37] |
| Belinostat | HDAC enzymes | Prolongation QT | [38] | |
| MoAbs | Rituximab | Anti-CD-20 | Infusion-related reactions, hypertension/hypotension, supraventricular tachycardia, AF | [39] |
| Ofatumumab | Anti-CD-20 | Infusion-related reactions, hypertension/hypotension, tachycardia | [40,41] | |
| Daratumumab | Anti-CD-38 | Infusion-related reactions, AF, cardiac failure, coronary disease | [42] | |
| Brentuximab vedotin | Anti-CD-30 | Sinus tachycardia hyperlipidemia, pulmonary embolism | [43,44,45] | |
| Obinutuzumab | Anti-CD-20 | Infusion-related reactions, ischemic vascular events, AF, HF, pulmonary embolism | [46,47] | |
| Isatuximab | Anti-CD-38 | Infusion-related reactions, AF, | [48] | |
| BsAbs | Blinatumomab | CD-19/CD-3 | CRS, tachycardia, AF, Ischemic vascular events, HF | [49,50,51] |
| Teclistamab | BCMA/CD3 | CRS, thrombocytopenia | [52] | |
| Talquetamab | GPRC5D/CD-3 | CRS | [53] | |
| Mosunetuzumab | CD-20/CD-3 | CRS | [49,54] | |
| Elranatamab | BCMA/CD-3 | CRS, injection site reactions, upper respiratory tract infections | [55,56] | |
| Glofitamab | CD-20/CD-3 | CRS | [49,57] | |
| Odronextamab | CD-20/CD-3 | CRS | [49,58] | |
| Epcoritamab | CD-20/CD-3 | CRS | [49,59] | |
| Alnuctamab | BCMA/CD3 | CRS | [60] | |
| CAR-T | Axicabtagene Ciloleucel | Anti-CD19 CAR T | CRS, atrial fibrillation, HF | [61,62] |
| Tisagenlecleucel | Anti-CD19 CAR T | CRS, ventricular arrhythmias, AF, venous thromboembolic event | [63,64,65] | |
| IMiDs | Lenalidomide | Cereblon protein | Ischemic vascular events, venous thromboembolic events, conduction abnormalities | [66] |
| Pomalidomide | Cereblon protein | Venous thromboembolic events | [67] | |
| Proteasome inhibitors | Bortezomib | Proteasome | Left ventricular dysfunction | [68,69] |
| Carfilzomib | Proteasome | Hypertension, HF, arrhythmias, cardiomyopathy | [68,70,71] | |
| Small molecule proteins | Venetoclax | Anti-BCL2 | Hypertension, AF, cardiomyopathy, cardiac arrhythmia | [72,73,74] |
2. Anthracycline and Liposomal Anthracyclines
3. Alkylating and Platinum-Based Agents
4. Tyrosine Kinase Inhibitors
4.1. BCR-ABL Inhibitors
4.2. Bruton’s Tyrosine Kinase Protein Inhibitors
4.2.1. Janus Kinase Inhibitors
4.2.2. PI3K Inhibitors
5. BCL2 Inhibitors
6. Histone Deacetylase Inhibitors
7. Immunomodulatory Drugs
8. Proteasome Inhibitors
9. Monoclonal Antibody
10. Chimeric Antigen Receptor (CAR-T)
11. Bispecific Antibodies
12. Conclusions
Funding
Conflicts of Interest
References
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef]
- Madanat, L.; Gupta, R.; Weber, P.; Kumar, N.; Chandra, R.; Ahaneku, H.; Bansal, Y.; Anderson, J.; Bilolikar, A.; Jaiyesimi, I. Cardiotoxicity of Biological Therapies in Cancer Patients: An In-depth Review. Curr. Cardiol. Rev. 2023, 19, e310522205428. [Google Scholar] [CrossRef] [PubMed]
- Schamroth Pravda, N.R.; Kornowski, R. Unmet Needs and Therapeutic Strategies in Cardio-Hemato-Oncology. Acta Haematol. 2018, 140, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Corrigendum. Eur. Heart J. 2018, 39, 839. [CrossRef]
- Sheppard, R.J.; Berger, J.; Sebag, I.A. Cardiotoxicity of cancer therapeutics: Current issues in screening, prevention, and therapy. Front. Pharmacol. 2013, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Barachini, S.; Ghelardoni, S.; Varga, Z.V.; Mehanna, R.A.; Montt-Guevara, M.M.; Ferdinandy, P.; Madonna, R. Antineoplastic drugs inducing cardiac and vascular toxicity—An update. Vasc. Pharmacol. 2023, 153, 107223. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxidative Med. Cell. Longev. 2018, 2018, 7582730. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, Y.; Xu, C.; An, P.; Luo, Y.; Jiao, L.; Luo, J.; Li, Y. Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 16053. [Google Scholar] [CrossRef]
- Sun, S.; Qin, J.; Liao, W.; Gao, X.; Shang, Z.; Luo, D.; Xiong, S. Mitochondrial Dysfunction in Cardiotoxicity Induced by BCR-ABL1 Tyrosine Kinase Inhibitors -Underlying Mechanisms, Detection, Potential Therapies. Cardiovasc. Toxicol. 2023, 23, 233–254. [Google Scholar] [CrossRef]
- Hsu, P.Y.; Mammadova, A.; Benkirane-Jessel, N.; Désaubry, L.; Nebigil, C.G. Updates on Anticancer Therapy-Mediated Vascular Toxicity and New Horizons in Therapeutic Strategies. Front. Cardiovasc. Med. 2021, 8, 694711. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [PubMed]
- Nagykálnai, T. Non-pegylated doxorubicin (Myocet®) as the less cardiotoxic alternative of free doxorubicin. Magy. Onkol. 2010, 54, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Davidson, N.; Juozaityte, E.; Erdkamp, F.; Pluzanska, A.; Azarnia, N.; Lee, L.W. Phase III trial of liposomal doxorubicin and cyclophosphamide compared with epirubicin and cyclophosphamide as first-line therapy for metastatic breast cancer. Ann. Oncol. 2004, 15, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Newell, L.F.; Stuart, R.K.; Michaelis, L.C.; Rubenstein, E.; Pentikis, H.S.; Callahan, T.; Alvarez, D.; Liboiron, B.D.; Mayer, L.D.; et al. A phase 2 study to assess the pharmacokinetics and pharmacodynamics of CPX-351 and its effects on cardiac repolarization in patients with acute leukemias. Cancer Chemother. Pharmacol. 2019, 84, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Eichhorst, B.; Hillmen, P.; Jurczak, W.; Kaźmierczak, M.; Lamanna, N.; O’Brien, S.M.; Tam, C.S.; Qiu, L.; Zhou, K.; et al. Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023, 388, 319–332. [Google Scholar] [CrossRef]
- Miklos, D.; Cutler, C.S.; Arora, M.; Waller, E.K.; Jagasia, M.; Pusic, I.; Flowers, M.E.; Logan, A.C.; Nakamura, R.; Blazar, B.R.; et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 2017, 130, 2243–2250. [Google Scholar] [CrossRef]
- Hillmen, P.; Eichhorst, B.; Brown, J.R.; Lamanna, N.; O’Brien, S.M.; Tam, C.S.; Qiu, L.; Kazmierczak, M.; Zhou, K.; Šimkovič, M.; et al. Zanubrutinib Versus Ibrutinib in Relapsed/Refractory Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma: Interim Analysis of a Randomized Phase III Trial. J. Clin. Oncol. 2023, 41, 1035–1045. [Google Scholar] [CrossRef]
- Wang, M.; Rule, S.; Zinzani, P.L.; Goy, A.; Casasnovas, O.; Smith, S.D.; Damaj, G.; Doorduijn, J.; Lamy, T.; Morschhauser, F.; et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): A single-arm, multicentre, phase 2 trial. Lancet 2018, 391, 659–667. [Google Scholar] [CrossRef]
- Quartermaine, C.; Ghazi, S.M.; Yasin, A.; Awan, F.T.; Fradley, M.; Wiczer, T.; Kalathoor, S.; Ferdousi, M.; Krishan, S.; Habib, A.; et al. Cardiovascular Toxicities of BTK Inhibitors in Chronic Lymphocytic Leukemia. JACC CardioOncol. 2023, 5, 570–590. [Google Scholar] [CrossRef]
- Brümmendorf, T.H.; Cortes, J.E.; Milojkovic, D.; Gambacorti-Passerini, C.; Clark, R.E.; le Coutre, P.; Garcia-Gutierrez, V.; Chuah, C.; Kota, V.; Lipton, J.H.; et al. Bosutinib versus imatinib for newly diagnosed chronic phase chronic myeloid leukemia: Final results from the BFORE trial. Leukemia 2022, 36, 1825–1833. [Google Scholar] [CrossRef]
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; le Coutre, P.; et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J. Clin. Oncol. 2018, 36, 231–237. [Google Scholar] [CrossRef]
- Hochhaus, A.; Saglio, G.; Hughes, T.P.; Larson, R.A.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.D.; Etienne, G.; Dorlhiac-Llacer, P.E.; Clark, R.E.; et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 2016, 30, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Hughes, T.P.; Larson, R.A.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Boquimpani, C.; Pasquini, R.; Clark, R.E.; et al. Long-term outcomes with frontline nilotinib versus imatinib in newly diagnosed chronic myeloid leukemia in chronic phase: ENESTnd 10-year analysis. Leukemia 2021, 35, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.D.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: Final 5-year results of the phase 2 PACE trial. Blood 2018, 132, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Short, N.J.; Jain, N.; Huang, X.; Montalban-Bravo, G.; Banerjee, P.; Rezvani, K.; Jiang, X.; Kim, K.H.; Kanagal-Shamanna, R.; et al. Ponatinib and blinatumomab for Philadelphia chromosome-positive acute lymphoblastic leukaemia: A US, single-centre, single-arm, phase 2 trial. Lancet Haematol. 2023, 10, e24–e34. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Bose, P.; Rampal, R.; Gerds, A.T.; Fleischman, A.; Verstovsek, S. Ten years after ruxolitinib approval for myelofibrosis: A review of clinical efficacy. Leuk. Lymphoma 2023, 64, 1063–1081. [Google Scholar] [CrossRef] [PubMed]
- von Bubnoff, N.; Ihorst, G.; Grishina, O.; Röthling, N.; Bertz, H.; Duyster, J.; Finke, J.; Zeiser, R. Ruxolitinib in GvHD (RIG) study: A multicenter, randomized phase 2 trial to determine the response rate of Ruxolitinib and best available treatment (BAT) versus BAT in steroid-refractory acute graft-versus-host disease (aGvHD) (NCT02396628). BMC Cancer 2018, 18, 1132. [Google Scholar] [CrossRef]
- Sapre, M.; Tremblay, D.; Wilck, E.; James, A.; Leiter, A.; Coltoff, A.; Koshy, A.G.; Kremyanskaya, M.; Hoffman, R.; Mascarenhas, J.O.; et al. Metabolic Effects of JAK1/2 Inhibition in Patients with Myeloproliferative Neoplasms. Sci. Rep. 2019, 9, 16609. [Google Scholar] [CrossRef]
- Patnaik, A.; Appleman, L.J.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Weiss, G.J.; Sachdev, J.C.; Chadha, M.; Fulk, M.; et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. 2016, 27, 1928–1940. [Google Scholar] [CrossRef]
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppä, S.; Follows, G.; Lenz, G.; Kim, W.S.; Nagler, A.; Dimou, M.; Demeter, J.; et al. Long-term safety and efficacy of the PI3K inhibitor copanlisib in patients with relapsed or refractory indolent lymphoma: 2-year follow-up of the CHRONOS-1 study. Am. J. Hematol. 2020, 95, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Hawkes, E.; Verhoef, G.; Haioun, C.; Thye Lim, S.; Seog Heo, D.; Ardeshna, K.; Chong, G.; Haaber, J.; Shi, W.; et al. Single-agent activity of phosphatidylinositol 3-kinase inhibition with copanlisib in patients with molecularly defined relapsed or refractory diffuse large B-cell lymphoma. Leukemia 2020, 34, 2184–2197. [Google Scholar] [CrossRef] [PubMed]
- Dreyling, M.; Morschhauser, F.; Bouabdallah, K.; Bron, D.; Cunningham, D.; Assouline, S.E.; Verhoef, G.; Linton, K.; Thieblemont, C.; Vitolo, U.; et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann. Oncol. 2017, 28, 2169–2178. [Google Scholar] [CrossRef]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Morschhauser, F.; Wilhelm, M.; Pinter-Brown, L.; et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: Pivotal study update demonstrates durable responses. J. Hematol. Oncol. 2014, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Bachy, E.; Camus, V.; Thieblemont, C.; Sibon, D.; Casasnovas, R.O.; Ysebaert, L.; Damaj, G.; Guidez, S.; Pica, G.M.; Kim, W.S.; et al. Romidepsin Plus CHOP Versus CHOP in Patients with Previously Untreated Peripheral T-Cell Lymphoma: Results of the Ro-CHOP Phase III Study (Conducted by LYSA). J. Clin. Oncol. 2022, 40, 242–251. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef]
- Matasar, M.J.; Capra, M.; Özcan, M.; Lv, F.; Li, W.; Yañez, E.; Sapunarova, K.; Lin, T.; Jin, J.; Jurczak, W.; et al. Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 678–689. [Google Scholar] [CrossRef]
- van Oers, M.H.; Kuliczkowski, K.; Smolej, L.; Petrini, M.; Offner, F.; Grosicki, S.; Levin, M.D.; Gupta, I.; Phillips, J.; Williams, V.; et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): An open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015, 16, 1370–1379. [Google Scholar] [CrossRef]
- van Oers, M.; Smolej, L.; Petrini, M.; Offner, F.; Grosicki, S.; Levin, M.D.; Davis, J.; Banerjee, H.; Stefanelli, T.; Hoever, P.; et al. Ofatumumab maintenance prolongs progression-free survival in relapsed chronic lymphocytic leukemia: Final analysis of the PROLONG study. Blood Cancer J. 2019, 9, 98. [Google Scholar] [CrossRef]
- Al-Yafeai, Z.; Ghoweba, M.; Ananthaneni, A.; Abduljabar, H.; Aziz, D. Cardiovascular complications of modern multiple myeloma therapy: A pharmacovigilance study. Br. J. Clin. Pharmacol. 2023, 89, 641–648. [Google Scholar] [CrossRef]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef]
- Prince, H.M.; Kim, Y.H.; Horwitz, S.M.; Dummer, R.; Scarisbrick, J.; Quaglino, P.; Zinzani, P.L.; Wolter, P.; Sanches, J.A.; Ortiz-Romero, P.L.; et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): An international, open-label, randomised, phase 3, multicentre trial. Lancet 2017, 390, 555–566. [Google Scholar] [CrossRef]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Trümper, L.; Iyer, S.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann. Oncol. 2022, 33, 288–298. [Google Scholar] [CrossRef]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Chua, N.; Mayer, J.; Dueck, G.; Trněný, M.; Bouabdallah, K.; Fowler, N.; Delwail, V.; Press, O.; Salles, G.; et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): A randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016, 17, 1081–1093. [Google Scholar] [CrossRef]
- Shen, F.; Shen, W. Isatuximab in the Treatment of Multiple Myeloma: A Review and Comparison with Daratumumab. Technol. Cancer Res. Treat. 2022, 21, 15330338221106563. [Google Scholar] [CrossRef]
- Bock, A.M.; Nowakowski, G.S.; Wang, Y. Bispecific Antibodies for Non-Hodgkin Lymphoma Treatment. Curr. Treat. Options Oncol. 2022, 23, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The landscape of bispecific T cell engager in cancer treatment. Biomark. Res. 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Martino, E.A.; Bruzzese, A.; Labanca, C.; Mendicino, F.; Lucia, E.; Olivito, V.; Neri, A.; Morabito, F.; Vigna, E.; Gentile, M. Teclistamab-cqyv in multiple myeloma. Eur. J. Haematol. 2023, 112, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Minnema, M.C.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.V.; Costa, L.J.; Caers, J.; et al. Talquetamab, a T-Cell-Redirecting GPRC5D Bispecific Antibody for Multiple Myeloma. N. Engl. J. Med. 2022, 387, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Kang, C. Mosunetuzumab: First Approval. Drugs 2022, 82, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Tomasson, M.H.; Arnulf, B.; Bahlis, N.J.; Miles Prince, H.; Niesvizky, R.; Rodrίguez-Otero, P.; Martinez-Lopez, J.; Koehne, G.; Touzeau, C.; et al. Elranatamab in relapsed or refractory multiple myeloma: Phase 2 MagnetisMM-3 trial results. Nat. Med. 2023, 29, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Rais, T.; Khan, A.; Riaz, R. Elrexfio™ (elranatamab-bcmm): The game-changer in treatment of multiple myeloma. Rare Tumors 2023, 15, 20363613231207483. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Glofitamab: First Approval. Drugs 2023, 83, 935–941. [Google Scholar] [CrossRef]
- Bannerji, R.; Arnason, J.E.; Advani, R.H.; Brown, J.R.; Allan, J.N.; Ansell, S.M.; Barnes, J.A.; O’Brien, S.M.; Chávez, J.C.; Duell, J.; et al. Odronextamab, a human CD20×CD3 bispecific antibody in patients with CD20-positive B-cell malignancies (ELM-1): Results from the relapsed or refractory non-Hodgkin lymphoma cohort in a single-arm, multicentre, phase 1 trial. Lancet Haematol. 2022, 9, e327–e339. [Google Scholar] [CrossRef]
- Thieblemont, C.; Phillips, T.; Ghesquieres, H.; Cheah, C.Y.; Clausen, M.R.; Cunningham, D.; Do, Y.R.; Feldman, T.; Gasiorowski, R.; Jurczak, W.; et al. Epcoritamab, a Novel, Subcutaneous CD3xCD20 Bispecific T-Cell-Engaging Antibody, in Relapsed or Refractory Large B-Cell Lymphoma: Dose Expansion in a Phase I/II Trial. J. Clin. Oncol. 2023, 41, 2238–2247. [Google Scholar] [CrossRef]
- Landgren, O.; Nadeem, O. Bispecific Monoclonal Antibodies in Multiple Myeloma: Data from ASH 2022: A Podcast. Adv. Ther. 2023, 40, 3291–3303. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.; Maor, E.; Bomze, D.; Liu, J.E.; Herrmann, J.; Fein, J.; Steingart, R.M.; Mahmood, S.S.; Schaffer, W.L.; Perales, M.A.; et al. Adverse Cardiovascular and Pulmonary Events Associated with Chimeric Antigen Receptor T-Cell Therapy. J. Am. Coll. Cardiol. 2021, 78, 1800–1813. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V. Immunotherapy: Tisagenlecleucel—The first approved CAR-T-cell therapy: Implications for payers and policy makers. Nat. Rev. Clin. Oncol. 2018, 15, 11–12. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Holstein, S.A.; Jung, S.H.; Richardson, P.G.; Hofmeister, C.C.; Hurd, D.D.; Hassoun, H.; Giralt, S.; Stadtmauer, E.A.; Weisdorf, D.J.; Vij, R.; et al. Updated analysis of CALGB (Alliance) 100104 assessing lenalidomide versus placebo maintenance after single autologous stem-cell transplantation for multiple myeloma: A randomised, double-blind, phase 3 trial. Lancet Haematol. 2017, 4, e431–e442. [Google Scholar] [CrossRef]
- Miguel, J.S.; Weisel, K.; Moreau, P.; Lacy, M.; Song, K.; Delforge, M.; Karlin, L.; Goldschmidt, H.; Banos, A.; Oriol, A.; et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 1055–1066. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hajek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, L.; Liu, M.; Wang, F.; Xiong, Y.; Tang, Z.; Li, Q.; Lu, Q.; Liang, S.; Niu, T.; et al. Myocardial Injury in Multiple Myeloma Patients with Preserved Left Ventricular Ejection Fraction: Noninvasive Left Ventricular Pressure-Strain Myocardial Work. Front. Cardiovasc. Med. 2021, 8, 782580. [Google Scholar] [CrossRef] [PubMed]
- Atrash, S.; Tullos, A.; Panozzo, S.; Bhutani, M.; Van Rhee, F.; Barlogie, B.; Usmani, S.Z. Cardiac complications in relapsed and refractory multiple myeloma patients treated with carfilzomib. Blood Cancer J. 2015, 5, e272. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.B.; Hobbs, M.A.; Abeykoon, J.P.; Kapoor, P. Common Adverse Effects of Novel Therapies for Multiple Myeloma (MM) and Their Management Strategies. Curr. Hematol. Malig. Rep. 2018, 13, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Mihalyova, J.; Jelinek, T.; Growkova, K.; Hrdinka, M.; Simicek, M.; Hajek, R. Venetoclax: A new wave in hematooncology. Exp. Hematol. 2018, 61, 10–25. [Google Scholar] [CrossRef] [PubMed]
- AlAsmari, A.F.; Alghamdi, A.; Ali, N.; Almeaikl, M.A.; Hakami, H.M.; Alyousef, M.K.; AlSwayyed, M.; Alharbi, M.; Alqahtani, F.; Alasmari, F.; et al. Venetoclax Induces Cardiotoxicity through Modulation of Oxidative-Stress-Mediated Cardiac Inflammation and Apoptosis via NF-κB and BCL-2 Pathway. Int. J. Mol. Sci. 2022, 23, 6260. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef]
- Mulrooney, D.A.; Hyun, G.; Ness, K.K.; Ehrhardt, M.J.; Yasui, Y.; Duprez, D.; Howell, R.M.; Leisenring, W.M.; Constine, L.S.; Tonorezos, E.; et al. Major cardiac events for adult survivors of childhood cancer diagnosed between 1970 and 1999: Report from the Childhood Cancer Survivor Study cohort. BMJ 2020, 368, l6794. [Google Scholar] [CrossRef]
- Meyer, M.; Seetharam, M. First-Line Therapy for Metastatic Soft Tissue Sarcoma. Curr. Treat. Options Oncol. 2019, 20, 6. [Google Scholar] [CrossRef]
- Giordano, S.H.; Lin, Y.L.; Kuo, Y.F.; Hortobagyi, G.N.; Goodwin, J.S. Decline in the use of anthracyclines for breast cancer. J. Clin. Oncol. 2012, 30, 2232–2239. [Google Scholar] [CrossRef]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef]
- Liesse, K.; Harris, J.; Chan, M.; Schmidt, M.L.; Chiu, B. Dexrazoxane Significantly Reduces Anthracycline-induced Cardiotoxicity in Pediatric Solid Tumor Patients: A Systematic Review. J. Pediatr. Hematol. Oncol. 2018, 40, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Mason, J.W.; Billingham, M.E.; Daniels, J.R. Doxorubicin cardiomyopathy: Evaluation by phonocardiography, endomyocardial biopsy, and cardiac catheterization. Ann. Intern. Med. 1978, 88, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Nousiainen, T.; Jantunen, E.; Vanninen, E.; Hartikainen, J. Early decline in left ventricular ejection fraction predicts doxorubicin cardiotoxicity in lymphoma patients. Br. J. Cancer 2002, 86, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol. Toxicol. 2003, 93, 105–115. [Google Scholar] [CrossRef]
- Santovito, D.; Steffens, S.; Barachini, S.; Madonna, R. Autophagy, innate immunity, and cardiac disease. Front. Cell Dev. Biol. 2023, 11, 1149409. [Google Scholar] [CrossRef]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef]
- Xu, H.; Yu, W.; Sun, S.; Li, C.; Zhang, Y.; Ren, J. Luteolin Attenuates Doxorubicin-Induced Cardiotoxicity Through Promoting Mitochondrial Autophagy. Front. Physiol. 2020, 11, 113. [Google Scholar] [CrossRef]
- Ghigo, A.; Li, M.; Hirsch, E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim. Biophys. Acta 2016, 1863 Pt B, 1916–1925. [Google Scholar] [CrossRef]
- Kitajima, N.; Numaga-Tomita, T.; Watanabe, M.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; et al. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep. 2016, 6, 37001. [Google Scholar] [CrossRef]
- Nishiyama, K.; Numaga-Tomita, T.; Fujimoto, Y.; Tanaka, T.; Toyama, C.; Nishimura, A.; Yamashita, T.; Matsunaga, N.; Koyanagi, S.; Azuma, Y.T.; et al. Ibudilast attenuates doxorubicin-induced cytotoxicity by suppressing formation of TRPC3 channel and NADPH oxidase 2 protein complexes. Br. J. Pharmacol. 2019, 176, 3723–3738. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2023, 8, e132747. [Google Scholar] [CrossRef]
- Shi, S.; Chen, Y.; Luo, Z.; Nie, G.; Dai, Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun. Signal. 2023, 21, 61. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Kashiwaya, Y.; Haskó, G.; Liaudet, L.; Szabó, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef]
- Clayton, Z.S.; Brunt, V.E.; Hutton, D.A.; VanDongen, N.S.; D’Alessandro, A.; Reisz, J.A.; Ziemba, B.P.; Seals, D.R. Doxorubicin-Induced Oxidative Stress and Endothelial Dysfunction in Conduit Arteries Is Prevented by Mitochondrial-Specific Antioxidant Treatment. JACC CardioOncol. 2020, 2, 475–488. [Google Scholar] [CrossRef]
- Li, X.; Gu, J.; Zhang, Y.; Feng, S.; Huang, X.; Jiang, Y.; Xia, Y.; Liu, Y.; Yang, X. l-arginine alleviates doxorubicin-induced endothelium-dependent dysfunction by promoting nitric oxide generation and inhibiting apoptosis. Toxicology 2019, 423, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Räsänen, M.; Degerman, J.; Nissinen, T.A.; Miinalainen, I.; Kerkelä, R.; Siltanen, A.; Backman, J.T.; Mervaala, E.; Hulmi, J.J.; Kivelä, R.; et al. VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc. Natl. Acad. Sci. USA 2016, 113, 13144–13149. [Google Scholar] [CrossRef] [PubMed]
- Faridvand, Y.; Haddadi, P.; Vahedian, V.; Nozari, S.; Nejabati, H.R.; Pezeshkian, M.; Afrasiabi, A.; Safaie, N.; Jodati, A.; Nouri, M. Human Amnion Membrane Proteins Prevent Doxorubicin-Induced Oxidative Stress Injury and Apoptosis in Rat H9c2 Cardiomyocytes. Cardiovasc. Toxicol. 2020, 20, 370–379. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, W.; Shou, W.; Field, L.J. P53 inhibition exacerbates late-stage anthracycline cardiotoxicity. Cardiovasc. Res. 2014, 103, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Klampfer, L. Signal transducers and activators of transcription (STATs): Novel targets of chemopreventive and chemotherapeutic drugs. Curr. Cancer Drug Targets 2006, 6, 107–121. [Google Scholar] [CrossRef]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-κB in the heart: To be or not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Q.; Li, W.; Zhang, Q.; Jiang, Y.; Guo, D.; Sun, X.; Lu, W.; Li, C.; Wang, Y. TFEB-NF-κB inflammatory signaling axis: A novel therapeutic pathway of Dihydrotanshinone I in doxorubicin-induced cardiotoxicity. J. Exp. Clin. Cancer Res. 2020, 39, 93. [Google Scholar] [CrossRef]
- Narezkina, A.; Narayan, H.K.; Zemljic-Harpf, A.E. Molecular mechanisms of anthracycline cardiovascular toxicity. Clin. Sci. 2021, 135, 1311–1332. [Google Scholar] [CrossRef]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflamm. 2005, 2, 29. [Google Scholar] [CrossRef]
- Wolfram, J.; Ferrari, M. Clinical Cancer Nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Orciuolo, E.; Buda, G.; Pelosini, M.; Petrini, M. Fludarabine, Bortezomib, Myocet and rituximab chemotherapy in relapsed and refractory mantle cell lymphoma. Br. J. Haematol. 2010, 148, 810–812. [Google Scholar] [CrossRef] [PubMed]
- Batist, G. Cardiac safety of liposomal anthracyclines. Cardiovasc. Toxicol. 2007, 7, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Swenson, C.E.; Bolcsak, L.E.; Batist, G.; Guthrie, T.H.; Tkaczuk, K.H.; Boxenbaum, H.; Welles, L.; Chow, S.C.; Bhamra, R.; Chaikin, P. Pharmacokinetics of doxorubicin administered i.v. as Myocet (TLC D-99; liposome-encapsulated doxorubicin citrate) compared with conventional doxorubicin when given in combination with cyclophosphamide in patients with metastatic breast cancer. Anticancer Drugs 2003, 14, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Raber, I.; Asnani, A. Cardioprotection in cancer therapy: Novel insights with anthracyclines. Cardiovasc. Res. 2019, 115, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Omland, T.; Heck, S.L.; Gulati, G. The Role of Cardioprotection in Cancer Therapy Cardiotoxicity. JACC CardioOncol. 2022, 4, 19–37. [Google Scholar] [CrossRef]
- Lankhorst, S.; Danser, A.H.; van den Meiracker, A.H. Endothelin-1 and antiangiogenesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R230–R234. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.C.; Touyz, R.M.; Lang, N.N. Vascular Complications of Cancer Chemotherapy. Can. J. Cardiol. 2016, 32, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Dugbartey, G.J.; Peppone, L.J.; de Graaf, I.A. An integrative view of cisplatin-induced renal and cardiac toxicities: Molecular mechanisms, current treatment challenges and potential protective measures. Toxicology 2016, 371, 58–66. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, E.-S.; Moustafa, Y.M.; Abo-Elmatty, D.M.; Radwan, A. Cisplatin-induced cardiotoxicity: Mechanisms and cardioprotective strategies. Eur. J. Pharmacol. 2011, 650, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Iqubal, M.K.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Ali, S.M.; Ali, J.; Haque, S.E. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: Old drug with a new vision. Life Sci. 2019, 218, 112–131. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.G.; Hunter, K.; Salloum, F.N. Cardiac complications of cancer therapies. Adv. Cancer Res. 2022, 155, 167–214. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Ky, B. Common Cardiovascular Complications of Cancer Therapy: Epidemiology, Risk Prediction, and Prevention. Annu. Rev. Med. 2018, 69, 97–111. [Google Scholar] [CrossRef]
- Kurauchi, K.; Nishikawa, T.; Miyahara, E.; Okamoto, Y.; Kawano, Y. Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res. Notes 2017, 10, 406. [Google Scholar] [CrossRef]
- Herradón, E.; González, C.; Uranga, J.A.; Abalo, R.; Martín, M.I.; López-Miranda, V. Characterization of Cardiovascular Alterations Induced by Different Chronic Cisplatin Treatments. Front. Pharmacol. 2017, 8, 196. [Google Scholar] [CrossRef]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Michels, K.R.; Lukacs, N.W.; Fonseca, W. TLR Activation and Allergic Disease: Early Life Microbiome and Treatment. Curr. Allergy Asthma Rep. 2018, 18, 61. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Li, L.; Sun, Y.; Yang, H.; Ye, Z.; Zhao, J. Effects of the TLR4/Myd88/NF-κB Signaling Pathway on NLRP3 Inflammasome in Coronary Microembolization-Induced Myocardial Injury. Cell Physiol. Biochem. 2018, 47, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, H.; Wang, D.; Zhang, L.; Ma, T. Peroxiredoxin2 (Prdx2) Reduces Oxidative Stress and Apoptosis of Myocardial Cells Induced by Acute Myocardial Infarction by Inhibiting the TLR4/Nuclear Factor kappa B (NF-κB) Signaling Pathway. Med. Sci. Monit. 2020, 26, e926281. [Google Scholar] [CrossRef]
- Chen, F.; Chen, Z.Q.; Zhong, G.L.; Zhu, J.J. Nicorandil inhibits TLR4/MyD88/NF-κB/NLRP3 signaling pathway to reduce pyroptosis in rats with myocardial infarction. Exp. Biol. Med. 2021, 246, 1938–1947. [Google Scholar] [CrossRef]
- Wang, X.Z.; Yu, Z.X.; Nie, B.; Chen, D.M. Perindopril inhibits myocardial apoptosis in mice with acute myocardial infarction through TLR4/NF-κB pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6672–6682. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: Molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 329. [Google Scholar] [CrossRef]
- Hehlmann, R. Innovation in hematology. Perspectives: CML 2016. Haematologica 2016, 101, 657–659. [Google Scholar] [CrossRef]
- Manouchehri, A.; Kanu, E.; Mauro, M.J.; Aday, A.W.; Lindner, J.R.; Moslehi, J. Tyrosine Kinase Inhibitors in Leukemia and Cardiovascular Events: From Mechanism to Patient Care. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 301–308. [Google Scholar] [CrossRef]
- Hoffmann, V.S.; Baccarani, M.; Hasford, J.; Castagnetti, F.; Di Raimondo, F.; Casado, L.F.; Turkina, A.; Zackova, D.; Ossenkoppele, G.; Zaritskey, A.; et al. Treatment and outcome of 2904 CML patients from the EUTOS population-based registry. Leukemia 2017, 31, 593–601. [Google Scholar] [CrossRef]
- Coutinho, A.D.; Makenbaeva, D.; Farrelly, E.; Landsman-Blumberg, P.B.; Lenihan, D. Elevated Cardiovascular Disease Risk in Patients with Chronic Myelogenous Leukemia Seen in Community-based Oncology Practices in the United States. Clin. Lymphoma Myeloma Leuk. 2017, 17, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Lamore, S.D.; Ahlberg, E.; Boyer, S.; Lamb, M.L.; Hortigon-Vinagre, M.P.; Rodriguez, V.; Smith, G.L.; Sagemark, J.; Carlsson, L.; Bates, S.M.; et al. Deconvoluting Kinase Inhibitor Induced Cardiotoxicity. Toxicol. Sci. 2017, 158, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Elmadani, M.; Raatikainen, S.; Mattila, O.; Alakoski, T.; Piuhola, J.; Åström, P.; Tenhunen, O.; Magga, J.; Kerkelä, R. Dasatinib targets c-Src kinase in cardiotoxicity. Toxicol. Rep. 2023, 10, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.L.; Wang, C.; Gu, J.L.; Ma, X.L.; Kumar, S.; Lee, J.C.; Feuerstein, G.Z.; Thomas, H.; Maleeff, B.; Ohlstein, E.H. Inhibition of extracellular signal-regulated kinase enhances Ischemia/Reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ. Res. 2000, 86, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.F.; Shannon, M.F.; Barry, S.; Phillips, J.A.; Cambareri, B.; Dottore, M.; Simmons, P.; Vadas, M.A. A human interleukin 3 analog with increased biological and binding activities. Proc. Natl. Acad. Sci. USA 1992, 89, 11842–11846. [Google Scholar] [CrossRef] [PubMed]
- Kerkelä, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Sukegawa, M.; Wang, X.; Nishioka, C.; Pan, B.; Xu, K.; Ohkawara, H.; Hamasaki, Y.; Mita, M.; Nakamura, K.; Okamoto, M.; et al. The BCR/ABL tyrosine kinase inhibitor, nilotinib, stimulates expression of IL-1β in vascular endothelium in association with downregulation of miR-3p. Leuk. Res. 2017, 58, 83–90. [Google Scholar] [CrossRef]
- Hadzijusufovic, E.; Albrecht-Schgoer, K.; Huber, K.; Hoermann, G.; Grebien, F.; Eisenwort, G.; Schgoer, W.; Herndlhofer, S.; Kaun, C.; Theurl, M.; et al. Nilotinib-induced vasculopathy: Identification of vascular endothelial cells as a primary target site. Leukemia 2017, 31, 2388–2397. [Google Scholar] [CrossRef]
- Alhawiti, N.; Burbury, K.L.; Kwa, F.A.; O’Malley, C.J.; Shuttleworth, P.; Alzard, M.; Hamadi, A.; Grigg, A.P.; Jackson, D.E. The tyrosine kinase inhibitor, nilotinib potentiates a prothrombotic state. Thromb. Res. 2016, 145, 54–64. [Google Scholar] [CrossRef]
- Caocci, G.; Mulas, O.; Annunziata, M.; Luciano, L.; Abruzzese, E.; Bonifacio, M.; Orlandi, E.M.; Albano, F.; Galimberti, S.; Iurlo, A.; et al. Long-term mortality rate for cardiovascular disease in 656 chronic myeloid leukaemia patients treated with second- and third-generation tyrosine kinase inhibitors. Int. J. Cardiol. 2020, 301, 163–166. [Google Scholar] [CrossRef]
- Latifi, Y.; Moccetti, F.; Wu, M.; Xie, A.; Packwood, W.; Qi, Y.; Ozawa, K.; Shentu, W.; Brown, E.; Shirai, T.; et al. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood 2019, 133, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor-Associated Hypertension and Vascular Disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef]
- Singh, A.P.; Umbarkar, P.; Tousif, S.; Lal, H. Cardiotoxicity of the BCR-ABL1 tyrosine kinase inhibitors: Emphasis on ponatinib. Int. J. Cardiol. 2020, 316, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Pieragostino, D.; Cufaro, M.C.; Doria, V.; Del Boccio, P.; Deidda, M.; Pierdomenico, S.D.; Dessalvi, C.C.; De Caterina, R.; Mercuro, G. Ponatinib Induces Vascular Toxicity through the Notch-1 Signaling Pathway. J. Clin. Med. 2020, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Barachini, S.; Moscato, S.; Ippolito, C.; Mattii, L.; Lenzi, C.; Balistreri, C.R.; Zucchi, R.; De Caterina, R. Sodium-glucose cotransporter type 2 inhibitors prevent ponatinib-induced endothelial senescence and disfunction: A potential rescue strategy. Vasc. Pharmacol. 2022, 142, 106949. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Glennon, M.S.; Umbarkar, P.; Gupte, M.; Galindo, C.L.; Zhang, Q.; Force, T.; Becker, J.R.; Lal, H. Ponatinib-induced cardiotoxicity: Delineating the signalling mechanisms and potential rescue strategies. Cardiovasc. Res. 2019, 115, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Dorer, D.J.; Knickerbocker, R.K.; Baccarani, M.; Cortes, J.E.; Hochhaus, A.; Talpaz, M.; Haluska, F.G. Impact of dose intensity of ponatinib on selected adverse events: Multivariate analyses from a pooled population of clinical trial patients. Leuk. Res. 2016, 48, 84–91. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Fazal, M.; Wei, C.; Chuy, K.L.; Hussain, K.; Gomez, S.E.; Ba, S.S.; Pietrasik, G.; Yadav, N.; Ghazizadeh, Z.; Kapoor, R.; et al. Tyrosine kinase inhibitor-associated ventricular arrhythmias: A case series and review of literature. J. Interv. Card. Electrophysiol. 2023, 66, 1165–1175. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Garcia-Sanz, R.; et al. Zanubrutinib Versus Ibrutinib in Symptomatic Waldenström Macroglobulinemia: Final Analysis From the Randomized Phase III ASPEN Study. J. Clin. Oncol. 2023, 41, 5099–5106. [Google Scholar] [CrossRef]
- Tuomi, J.M.; Bohne, L.J.; Dorey, T.W.; Jansen, H.J.; Liu, Y.; Jones, D.L.; Rose, R.A. Distinct Effects of Ibrutinib and Acalabrutinib on Mouse Atrial and Sinoatrial Node Electrophysiology and Arrhythmogenesis. J. Am. Heart Assoc. 2021, 10, e022369. [Google Scholar] [CrossRef]
- Xiao, L.; Salem, J.E.; Clauss, S.; Hanley, A.; Bapat, A.; Hulsmans, M.; Iwamoto, Y.; Wojtkiewicz, G.; Cetinbas, M.; Schloss, M.J.; et al. Ibrutinib-Mediated Atrial Fibrillation Attributable to Inhibition of C-Terminal Src Kinase. Circulation 2020, 142, 2443–2455. [Google Scholar] [CrossRef]
- de Jong, J.; Hellemans, P.; Jiao, J.J.; Huang, Y.; Mesens, S.; Sukbuntherng, J.; Ouellet, D. Ibrutinib does not prolong the corrected QT interval in healthy subjects: Results from a thorough QT study. Cancer Chemother. Pharmacol. 2017, 80, 1227–1237. [Google Scholar] [CrossRef]
- Giudice, V.; Vecchione, C.; Selleri, C. Cardiotoxicity of Novel Targeted Hematological Therapies. Life 2020, 10, 344. [Google Scholar] [CrossRef]
- Tarnowski, D.; Feder, A.L.; Trum, M.; Kreitmeier, K.G.; Stengel, L.; Maier, L.S.; Sag, C.M. Ibrutinib impairs IGF-1-dependent activation of intracellular Ca handling in isolated mouse ventricular myocytes. Front. Cardiovasc. Med. 2023, 10, 1190099. [Google Scholar] [CrossRef]
- Pretorius, L.; Du, X.J.; Woodcock, E.A.; Kiriazis, H.; Lin, R.C.; Marasco, S.; Medcalf, R.L.; Ming, Z.; Head, G.A.; Tan, J.W.; et al. Reduced phosphoinositide 3-kinase (p110alpha) activation increases the susceptibility to atrial fibrillation. Am. J. Pathol. 2009, 175, 998–1009. [Google Scholar] [CrossRef]
- Natarajan, G.; Terrazas, C.; Oghumu, S.; Varikuti, S.; Dubovsky, J.A.; Byrd, J.C.; Satoskar, A.R. Ibrutinib enhances IL-17 response by modulating the function of bone marrow derived dendritic cells. Oncoimmunology 2016, 5, e1057385. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e876–e894. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Janus kinase (JAK) inhibitors in the treatment of neoplastic and inflammatory disorders. Pharmacol. Res. 2022, 183, 106362. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Tefferi, A. How I treat myelofibrosis after failure of JAK inhibitors. Blood 2018, 132, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef]
- Ross, D.M.; Babon, J.J.; Tvorogov, D.; Thomas, D. Persistence of myelofibrosis treated with ruxolitinib: Biology and clinical implications. Haematologica 2021, 106, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Boehm, M.; Egemnazarov, B.; Grimminger, F.; Savai Pullamsetti, S.; Kwapiszewska, G.; Schermuly, R.T. Kinases as potential targets for treatment of pulmonary hypertension and right ventricular dysfunction. Br. J. Pharmacol. 2021, 178, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Low, A.T.; Howard, L.; Harrison, C.; Tulloh, R.M. Pulmonary arterial hypertension exacerbated by ruxolitinib. Haematologica 2015, 100, e244–e245. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yin, C.; Wang, S.; Xiao, Y. JAK-STAT in lipid metabolism of adipocytes. JAKSTAT 2013, 2, e27203. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Markham, A. Idelalisib: First global approval. Drugs 2014, 74, 1701–1707. [Google Scholar] [CrossRef]
- Kumar, A.; Bhatia, R.; Chawla, P.; Anghore, D.; Saini, V.; Rawal, R.K. Copanlisib: Novel PI3K Inhibitor for the Treatment of Lymphoma. Anticancer Agents Med. Chem. 2020, 20, 1158–1172. [Google Scholar] [CrossRef]
- Kienle, D.L.; Stilgenbauer, S. Approved and emerging PI3K inhibitors for the treatment of chronic lymphocytic leukemia and non-Hodgkin lymphoma. Expert. Opin. Pharmacother. 2020, 21, 917–929. [Google Scholar] [CrossRef]
- Reinwald, M.; Silva, J.T.; Mueller, N.J.; Fortún, J.; Garzoni, C.; de Fijter, J.W.; Fernández-Ruiz, M.; Grossi, P.; Aguado, J.M. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: An infectious diseases perspective (Intracellular signaling pathways: Tyrosine kinase and mTOR inhibitors). Clin. Microbiol. Infect. 2018, 24 (Suppl. S2), S53–S70. [Google Scholar] [CrossRef]
- Juárez-Salcedo, L.M.; Desai, V.; Dalia, S. Venetoclax: Evidence to date and clinical potential. Drugs Context 2019, 8, 212574. [Google Scholar] [CrossRef] [PubMed]
- Ploumaki, I.; Triantafyllou, E.; Koumprentziotis, I.A.; Karampinos, K.; Drougkas, K.; Karavolias, I.; Trontzas, I.; Kotteas, E.A. Bcl-2 pathway inhibition in solid tumors: A review of clinical trials. Clin. Transl. Oncol. 2023, 25, 1554–1578. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Stilgenbauer, S.; Seymour, J.F.; Huang, D.C.S. Venetoclax in Patients with Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 4527–4533. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Lowe, S.W. Bcl-2 mediates chemoresistance in matched pairs of primary E(mu)-myc lymphomas in vivo. Blood Cells Mol. Dis. 2001, 27, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef]
- Perini, G.F.; Feres, C.C.P.; Teixeira, L.L.C.; Hamerschlak, N. BCL-2 Inhibition as Treatment for Chronic Lymphocytic Leukemia. Curr. Treat. Options Oncol. 2021, 22, 66. [Google Scholar] [CrossRef]
- Davids, M.S.; Hallek, M.; Wierda, W.; Roberts, A.W.; Stilgenbauer, S.; Jones, J.A.; Gerecitano, J.F.; Kim, S.Y.; Potluri, J.; Busman, T.; et al. Comprehensive Safety Analysis of Venetoclax Monotherapy for Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2018, 24, 4371–4379. [Google Scholar] [CrossRef]
- Wanchoo, R.; Bernabe Ramirez, C.; Barrientos, J.; Jhaveri, K.D. Renal involvement in chronic lymphocytic leukemia. Clin. Kidney J. 2018, 11, 670–680. [Google Scholar] [CrossRef]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef]
- El Omari, N.; Lee, L.H.; Bakrim, S.; Makeen, H.A.; Alhazmi, H.A.; Mohan, S.; Khalid, A.; Ming, L.C.; Bouyahya, A. Molecular mechanistic pathways underlying the anticancer therapeutic efficiency of romidepsin. Biomed. Pharmacother. 2023, 164, 114774. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Canestraro, M.; Savli, H.; Palumbo, G.A.; Tibullo, D.; Nagy, B.; Piaggi, S.; Guerrini, F.; Cine, N.; Metelli, M.R.; et al. ITF2357 interferes with apoptosis and inflammatory pathways in the HL-60 model: A gene expression study. Anticancer Res. 2010, 30, 4525–4535. [Google Scholar] [PubMed]
- Vinodhkumar, R.; Song, Y.S.; Devaki, T. Romidepsin (depsipeptide) induced cell cycle arrest, apoptosis and histone hyperacetylation in lung carcinoma cells (A549) are associated with increase in p21 and hypophosphorylated retinoblastoma proteins expression. Biomed. Pharmacother. 2008, 62, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Panicker, J.; Li, Z.; McMahon, C.; Sizer, C.; Steadman, K.; Piekarz, R.; Bates, S.E.; Thiele, C.J. Romidepsin (FK228/depsipeptide) controls growth and induces apoptosis in neuroblastoma tumor cells. Cell Cycle 2010, 9, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Li, L.H.; Zhang, P.R.; Cai, P.Y.; Li, Z.C. Histone deacetylase inhibitor, Romidepsin (FK228) inhibits endometrial cancer cell growth through augmentation of p53-p21 pathway. Biomed. Pharmacother. 2016, 82, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Sutheesophon, K.; Kobayashi, Y.; Takatoku, M.A.; Ozawa, K.; Kano, Y.; Ishii, H.; Furukawa, Y. Histone deacetylase inhibitor depsipeptide (FK228) induces apoptosis in leukemic cells by facilitating mitochondrial translocation of Bax, which is enhanced by the proteasome inhibitor bortezomib. Acta Haematol. 2006, 115, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, A.R.; Wright, J.J.; Steinberg, S.M.; Liewehr, D.J.; Rosing, D.R.; Sachdev, V.; Fojo, T.; Bates, S.E. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clin. Cancer Res. 2006, 12, 3762–3773. [Google Scholar] [CrossRef]
- Sager, P.T.; Balser, B.; Wolfson, J.; Nichols, J.; Pilot, R.; Jones, S.; Burris, H.A. Electrocardiographic effects of class 1 selective histone deacetylase inhibitor romidepsin. Cancer Med. 2015, 4, 1178–1185. [Google Scholar] [CrossRef]
- Spence, S.; Deurinck, M.; Ju, H.; Traebert, M.; McLean, L.; Marlowe, J.; Emotte, C.; Tritto, E.; Tseng, M.; Shultz, M.; et al. Histone Deacetylase Inhibitors Prolong Cardiac Repolarization through Transcriptional Mechanisms. Toxicol. Sci. 2016, 153, 39–54. [Google Scholar] [CrossRef]
- Moskowitz, A.J.; Horwitz, S.M. Targeting histone deacetylases in T-cell lymphoma. Leuk. Lymphoma 2017, 58, 1306–1319. [Google Scholar] [CrossRef]
- Noonan, A.M.; Eisch, R.A.; Liewehr, D.J.; Sissung, T.M.; Venzon, D.J.; Flagg, T.P.; Haigney, M.C.; Steinberg, S.M.; Figg, W.D.; Piekarz, R.L.; et al. Electrocardiographic studies of romidepsin demonstrate its safety and identify a potential role for K(ATP) channel. Clin. Cancer Res. 2013, 19, 3095–3104. [Google Scholar] [CrossRef]
- Rivers, Z.T.; Oostra, D.R.; Westholder, J.S.; Vercellotti, G.M. Romidepsin-associated cardiac toxicity and ECG changes: A case report and review of the literature. J. Oncol. Pharm. Pract. 2018, 24, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef] [PubMed]
- El-Cheikh, J.; Moukalled, N.; Malard, F.; Bazarbachi, A.; Mohty, M. Cardiac toxicities in multiple myeloma: An updated and a deeper look into the effect of different medications and novel therapies. Blood Cancer J. 2023, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Garcia, D.; Cornell, R.F.; Gailani, D.; Laubach, J.; Maglio, M.E.; Richardson, P.G.; Moslehi, J. Cardiovascular and Thrombotic Complications of Novel Multiple Myeloma Therapies: A Review. JAMA Oncol. 2017, 3, 980–988. [Google Scholar] [CrossRef]
- Delforge, M.; Bladé, J.; Dimopoulos, M.A.; Facon, T.; Kropff, M.; Ludwig, H.; Palumbo, A.; Van Damme, P.; San-Miguel, J.F.; Sonneveld, P. Treatment-related peripheral neuropathy in multiple myeloma: The challenge continues. Lancet Oncol. 2010, 11, 1086–1095. [Google Scholar] [CrossRef]
- Das, A.; Dasgupta, S.; Gong, Y.; Shah, U.A.; Fradley, M.G.; Cheng, R.K.; Roy, B.; Guha, A. Cardiotoxicity as an adverse effect of immunomodulatory drugs and proteasome inhibitors in multiple myeloma: A network meta-analysis of randomized clinical trials. Hematol. Oncol. 2022, 40, 233–242. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Chen, C.; Spencer, A.; Niesvizky, R.; Attal, M.; Stadtmauer, E.A.; Petrucci, M.T.; Yu, Z.; Olesnyckyj, M.; Zeldis, J.B.; et al. Long-term follow-up on overall survival from the MM-009 and MM-010 phase III trials of lenalidomide plus dexamethasone in patients with relapsed or refractory multiple myeloma. Leukemia 2009, 23, 2147–2152. [Google Scholar] [CrossRef]
- Jacob, R.; Strati, P.; Palaskas, N.; Lopez-Mattei, J.C.; Marmagkiolis, K.; Buja, L.M.; Deswal, A.; Iliescu, C. Lenalidomide-Induced Myocarditis, Rare But Possibly Fatal Toxicity of a Commonly Used Immunotherapy. JACC Case Rep. 2020, 2, 2095–2100. [Google Scholar] [CrossRef]
- Verbesselt, M.; Meekers, E.; Vandenberghe, P.; Delforge, M.; Vandenbriele, C. Combined lenalidomide/bortezomib for multiple myeloma complicated by fulminant myocarditis: A rare case report of widely used chemotherapy. Eur. Heart J. Case Rep. 2022, 6, ytac093. [Google Scholar] [CrossRef]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Matsumoto, M.; Suzuki, K.; Kuroda, J.; Taniwaki, M.; Sunami, K.; Kosugi, H.; Ando, K.; Maruyama, D.; Tobinai, K.; Kher, U.; et al. Pembrolizumab plus pomalidomide and dexamethasone for relapsed or refractory multiple myeloma (KEYNOTE-183): Subgroup analysis in Japanese patients. Int. J. Hematol. 2021, 113, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Kim, C.; Thompson, J.; McGuire, C.; Kallakury, B.; Chahine, J.J.; Manning, M.; Mogg, R.; Blumenschein, W.M.; Tan, M.T.; et al. Pembrolizumab in patients with thymic carcinoma: A single-arm, single-centre, phase 2 study. Lancet Oncol. 2018, 19, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Petrini, I.; Sollini, M.; Bartoli, F.; Barachini, S.; Montali, M.; Pardini, E.; Burzi, I.S.; Erba, P.A. ED-B-Containing Isoform of Fibronectin in Tumor Microenvironment of Thymomas: A Target for a Theragnostic Approach. Cancers 2022, 14, 2592. [Google Scholar] [CrossRef]
- Wu, P.; Oren, O.; Gertz, M.A.; Yang, E.H. Proteasome Inhibitor-Related Cardiotoxicity: Mechanisms, Diagnosis, and Management. Curr. Oncol. Rep. 2020, 22, 66. [Google Scholar] [CrossRef] [PubMed]
- Birks, E.J.; Latif, N.; Enesa, K.; Folkvang, T.; Luong, L.A.; Sarathchandra, P.; Khan, M.; Ovaa, H.; Terracciano, C.M.; Barton, P.J.; et al. Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc. Res. 2008, 79, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Spänig, S.; Kellermann, K.; Dieterlen, M.T.; Noack, T.; Lehmann, S.; Borger, M.A.; Garbade, J.; Barac, Y.D.; Emrich, F. The Ubiquitin Proteasome System in Ischemic and Dilated Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 6354. [Google Scholar] [CrossRef] [PubMed]
- Enrico, O.; Gabriele, B.; Nadia, C.; Sara, G.; Daniele, V.; Giulia, C.; Antonio, S.; Mario, P. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br. J. Haematol. 2007, 138, 396–397. [Google Scholar] [CrossRef]
- Pancheri, E.; Guglielmi, V.; Wilczynski, G.M.; Malatesta, M.; Tonin, P.; Tomelleri, G.; Nowis, D.; Vattemi, G. Non-Hematologic Toxicity of Bortezomib in Multiple Myeloma: The Neuromuscular and Cardiovascular Adverse Effects. Cancers 2020, 12, 2540. [Google Scholar] [CrossRef] [PubMed]
- Forghani, P.; Rashid, A.; Sun, F.; Liu, R.; Li, D.; Lee, M.R.; Hwang, H.; Maxwell, J.T.; Mandawat, A.; Wu, R.; et al. Carfilzomib Treatment Causes Molecular and Functional Alterations of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. J. Am. Heart Assoc. 2021, 10, e022247. [Google Scholar] [CrossRef] [PubMed]
- Tantawy, M.; Chekka, L.M.; Huang, Y.; Garrett, T.J.; Singh, S.; Shah, C.P.; Cornell, R.F.; Baz, R.C.; Fradley, M.G.; Waheed, N.; et al. Lactate Dehydrogenase B and Pyruvate Oxidation Pathway Associated with Carfilzomib-Related Cardiotoxicity in Multiple Myeloma Patients: Result of a Multi-Omics Integrative Analysis. Front. Cardiovasc. Med. 2021, 8, 645122. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.; Quach, H.; Mateos, M.V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Versari, D.; Herrmann, J.; Gössl, M.; Mannheim, D.; Sattler, K.; Meyer, F.B.; Lerman, L.O.; Lerman, A. Dysregulation of the ubiquitin-proteasome system in human carotid atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2132–2139. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Mateos, C.; Alcaraz-Serna, A.; Somovilla-Crespo, B.; Muñoz-Calleja, C. Monoclonal Antibody Therapies for Hematological Malignancies: Not Just Lineage-Specific Targets. Front. Immunol. 2017, 8, 1936. [Google Scholar] [CrossRef]
- Weiner, G.J. Monoclonal antibody mechanisms of action in cancer. Immunol. Res. 2007, 39, 271–278. [Google Scholar] [CrossRef]
- Spasevska, I.; Matera, E.L.; Chettab, K.; Ville, J.; Potier-Cartereau, M.; Jordheim, L.P.; Thieblemont, C.; Sahin, D.; Klein, C.; Dumontet, C. Calcium Channel Blockers Impair the Antitumor Activity of Anti-CD20 Monoclonal Antibodies by Blocking EGR-1 Induction. Mol. Cancer Ther. 2020, 19, 2371–2381. [Google Scholar] [CrossRef]
- Vacher, P.; Vacher, A.M.; Pineau, R.; Latour, S.; Soubeyran, I.; Pangault, C.; Tarte, K.; Soubeyran, P.; Ducret, T.; Bresson-Bepoldin, L. Localized Store-Operated Calcium Influx Represses CD95-Dependent Apoptotic Effects of Rituximab in Non-Hodgkin B Lymphomas. J. Immunol. 2015, 195, 2207–2215. [Google Scholar] [CrossRef]
- Latour, S.; Zanese, M.; Le Morvan, V.; Vacher, A.M.; Menard, N.; Bijou, F.; Durrieu, F.; Soubeyran, P.; Savina, A.; Vacher, P.; et al. Role of Calcium Signaling in GA101-Induced Cell Death in Malignant Human B Cells. Cancers 2019, 11, 291. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; González Barca, E.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, A.; Sureda, A.; Stiff, P.; Holowiecki, J.; Abidi, M.; Hunder, N.; Pecsok, M.; Uttarwar, M.; Purevjal, I.; Sweetenham, J. Safety Analysis of Brentuximab Vedotin from the Phase III AETHERA Trial in Hodgkin Lymphoma in the Post-Transplant Consolidation Setting. Biol. Blood Marrow Transplant. 2018, 24, 2354–2359. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Burstein, D.S.; Maude, S.; Grupp, S.; Griffis, H.; Rossano, J.; Lin, K. Cardiac Profile of Chimeric Antigen Receptor T Cell Therapy in Children: A Single-Institution Experience. Biol. Blood Marrow Transplant. 2018, 24, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Alvi, R.M.; Frigault, M.J.; Fradley, M.G.; Jain, M.D.; Mahmood, S.S.; Awadalla, M.; Lee, D.H.; Zlotoff, D.A.; Zhang, L.; Drobni, Z.D.; et al. Cardiovascular Events Among Adults Treated with Chimeric Antigen Receptor T-Cells (CAR-T). J. Am. Coll. Cardiol. 2019, 74, 3099–3108. [Google Scholar] [CrossRef]
- Ganatra, S.; Redd, R.; Hayek, S.S.; Parikh, R.; Azam, T.; Yanik, G.A.; Spendley, L.; Nikiforow, S.; Jacobson, C.; Nohria, A. Chimeric Antigen Receptor T-Cell Therapy-Associated Cardiomyopathy in Patients with Refractory or Relapsed Non-Hodgkin Lymphoma. Circulation 2020, 142, 1687–1690. [Google Scholar] [CrossRef]
- Hashmi, H.; Mirza, A.S.; Darwin, A.; Logothetis, C.; Garcia, F.; Kommalapati, A.; Mhaskar, R.S.; Bachmeier, C.; Chavez, J.C.; Shah, B.; et al. Venous thromboembolism associated with CD19-directed CAR T-cell therapy in large B-cell lymphoma. Blood Adv. 2020, 4, 4086–4090. [Google Scholar] [CrossRef]
- Lefebvre, B.; Kang, Y.; Smith, A.M.; Frey, N.V.; Carver, J.R.; Scherrer-Crosbie, M. Cardiovascular Effects of CAR T Cell Therapy: A Retrospective Study. JACC CardioOncol. 2020, 2, 193–203. [Google Scholar] [CrossRef]
- Wudhikarn, K.; Pennisi, M.; Garcia-Recio, M.; Flynn, J.R.; Afuye, A.; Silverberg, M.L.; Maloy, M.A.; Devlin, S.M.; Batlevi, C.L.; Shah, G.L.; et al. DLBCL patients treated with CD19 CAR T cells experience a high burden of organ toxicities but low nonrelapse mortality. Blood Adv. 2020, 4, 3024–3033. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef]
- Ganatra, S.; Carver, J.R.; Hayek, S.S.; Ky, B.; Leja, M.J.; Lenihan, D.J.; Lenneman, C.; Mousavi, N.; Park, J.H.; Perales, M.A.; et al. Chimeric Antigen Receptor T-Cell Therapy for Cancer and Heart: JACC Council Perspectives. J. Am. Coll. Cardiol. 2019, 74, 3153–3163. [Google Scholar] [CrossRef] [PubMed]
- Obstfeld, A.E.; Frey, N.V.; Mansfield, K.; Lacey, S.F.; June, C.H.; Porter, D.L.; Melenhorst, J.J.; Wasik, M.A. Cytokine release syndrome associated with chimeric-antigen receptor T-cell therapy: Clinicopathological insights. Blood 2017, 130, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Ganatra, S.; Dani, S.S.; Yang, E.H.; Zaha, V.G.; Nohria, A. Cardiotoxicity of T-Cell Antineoplastic Therapies. JACC CardioOncol. 2022, 4, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Comini, L.; Pasini, E.; Bachetti, T.; Dreano, M.; Garotta, G.; Ferrari, R. Acute haemodynamic effects of IL-6 treatment in vivo: Involvement of vagus nerve in NO-mediated negative inotropism. Cytokine 2005, 30, 236–242. [Google Scholar] [CrossRef]
- Yu, X.; Kennedy, R.H.; Liu, S.J. JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J. Biol. Chem. 2003, 278, 16304–16309. [Google Scholar] [CrossRef]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef]
- Freyer, C.W.; Porter, D.L. Cytokine release syndrome and neurotoxicity following CAR T-cell therapy for hematologic malignancies. J. Allergy Clin. Immunol. 2020, 146, 940–948. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Schuster, S.J.; Tam, C.S.; Borchmann, P.; Worel, N.; McGuirk, J.P.; Holte, H.; Waller, E.K.; Jaglowski, S.; Bishop, M.R.; Damon, L.E.; et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 1403–1415. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Bachy, E.; Le Gouill, S.; Di Blasi, R.; Sesques, P.; Manson, G.; Cartron, G.; Beauvais, D.; Roulin, L.; Gros, F.X.; Rubio, M.T.; et al. A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nat. Med. 2022, 28, 2145–2154. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Budde, L.E.; Assouline, S.; Sehn, L.H.; Schuster, S.J.; Yoon, S.S.; Yoon, D.H.; Matasar, M.J.; Bosch, F.; Kim, W.S.; Nastoupil, L.J.; et al. Single-Agent Mosunetuzumab Shows Durable Complete Responses in Patients with Relapsed or Refractory B-Cell Lymphomas: Phase I Dose-Escalation Study. J. Clin. Oncol. 2022, 40, 481–491. [Google Scholar] [CrossRef]
- Hutchings, M.; Mous, R.; Clausen, M.R.; Johnson, P.; Linton, K.M.; Chamuleau, M.E.D.; Lewis, D.J.; Sureda Balari, A.; Cunningham, D.; Oliveri, R.S.; et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: An open-label, phase 1/2 study. Lancet 2021, 398, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.J.; Carlo-Stella, C.; Morschhauser, F.; Bachy, E.; Corradini, P.; Iacoboni, G.; Khan, C.; Wróbel, T.; Offner, F.; Trněný, M.; et al. Glofitamab for Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 387, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Budde, L.E.; Sehn, L.H.; Matasar, M.; Schuster, S.J.; Assouline, S.; Giri, P.; Kuruvilla, J.; Canales, M.; Dietrich, S.; Fay, K.; et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: A single-arm, multicentre, phase 2 study. Lancet Oncol. 2022, 23, 1055–1065. [Google Scholar] [CrossRef]
- Gutierrez, C.; Neilan, T.G.; Grover, N.S. How I approach optimization of patients at risk of cardiac and pulmonary complications after CAR T-cell therapy. Blood 2023, 141, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barachini, S.; Buda, G.; Petrini, I. Cardiovascular Toxicity of Antineoplastic Treatments in Hematological Diseases: Focus on Molecular Mechanisms to Improve Therapeutic Management. J. Clin. Med. 2024, 13, 1574. https://doi.org/10.3390/jcm13061574
Barachini S, Buda G, Petrini I. Cardiovascular Toxicity of Antineoplastic Treatments in Hematological Diseases: Focus on Molecular Mechanisms to Improve Therapeutic Management. Journal of Clinical Medicine. 2024; 13(6):1574. https://doi.org/10.3390/jcm13061574
Chicago/Turabian StyleBarachini, Serena, Gabriele Buda, and Iacopo Petrini. 2024. "Cardiovascular Toxicity of Antineoplastic Treatments in Hematological Diseases: Focus on Molecular Mechanisms to Improve Therapeutic Management" Journal of Clinical Medicine 13, no. 6: 1574. https://doi.org/10.3390/jcm13061574
APA StyleBarachini, S., Buda, G., & Petrini, I. (2024). Cardiovascular Toxicity of Antineoplastic Treatments in Hematological Diseases: Focus on Molecular Mechanisms to Improve Therapeutic Management. Journal of Clinical Medicine, 13(6), 1574. https://doi.org/10.3390/jcm13061574