Screening for Fabry Disease-Related Mutations Among 829 Kidney Transplant Recipients

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
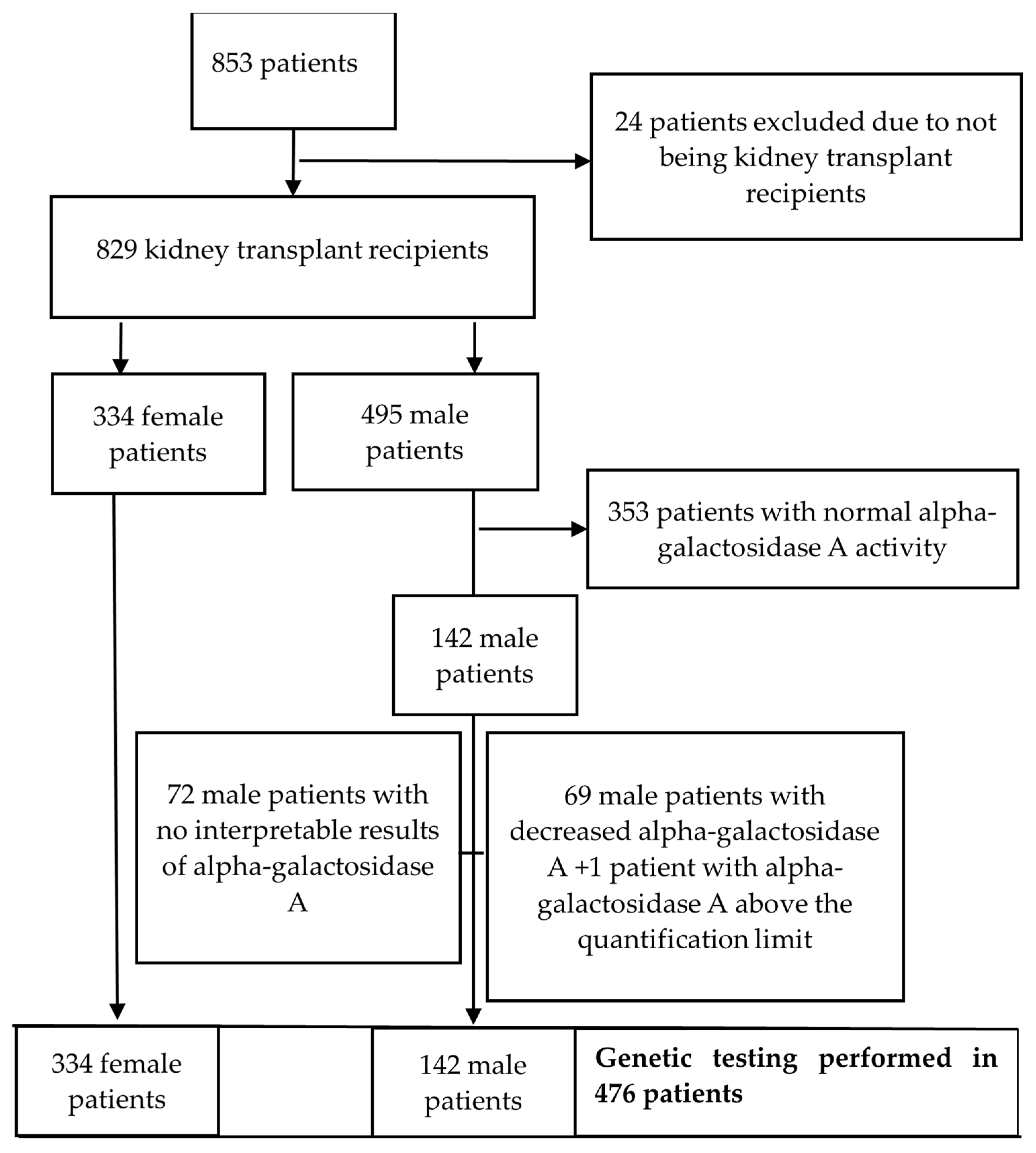
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michaud, M.; Mauhin, W.; Belmatoug, N.; Bedreddine, N.; Garnotel, R.; Catros, F.; Lidove, O.; Gaches, F. Maladie de Fabry: Quand y penser? [Fabry disease: A review]. Rev. Med. Interne 2021, 42, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Mayatepek, E. Fabry Disease—Often Seen, Rarely Diagnosed. Dtsch. Arztebl. Int. 2009, 106, 440–447. [Google Scholar] [PubMed]
- Kermond-Marino, A.; Weng, A.; Zhang, S.K.X.; Tran, Z.; Huang, M.; Savige, J. Population Frequency of Undiagnosed Fabry Disease in the General Population. Kidney Int. Rep. 2023, 8, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Feriozzi, S.; Rozenfeld, P. Pathology and pathogenic pathways in fabry nephropathy. Clin. Exp. Nephrol. 2021, 25, 925–934. [Google Scholar] [CrossRef]
- Pećin, I.; Merćep, I.; Bašić-Kes, V.; Bilić, E.; Borovečki, F.; Bradamante, M.; Čikeš, M.; Fumić, K.; Godan Hauptman, A.; Jelaković, B.; et al. Smjernice za dijagnostiku i liječenje odraslih bolesnika s Fabryjevom bolesti. Liječnički Vjesnik 2024, 146, 157–169. [Google Scholar]
- HGMD®. Home Page. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 24 February 2022).
- Amodio, F.; Caiazza, M.; Monda, E.; Rubino, M.; Capodicasa, L.; Chiosi, F.; Simonelli, V.; Dongiglio, F.; Fimiani, F.; Pepe, N.; et al. An Overview of Molecular Mechanisms in Fabry Disease. Biomolecules 2022, 12, 1460. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- The Genome Aggregation Database (gnomAD)|gnomAD Browser [Internet]. Available online: https://gnomad.broadinstitute.org/news/2017-02-the-genome-aggregation-database/ (accessed on 28 July 2024).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 3 March 2020).
- Simoncini, C.; Torri, S.; Montano, V.; Chico, L.; Gruosso, F.; Tuttolomondo, A.; Pinto, A.; Simonetta, I.; Cianci, V.; Salviati, A.; et al. Oxidative stress biomarkers in Fabry disease: Is there a room for them? J. Neurol. 2020, 267, 3741–3752. [Google Scholar] [CrossRef]
- Romani, I.; Sarti, C.; Nencini, P.; Pracucci, G.; Zedde, M.; Cianci, V.; Nucera, A.; Moller, J.; Orsucci, D.; Toni, D.; et al. Prevalence of Fabry disease and GLA variants in young patients with acute stroke: The challenge to widen the screening. The Fabry-Stroke Italian Registry. J. Neurol. Sci. 2024, 457, 122905. [Google Scholar] [CrossRef]
- Skrinska, V.; Khneisser, I.; Schielen, P.; Loeber, G. Introducing and Expanding Newborn Screening in the MENA Region. Int. J. Neonatal Screen. 2020, 6, 12. [Google Scholar] [CrossRef]
- Živná, M.; Dostálová, G.; Barešová, V.; Mušálková, D.; Kuchař, L.; Asfaw, B.; Poupětová, H.; Vlášková, H.; Kmochová, T.; Vyletal, P.; et al. AGAL misprocessing-induced ER stress and the unfolded protein response: Lysosomal storage-independent mechanism of Fabry disease pathogenesis? bioRxiv 2022, bioRxiv:509714. [Google Scholar]
- Nance, C.S.; Klein, C.J.; Banikazemi, M.; Dikman, S.H.; Phelps, R.G.; McArthur, J.C.; Rodriguez, M.; Desnick, R.J. Later-onset Fabry disease: An adult variant presenting with the cramp-fasciculation syndrome. Arch. Neurol. 2006, 63, 453–457. [Google Scholar] [CrossRef]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Terryn, W.; Deschoenmakere, G.; De Keyser, J.; Meersseman, W.; Van Biesen, W.; Wuyts, B.; Hemelsoet, D.; Pascale, H.; De Backer, J.; De Paepe, A.; et al. Prevalence of Fabry disease in a predominantly hypertensive population with left ventricular hypertrophy. Int. J. Cardiol. 2012, 167, 2555–2560. [Google Scholar] [CrossRef] [PubMed]
- Terryn, W.; Vanholder, R.; Hemelsoet, D.; Leroy, B.P.; Van Biesen, W.; De Schoenmakere, G.; Wuyts, B.; Claes, K.; De Backer, J.; De Paepe, G.; et al. Questioning the Pathogenic Role of the GLA p.Ala143Thr “Mutation” in Fabry Disease: Implications for Screening Studies and ERT. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2013; Volume 8, pp. 101–108. [Google Scholar]
- Smid, B.E.; Hollak, C.E.; Poorthuis, B.J.; van den Bergh Weerman, M.A.; Florquin, S.; Kok, W.E.; Lekanne Deprez, R.H.; Timmermans, J.; Linthorst, G.E. Diagnostic dilemmas in Fabry disease: A case series study on GLA mutations of unknown clinical significance. Clin. Genet. 2015, 88, 161–166. [Google Scholar] [CrossRef]
- Lenders, M.; Weidemann, F.; Kurschat, C.; Canaan-Kühl, S.; Duning, T.; Stypmann, J.; Schmitz, B.; Reiermann, S.; Krämer, J.; Blaschke, D.; et al. Alpha-Galactosidase A p.A143T, a non-Fabry disease-causing variant. Orphanet J. Rare Dis. 2016, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Morais, P.; Santos, A.L.; Baudrier, T.; Mota, A.V.; Oliveira, J.P.; Azevedo, F. Angiokeratomas of Fabry successfully treated with intense pulsed light. J. Cosmet. Laser Ther. 2008, 10, 218–222. [Google Scholar] [CrossRef]
- Baptista, M.V.; Ferreira, S.; Pinho-E-Melo, T.; Carvalho, M.; Cruz, V.T.; Carmona, C.; Silva, F.A.; Tuna, A.; Rodrigues, M.; Ferreira, C.; et al. Mutations of the GLA gene in young patients with stroke: The PORTYSTROKE study—Screening genetic conditions in Portuguese young stroke patients. Stroke 2010, 41, 431–436. [Google Scholar] [CrossRef]
- Lanthier, S.; Saposnik, G.; Lebovic, G.; Pope, K.; Selchen, D.; Moore, D.F.; Selchen, D.; Boulanger, J.-M.; Buck, B.; Butcher, K.; et al. Prevalence of Fabry Disease and Outcomes in Young Canadian Patients with Cryptogenic Ischemic Cerebrovascular Events. Stroke 2017, 48, 1766–1772. [Google Scholar] [CrossRef]
- Genoni, G.; Demarchi, I.; Bellone, S.; Petri, A.; Settanni, F.; Dondi, E.; Negro, M.; Cortese, L.; Prodam, F.; Bona, G. Early diagnosis of Fabry disease in children. Minerva Pediatr. 2011, 63, 425–430. [Google Scholar]
- Gaspar, P.; Herrera, J.; Rodrigues, D.; Cerezo, S.; Delgado, R.; Andrade, C.F.; Forascepi, R.; Macias, J.; del Pino, M.D.; Prados, M.D.; et al. Frequency of Fabry disease in male and female haemodialysis patients in Spain. BMC Med. Genet. 2010, 11, 19. [Google Scholar] [CrossRef]
- Connaughton, D.M.; Kennedy, C.; Shril, S.; Mann, N.; Murray, S.L.; Williams, P.A.; Conlon, E.; Nakayama, M.; van der Ven, A.T.; Ityel, H.; et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019, 95, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Capuano, I.; Buonanno, P.; Riccio, E.; Crocetto, F.; Pisani, A. Parapelvic Cysts: An Imaging Marker of Kidney Disease Potentially Leading to the Diagnosis of Treatable Rare Genetic Disorders? A Narrative Review of the Literature. J. Nephrol. 2022, 35, 2035–2046. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Bettis, K.E.; Choyke, P.; Kopp, J.B.; Austin, H.A., III; Brady, R.O.; Schiffmann, R. Parapelvic kidney cysts: A distinguishing feature with high prevalence in Fabry disease. Kidney Int. 2004, 66, 978–982. [Google Scholar] [CrossRef]
- Cerón-Rodríguez, M.; Ramón-García, G.; Barajas-Colón, E.; Franco-Álvarez, I.; Salgado-Loza, J.L. Renal globotriaosylceramide deposits for Fabry disease linked to uncertain pathogenicity gene variant c.352C>T/p.Arg118Cys: A family study. Mol. Genet. Genom. Med. 2019, 7, e981. [Google Scholar] [CrossRef]
- Ferreira, S.; Ortiz, A.; Germain, D.P.; Viana-Baptista, M.; Caldeira-Gomes, A.; Camprecios, M.; Fenollar-Cortés, M.; Gallegos-Villalobos, Á.; Garcia, D.; García-Robles, J.A.; et al. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: Data from individual patients and family studies. Mol. Genet. Metab. 2015, 114, 248–258. [Google Scholar] [CrossRef]
- Caetano, F.; Botelho, A.; Mota, P.; Silva, J.; Leitão Marques, A. Fabry disease presenting as apical left ventricular hypertrophy in a patient carrying the missense mutation R118C. Rev. Port. Cardiol. 2014, 33, 183.e1–183.e5. [Google Scholar] [CrossRef]
- Chaves-Markman, Â.V.; Markman, M.; Calado, E.B.; Pires, R.F.; Santos-Veloso, M.A.O.; Pereira, C.M.F.; Lordsleem, A.B.; Lima, S.G.; Markman Filho, B.; Oliveira, D.C. GLA Gene Mutation in Hypertrophic Cardiomyopathy with a New Variant Description: Is it Fabry’s Disease? Arq. Bras. Cardiol. 2019, 113, 77–84. [Google Scholar] [CrossRef]
- Azevedo, O.; Marques, N.; Reis, L.; Cruz, I.; Craveiro, N.; Antunes, H.; Lourenço, C.; Gomes, R.; Guerreiro, R.A.; Faria, R.; et al. Predictors of Fabry disease in patients with hypertrophic cardiomyopathy: How to guide the diagnostic strategy? Am. Heart J. 2020, 226, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Talbot, A.; Nicholls, K. Elevated Lyso-Gb3 Suggests the R118C GLA Mutation Is a Pathological Fabry Variant. In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; Volume 45, pp. 95–98. [Google Scholar] [CrossRef]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163. [Google Scholar] [CrossRef]
- Kang, J.J.; Kaissarian, N.M.; Desch, K.C.; Kelly, R.J.; Shu, L.; Bodary, P.F.; Shayman, J.A. α-galactosidase A deficiency promotes von Willebrand factor secretion in models of Fabry disease. Kidney Int. 2018, 95, 149–159. [Google Scholar] [CrossRef]
- Waldek, S.; Feriozzi, S. Fabry nephropathy: A review—How can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Palaiodimou, L.; Kokotis, P.; Zompola, C.; Papagiannopoulou, G.; Bakola, E.; Papadopoulou, M.; Zouvelou, V.; Petras, D.; Vlachopoulos, C.; Tsivgoulis, G. Fabry Disease: Current and Novel Therapeutic Strategies. A Narrative Review. Curr. Neuropharmacol. 2023, 21, 440–456. [Google Scholar]
- El Dib, R.; Gomaa, H.; Carvalho, R.P.; Camargo, S.E.; Bazan, R.; Barretti, P.; Barreto, F.C. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst. Rev. 2016, 2017, CD006663. [Google Scholar] [CrossRef]
- Tsuboi, K.; Yamamoto, H. Efficacy and safety of enzyme-replacement-therapy with agalsidase alfa in 36 treatment-naïve Fabry disease patients. BMC Pharmacol. Toxicol. 2017, 18, 43. [Google Scholar] [CrossRef]
- Alegra, T.; Vairo, F.; de Souza, M.V.; Krug, B.C.; Schwartz, I.V. Enzyme replacement therapy for Fabry disease: A systematic review and meta-analysis. Genet. Mol. Biol. 2012, 35, 947–954. [Google Scholar] [CrossRef]
- West, M.; Nicholls, K.; Mehta, A.; Clarke, J.T.; Steiner, R.; Beck, M.; Barshop, B.A.; Rhead, W.; Mensah, R.; Ries, M.; et al. Agalsidase Alfa and Kidney Dysfunction in Fabry Disease. J. Am. Soc. Nephrol. 2009, 20, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.; Cruz, J.; Brignani, E.; Acuna, L.; Lazaro, E.; Soria, C. Quality of life and unmet needs in patients with fabry disease: A qualitative study. Orphanet J. Rare Dis. 2024, 19, 389. [Google Scholar] [CrossRef] [PubMed]
- Laney, D.A.; Bennett, R.L.; Clarke, V.; Fox, A.; Hopkin, R.J.; Johnson, J.; O’Rourke, E.; Sims, K.; Walter, G. Fabry disease practice guidelines: Recommendations of the National Society of Genetic Counselors. J. Genet. Couns. 2013, 22, 555–564. [Google Scholar] [CrossRef]
- Capelli, I.; Aiello, V.; Gasperoni, L.; Comai, G.; Corradetti, V.; Ravaioli, M.; Biagini, E.; Graziano, C.; La Manna, G. Kidney Transplant in Fabry Disease: A Revision of the Literature. Medicina 2020, 56, 284. [Google Scholar] [CrossRef]
- Linares, D.; Luna, B.; Loayza, E.; Taboada, G.; Ramaswami, U. Prevalence of Fabry disease in patients with chronic kidney disease: A systematic review and meta-analysis. Mol. Genet. Metab. 2023, 140, 107714. [Google Scholar] [CrossRef]
- Erdogmus, S.; Kutlay, S.; Kumru, G.; Sendogan, D.O.; Erturk, S.; Keven, K.; Ceylaner, G.; Sengul, S. Fabry Disease Screening in Patients With Kidney Transplant: A Single-Center Study in Turkey. Exp. Clin. Transplant. 2020, 18, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Hasbal, N.B.; Caglayan, F.B.; Sakaci, T.; Ahbap, E.; Koc, Y.; Sevinc, M.; Ucar, Z.A.; Unsal, A.; Basturk, T. Unexpectedly High Prevalence of Low Alpha-Galactosidase A Enzyme Activity in Patients with Focal Segmental Glomerulosclerosis. Clinics 2020, 75, e1811. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Sex [n (%)] | ||
|---|---|---|
| Male | 495 (59.7) | |
| Female | 334 (40.3) | |
| Age (years) [Median (IQR)] | 59 (45–68) | min 19 max 87 |
| PHD of native kidney disease (n = 798) [n (%)] | ||
| No | 492 (61.7) | |
| Yes | 306 (38.3) | |
| Donor type—1st Tx (n = 822) [n (%)] | ||
| Deceased | 759 (92.3) | |
| Living related | 61 (7.4) | |
| Living unrelated | 2 (0.3) | |
| Donor type—2nd Tx (n = 59) [n (%)] | ||
| Deceased | 55/59 (93.2) | |
| Living related | 4/59 (6.8) | |
| Donor type—3rd Tx (n = 2) | ||
| Deceased | 1/2 | |
| Living unrelated | 1/2 | |
| Type of dialysis [n (%)] (n = 829) | ||
| Pre-emptive | 28 (3.4) | |
| HD | 574 (69.2) | |
| PD | 132 (16.0) | |
| HD + PD | 95 (11.4) | |
| Dialysis vintage (months) [Median (IQR)] | 33 (16–56) | min 0 max 300 |
| eGFR [Median (IQR)] | 51 (35–67) | min < 15 max 138 |
| Proteinuria/24 h [Median (IQR)] | 213 (113–428.3) | min 25 max 10,072 |
| CNI [n (%)] (n = 724) | ||
| Tacrolimus | 532 (73.5) | |
| Cyclosporine | 192 (26.5) | |
| mTORi [n (%)] (n = 173) | ||
| Everolimus | 166 (96) | |
| Sirolimus | 7 (4) | |
| Antimetabolites [n (%)] (n = 703) | ||
| Mycophenolate | 661 (94) | |
| Azathioprine | 42 (6) | |
| Corticosteroid dose (n = 829) [Median (IQR)] | 5 (5–5) | min 2.5 max 20 |
| Alpha-gal A activity (µmol/L/h) (n = 422) | 19 (16.1–23.5) | min 6.8 max 101.0 |
| Alpha-gal A [n (%)] (n = 423) | ||
| Reference value (≥15.3 µmol/L/h) | 353 (83.5) | |
| <15.3 µmol/L/h | 69 (16.3) | |
| Lyso Gb3 level (ng/mL) (n = 142) | 1.25 (1.10–1.43) | min 0.60 max 1.90 |
| Lyso Gb3 [n (%)] (n = 142) | ||
| Reference value (≤1.8 ng/mL) | 141 (99.3) | |
| >1.8 ng/mL | 1 (0.7) | |
| Fabry disease-related mutation [n (%)] | 3 (0.4) | |
| GLA mutation testing [n (%)] (n = 476) | ||
| no clinically relevant variant | 473 (99.4) | |
| GLA, c.427G>A p.(Ala143Thr) | 1 (0.2) | |
| GLA, c.1181T>C p.(Leu394Pro) | 1 (0.2) | |
| GLA, c.352C>T p.(Arg118Cys) | 1 (0.2) |
| Patient | Sex | Age | Alpha-Galactosidase A (µmol/L/h) | Lyso-Gb3 (ng/mL) | GLA Mutation | Kidney Biopsy | Diagnosis Prior to Testing | Other Non-Renal Manifestations | Follow Up | Centogene’s Classification of Variant Based on ACMG Guidelines |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 61 | >100 | 0.9 | c.1181T>C p.(Leu394Pro) | No | Chronic GN | LVH, paraesthesia, angiokeratomas, deafness | Preserved graft function, on regular ERT. | Likely pathogenic |
| 2 | M | 32 | 6.8 | 1.5 | c.427G>A p.(Ala143Thr) | Yes | IgAN | Headaches | Preserved graft function, not on ERT. | VUS |
| 3 | M | 45 | 7.8 | 1.4 | c.352C>T p.(Arg118Cys) | Yes | Renal carcinoma | PE in 2024, LVH, muscle/joint pain | Preserved graft function, not on ERT. | VUS |
| Pearson’s Correlation Coefficient R (p Value) | 95% Confidence Interval (CI) | |||
|---|---|---|---|---|
| Alpha-Galactosidase A Activity | Lyso Gb3 Level | Lower | Upper | |
| Age | −0.094 (0.05) | 0.154 (0.08) | −1488 | 2550 |
| Dialysis vintage (months) | 0.002 (0.97) | 0.002 (0.98) | −10,873 | 1166 |
| eGFR | −0.092 (0.06) | 0.088 (0.32) | −4657 | 1620 |
| Proteinuria | 0.054 (0.26) | −0.055 (0.54) | 40,956 | 282,714 |
| Corticosteroid dose | 0.158 (0.001) | −0.189 (0.03) | 0.1075 | 0.8724 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kljajic, M.; Atic, A.; Pecin, I.; Jelakovic, B.; Basic-Jukic, N. Screening for Fabry Disease-Related Mutations Among 829 Kidney Transplant Recipients. J. Clin. Med. 2024, 13, 7069. https://doi.org/10.3390/jcm13237069
Kljajic M, Atic A, Pecin I, Jelakovic B, Basic-Jukic N. Screening for Fabry Disease-Related Mutations Among 829 Kidney Transplant Recipients. Journal of Clinical Medicine. 2024; 13(23):7069. https://doi.org/10.3390/jcm13237069
Chicago/Turabian StyleKljajic, Marina, Armin Atic, Ivan Pecin, Bojan Jelakovic, and Nikolina Basic-Jukic. 2024. "Screening for Fabry Disease-Related Mutations Among 829 Kidney Transplant Recipients" Journal of Clinical Medicine 13, no. 23: 7069. https://doi.org/10.3390/jcm13237069
APA StyleKljajic, M., Atic, A., Pecin, I., Jelakovic, B., & Basic-Jukic, N. (2024). Screening for Fabry Disease-Related Mutations Among 829 Kidney Transplant Recipients. Journal of Clinical Medicine, 13(23), 7069. https://doi.org/10.3390/jcm13237069