

Therapeutic Drug Monitoring in Patients with Systemic Lupus Erythematosus: Utility and Gaps

 , , and
, , and
Abstract
1. Introduction
2. Methods
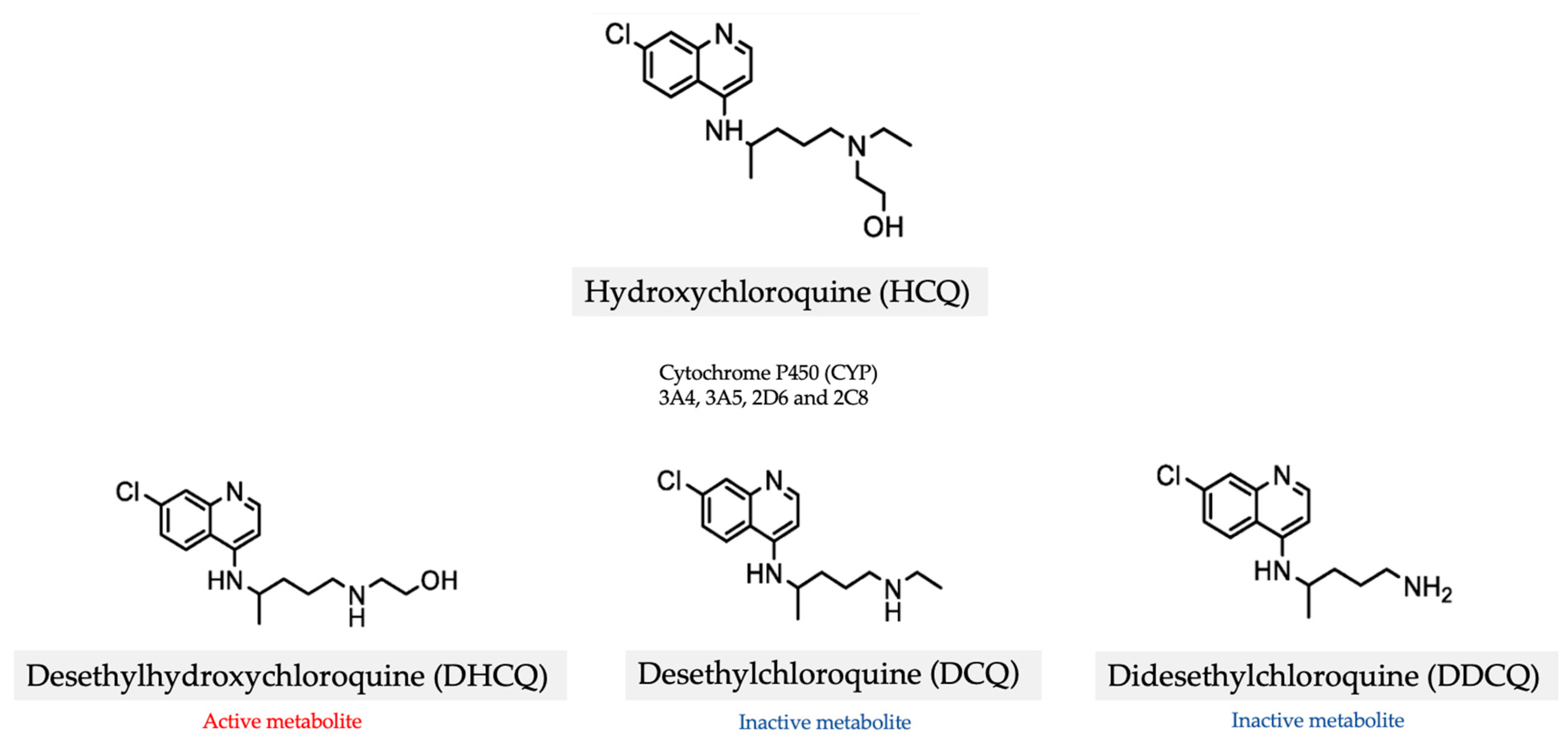
3. Hydroxychloroquine (HCQ)
4. Mycophenolate
5. Calcineurin Inhibitors
6. Azathioprine (AZA)
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cornet, A.; Andersen, J.; Myllys, K.; Edwards, A.; Arnaud, L. Living with systemic lupus erythematosus in 2020: A European patient survey. Lupus Sci. Med. 2021, 8, e000469. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.H.; Chan, T.M. Lupus nephritis in Asia: Clinical features and management. Kidney Dis. 2015, 1, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.; Jawad, A. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology 2016, 56, i67–i77. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Shen, L.; Huq, M.; Kandane-Rathnayake, R.; Golder, V.; Louthrenoo, W.; Chen, Y.-H.; Hamijoyo, L.; Luo, S.-F.; Wu, Y.-J.J.; et al. Impact of low disease activity, remission, and complete remission on flares following tapering of corticosteroids and immunosuppressive therapy in patients with systemic lupus erythematosus: A multinational cohort study. Lancet Rheumatol. 2023, 5, E584–E593. [Google Scholar]
- Alarcón, G.S.; McGwin, G.; Bertoli, A.M.; Fessler, B.J.; Calvo-Alén, J.; Bastian, H.M.; Vila, L.M.; Reveille, J.D. Effect of hydroxychloroquine on the survival of patients with systemic lupus erythematosus: Data from LUMINA, a multiethnic US cohort (LUMINA L). Ann. Rheum. Dis. 2007, 66, 1168–1172. [Google Scholar] [CrossRef]
- Tsakonas, E.; Joseph, L.; Esdaile, J.M.; Choquette, D.; Senécal, J.Ļ.; Cividino, A.; Danoff, D.; Osterland, C.K.; Yeadon, C.; Smith, C.D. A long-term study of hydroxychloroquine withdrawal on exacerbations in systemic lupus erythematosus. The Canadian hydroxychloroquine study group. Lupus 1998, 7, 80–85. [Google Scholar]
- Rao, I.R.; Kolakemar, A.; Shenoy, S.V.; Attur, R.P.; Nagaraju, S.P.; Rangaswamy, D.; Bhojaraja, M.V. Hydroxychloroquine in nephrology: Current status and future directions. J. Nephrol. 2023, 36, 2191–2208. [Google Scholar] [CrossRef]
- Akhavan, P.; Su, J.; Lou, W.; Gladman, D.D.; Urowitz, M.B.; Fortin, P.R. The early protective effect of hydroxychloroquine on the risk of cumulative damage in patients with systemic lupus erythematosus. J. Rheumatol. 2013, 40, 831–841. [Google Scholar] [CrossRef]
- Kravvariti, E.; Koutsogianni, A.; Samoli, E.; Sfikakis, P.P.; Tektonidou, M.G. The effect of hydroxychloroquine on thrombosis prevention and antiphospholipid antibody levels in primary antiphospholipid syndrome: A pilot open label randomized prospective study. Autoimmun. Rev. 2020, 19, 102491. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Andersen, J.; Aringer, M.; Arnaud, L.; Bae, S.C.; Boletis, J.; Bruce, I.N.; Cervera, R.; Doria, A.; et al. EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann. Rheum. Dis. 2023, 83, 15–29. [Google Scholar] [CrossRef]
- Hahn, B.H.; McMahon, M.; Wilkinson, A.H.; Wallace, W.D.; Daikh, D.I.; Fitzgerald, J.; Karpouzas, G.A.; Merrill, J.T.; Wallace, D.J.; Yazdany, J.; et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res. 2012, 64, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Kellner, U.; Lai, T.Y.Y.; Melles, R.B.; Mieler, W.F. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Am. Acad. Ophthalmol. Statement 2016, 123, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Melles, R.B.; Marmor, M.F. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol. 2014, 132, 1453. [Google Scholar] [CrossRef] [PubMed]
- Huybrechts, K.F.; Bateman, B.T.; Zhu, Y.; Straub, L.; Mogun, H.; Kim, S.Y.; Desai, R.J.; Hernandez-Diaz, S. Hydroxychloroquine early in pregnancy and risk of birth defects. Am. J. Obstet. Gynecol. 2021, 224, 290.e1–290.e22. [Google Scholar] [CrossRef] [PubMed]
- Soichot, M.; Mégarbane, B.; Houzé, P.; Chevillard, L.; Fonsart, J.; Baud, F.J.; Laprevote, O.; Bourgogne, E. Development, validation and clinical application of a LC-MS/MS method for the simultaneous quantification of hydroxychloroquine and its active metabolites in human whole blood. J. Pharm. Biomed. Anal. 2014, 100, 131–137. [Google Scholar] [CrossRef]
- Blanchet, B.; Jallouli, M.; Allard, M.; Ghillani-Dalbin, P.; Galicier, L.; Aumaître, O.; Chasset, F.; Guern, V.L.; Liote, F.; Smail, A.; et al. Hydroxychloroquine levels in patients with systemic lupus erythematosus: Whole blood is preferable but serum levels also detect non-adherence. Arthritis Res. Ther. 2020, 22, 223. [Google Scholar] [CrossRef]
- Rombo, L.; Ericsson, Ö.; Alvän, G.; Lindström, B.; Gustafsson, L.L.; Sjöqvist, F. Chloroquine and desethylchloroquine in plasma, serum, and whole blood. Ther. Drug Monit. 1985, 7, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Highley, M.S.; De Bruijn, E.A. Erythrocytes and the transport of drugs and endogenous compounds. Pharm. Res. 1996, 13, 186–195. [Google Scholar] [CrossRef]
- Rendić, S.; Guengerich, F.P. Metabolism and interactions of chloroquine and hydroxychloroquine with human cytochrome P450 enzymes and drug transporters. Curr. Drug Metab. 2020, 21, 1127–1135. [Google Scholar]
- Dima, A.; Jurcuț, C.; Chasset, F.; Felten, R.; Arnaud, L. Hydroxychloroquine in systemic lupus erythematosus: Overview of current knowledge. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X2110730. [Google Scholar] [CrossRef]
- Paludetto, M.N.; Kurkela, M.; Kahma, H.; Backman, J.T.; Niemi, M.; Filppula, A.M. Hydroxychloroquine is metabolized by cytochrome P450 2D6, 3A4, and 2C8, and inhibits cytochrome P450 2D6, while its metabolites also inhibit cytochrome P450 3A in vitro. Drug Metab. Dispos. 2022, 51, 293–305. [Google Scholar] [CrossRef]
- Yang, J.; Holmquist, B.; Punzalan, R.; Chun, K. Therapeutic drug monitoring (TDM) of hydroxychloroquine in whole blood: Analysis of over 10,000 patient results using lab developed liquid chromatography tandem mass spectrometry. Arthritis Rheumatol. 2023, 75. [Google Scholar]
- Jallouli, M.; Galicier, L.; Zahr, N.; Aumaître, O.; Françès, C.; Guern, V.L.; Liote, F.; Smail, A.; Limal, N.; Perard, L.; et al. Determinants of hydroxychloroquine blood concentration variations in systemic lupus erythematosus. Arthritis Rheumatol. 2015, 67, 2176–2184. [Google Scholar] [CrossRef] [PubMed]
- Leroux, G.; Costedoat-Chalumeau, N.; Hulot, J.; Amoura, Z.; Françès, C.; Aymard, G.; Lechat, P.; Piette, J.-C. Relationship between blood hydroxychloroquine and desethylchloroquine concentrations and cigarette smoking in treated patients with connective tissue diseases. Ann. Rheum. Dis. 2007, 66, 1547–1548. [Google Scholar] [CrossRef] [PubMed]
- Dervieux, T.; Brady, K.; Thomas, D.; Conklin, J.; Fung, E.; Ibarra, C.; Petri, M. Distribution and predictors of whole blood hydroxychloroquine levels in clinical rheumatology practices in the United States. Arthritis Rheumatol. 2019, 71. [Google Scholar]
- Lee, J.Y.; Vinayagamoorthy, N.; Han, K.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Jung, S.-H.; Park, S.-W.; Chung, Y.-J.; Park, S.-H. Association of polymorphisms of cytochrome P450 2D6 with blood hydroxychloroquine levels in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2015, 68, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Tan, T.; Cao, X.; Pan, M.; Yang, C.; Wang, J.; Shuai, Z.; Xia, Q. Relationship of cytochrome P450 gene polymorphisms with blood concentrations of hydroxychloroquine and its metabolites and adverse drug reactions. BMC Med. Genom. 2022, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Pons-Estel, G.J.; Alarcón, G.S.; McGwin, G.; Danila, M.I.; Zhang, J.; Bastian, H.M.; Reveille, J.D.; Vila, L.M. Protective effect of hydroxychloroquine on renal damage in patients with lupus nephritis: LXV, data from a multiethnic US cohort. Arthritis Care Res. 2009, 61, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Bose, C.; Karaduta, O.; Apostolov, E.O.; Kaushal, G.P.; Fahmi, T.; Segal, M.S.; Shah, S.V. Impact of hydroxychloroquine on atherosclerosis and vascular stiffness in the presence of chronic kidney disease. PLoS ONE 2015, 10, e0139226. [Google Scholar] [CrossRef]
- Zhong, X.; Jin, Y.; Zhang, Q.; Liu, S.; He, J. Low estimated glomerular filtration rate is an independent risk factor for higher hydroxychloroquine concentration. Clin. Rheumatol. 2023, 42, 1943–1950. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Cheema, K.; Anders, H.J.; Aringer, M.; Bajema, I.; Boletis, J.; Frangou, E.; Houssiau, F.A.; Hollis, J.; et al. 2019, update of the joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann. Rheum. Dis. 2020, 79, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Durcan, L.; Clarke, W.; Magder, L.S.; Petri, M. Hydroxychloroquine blood levels in systemic lupus erythematosus: Clarifying dosing controversies and improving adherence. J. Rheumatol. 2015, 42, 2092–2097. [Google Scholar] [CrossRef]
- Shimizu, M.; Furudate, S.; Nagai, Y.; Shimada, K.; Ohshima, M.; Setoguchi, K.; Hashiguchi, M.; Yokogawa, N. Pharmacokinetics of hydroxychloroquine in Japanese systemic lupus erythematosus patients with renal impairment. Mod. Rheumatol. 2022, 33, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, J.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Park, S.H. Factors related to blood hydroxychloroquine concentration in patients with systemic lupus erythematosus. Arthritis Care Res. 2017, 69, 536–542. [Google Scholar]
- Fasano, S.; Messiniti, V.; Ludici, M.; Coscia, M.A.; Ciccia, F. Hydroxychloroquine daily dose, hydroxychloroquine blood levels and the risk of flares in patients with systemic lupus erythematosus. Lupus Sci. Med. 2023, 10, e000841. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Unnithan, R.; Hansen, K.E.; Costedoat-Chalumeau, N.; Bartels, C.M. Clinical significance of monitoring hydroxychloroquine levels in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Arthritis Care Res. 2021, 73, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Petri, M. Drug monitoring in systemic lupus erythematosus. Curr. Opin. Pharmacol. 2022, 64, 102225. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Chewning, B.; Astor, B.; Bartels, C.M. Therapeutic range of hydroxychloroquine blood levels may reduce odds of high lupus disease activity. Arthritis Rheumatol. 2023, 75. [Google Scholar]
- Nguyen, Y.; Blanchet, B.; Urowitz, M.; Hanly, J.; Gordon, C.; Bae, S.; Romero-Diaz, J.; Sanchez-Guerrero, J.; Clarke, A.E.; Bernatsky, S.; et al. Severe non-adherence to hydroxychloroquine is associated with flares, early damage, and mortality in systemic lupus erythematosus: Data from 660 patients from the Systemic Lupus International Collaborating Clinics (SLICC) Inception Cohort. Arthritis Rheumatol. 2022, 74. [Google Scholar]
- Petri, M.; Konig, M.F.; Li, J.; Goldman, D.W. Association of higher hydroxychloroquine blood levels with reduced thrombosis risk in systemic lupus erythematosus. Arthritis Rheumatol. 2021, 73, 997–1004. [Google Scholar] [CrossRef]
- Petri, M.; Elkhalifa, M.; Li, J.; Magder, L.S. Hydroxychloroquine blood levels predict hydroxychloroquine retinopathy. Arthritis Rheumatol. 2020, 72, 448–453. [Google Scholar] [CrossRef]
- Belmont, H.M.; Haj-Ali, M. Whole blood drug levels do not correlate with QTc intervals in hydroxychloroquine-treated systemic lupus erythematosus patients. Rheumatology 2022, 62, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Balevic, S.J.; Cohen-Wolkowiez, M.; Eudy, A.M.; Green, T.P.; Schanberg, L.E.; Clowse, M.E.B. Hydroxychloroquine Levels throughout Pregnancies Complicated by Rheumatic Disease: Implications for Maternal and Neonatal Outcomes. J. Rheumatol. 2018, 46, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Allison, A. Mechanisms of action of mycophenolate mofetil. Lupus 2005, 14 (Suppl. S3), 2–8. [Google Scholar] [CrossRef] [PubMed]
- Gabardi, S.; Tran, J.L.; Clarkson, M.R. Enteric-coated mycophenolate sodium. Ann. Pharmacother. 2003, 37, 1685–1693. [Google Scholar] [CrossRef]
- Van Gelder, T.; Berden, J.H.M.; Berger, S.P. To TDM or not to TDM in lupus nephritis patients treated with MMF? Nephrol. Dial. Transplant. 2014, 30, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Pawiński, T. Therapeutic drug monitoring of mycophenolic acid: A potential treatment for lupus nephritis. Kidney Int. 2010, 78, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Genvigir, F.D.V.; Cerda, A.; Hirata, T.D.C.; Hirata, M.H.; Hirata, R.D.C. Mycophenolic acid pharmacogenomics in kidney transplantation. J. Transl. Genet. Genom. 2020, 4, 320–355. [Google Scholar] [CrossRef]
- Yap, D.Y.H.; Tam, C.H.; Yung, S.; Wong, S.P.; Tang, C.L.; Mok, T.M.Y.; Yuen, C.K.Y.; Ma, M.K.M.; Lau, C.S.; Chan, T.M. Pharmacokinetics and pharmacogenomics of mycophenolic acid and its clinical correlations in maintenance immunosuppression for lupus nephritis. Nephrol. Dial. Transplant. 2018, 35, 810–818. [Google Scholar] [CrossRef]
- Shaw, L.M.; Korecka, M.; Venkataramanan, R.; Goldberg, L.R.; Bloom, R.D.; Brayman, K.L. Mycophenolic acid pharmacodynamics and pharmacokinetics provide a basis for rational monitoring strategies. Am. J. Transplant. 2003, 3, 534–542. [Google Scholar] [CrossRef]
- Hesselink, D.A.; Van Gelder, T. Genetic and nongenetic determinants of between-patient variability in the pharmacokinetics of mycophenolic acid. Clin. Pharmacol. Ther. 2005, 78, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Shuker, N.; Hesselink, D.A.; Van Schaik, R.H.N.; Zhang, X.; Van Gelder, T. Do Asian renal transplant patients need another mycophenolate mofetil dose compared with Caucasian or African American patients? Transpl. Int. 2014, 27, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M.; Mick, R.; Nowak, I.; Korecka, M.; Brayman, K.L. Pharmacokinetics of mycophenolic acid in renal transplant patients with delayed graft function. J. Clin. Pharmacol. 1998, 38, 268–275. [Google Scholar] [CrossRef]
- Lertdumrongluk, P.; Somparn, P.; Kittanamongkolchai, W.; Traitanon, O.; Vadcharavivad, S.; Avihingsanon, Y. Pharmacokinetics of mycophenolic acid in severe lupus nephritis. Kidney Int. 2010, 78, 389–395. [Google Scholar] [CrossRef]
- Kumar, S.; Kirubakaran, N.; Punnen, A.; Prabha, R.; Agarwal, I. Therapeutic drug monitoring of mycophenolate mofetil for the treatment of pediatric lupus nephritis: A cross-sectional study. Indian J. Rheumatol. 2022, 17, 124. [Google Scholar] [CrossRef]
- Mizaki, T.; Nobata, H.; Banno, S.; Yamaguchi, M.; Kinashi, H.; Iwagaitsu, S.; Ishimoto, T.; Kuru, Y.; Ohnishi, M.; Sako, K.-I.; et al. Population pharmacokinetics and limited sampling strategy for therapeutic drug monitoring of mycophenolate mofetil in Japanese patients with lupus nephritis. J. Pharm. Health Care Sci. 2023, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, D.; Meur, Y.L.; Cantarovich, M.; Tredger, M.; Tett, S.E.; Cattaneo, D.; Tonshoff, B.; Holt, D.W.; Chapman, J.; Gelder, T. Consensus report on therapeutic drug monitoring of mycophenolic acid in solid organ transplantation. Clin. J. Am. Soc. Nephrol. 2010, 5, 341–358. [Google Scholar] [CrossRef]
- Kuypers, D.; Claes, K.; Evenepoel, P.; Maes, B.; Coosemans, W.; Pirenne, J.; Vanrenterghem, Y. Long-term changes in mycophenolic acid exposure in combination with tacrolimus and corticosteroids are dose dependent and not reflected by trough plasma concentration: A prospective study in 100 de novo renal allograft recipients. J. Clin. Pharmacol. 2003, 43, 866–880. [Google Scholar] [CrossRef]
- Okamoto, M.; Wakabayashi, Y.; Higuchi, A.; Kadotani, Y.; Ogino, S.; Ushigome, H.; Akioka, K.; Kaihara, S.; Yoshimura, N. Therapeutic drug monitoring of mycophenolic acid in renal transplant recipients. Transplant. Proc. 2005, 37, 859–860. [Google Scholar] [CrossRef]
- Kuriyama, T.; Ozaki, T.; Ozeki, T.; Hachiya, A.; Kim, H.; Kato, N.; Ishimoto, T.; Kato, S.; Kosugi, T.; Tsuboi, N.; et al. Investigation on the benefits of mycophenolate mofetil and therapeutic drug monitoring in the treatment of Japanese patients with lupus nephritis. Clin. Exp. Nephrol. 2018, 22, 1341–1350. [Google Scholar]
- Pourafshar, N.; Karimi, A.; Wen, X.; Sobel, E.S.; Pourafshar, S.; Agrawal, N.; Segal, E.; Mohandas, R.; Segal, M.S. The utility of trough mycophenolic acid levels for the management of lupus nephritis. Nephrol. Dial. Transplant. 2018, 34, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Staatz, C.E.; Tett, S.E. Clinical pharmacokinetics and pharmacodynamics of mycophenolate in solid organ transplant recipients. Clin. Pharmacokinet. 2007, 46, 13–58. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.; Fleming, D.H.; Mathew, B.S.; Varughese, S.; Jeyaseelan, V.; Tamilarasi, V.; Jacob, C.K.; John, G.T. Pharmacokinetics of concentration-controlled mycophenolate mofetil in proliferative lupus nephritis. Ther. Drug Monit. 2014, 36, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Daleboudt, G.M.N.; Reinders, M.E.J.; Hartigh, R.D.; Huizinga, T.W.J.; Rabelink, T.J.; De Fijter, J.W.; Berger, S.P. Concentration-controlled treatment of lupus nephritis with mycophenolate mofetil. Lupus 2012, 22, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Łuszczyńska, P.; Pawiński, T. Therapeutic drug monitoring of mycophenolic acid in lupus nephritis. Ther. Drug Monit. 2015, 37, 711–717. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, H.; Liu, Z.; Xing, C.; Fu, P.; Ni, Z.; Chen, J.; Lin, H.; Liu, F.; He, Y.; et al. Multitarget therapy for induction treatment of lupus nephritis. Ann. Intern. Med. 2015, 162, 18–26. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Z.; Zhou, M.; Liu, Z.; Chen, J.; Xing, C.; Lin, H.; Ni, Z.; Fu, P.; Liu, F.; et al. Multitarget therapy for maintenance treatment of lupus nephritis. J. Am. Soc. Nephrol. 2017, 28, 3671–3678. [Google Scholar] [CrossRef]
- Hardinger, K.; Magee, C.C. Pharmacology of cyclosporine and tacrolimus. In UpToDate [Internet]; Furst, D.E., Brennan, D.C., Eds.; Wolters Kluwer: Waltman, DC, USA, 2020. [Google Scholar]
- Safarini, O.A.; Keshavamurthy, C.; Patel, P. Calcineurin inhibitors. In Treasure Island (FL); StatPearls Publishing: St. Petersburg, FL, USA, 2022. [Google Scholar]
- Birdwell, K.A.; Decker, B.S.; Barbarino, J.M.; Peterson, J.; Stein, C.M.; Sadée, W.; Wang, D.; Vinks, A.A.; He, Y.; Swen, J.J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin. Pharmacol. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef]
- Farhat, R.; Mendel, A.; Malhamé, I.; Lee, J.Y.E.; Ciofani, L.; Bernatsky, S.; Vinet, E. Therapeutic drug monitoring of azathioprine and tacrolimus in SLE pregnancies: Preliminary results from the LEGACY cohort. Ann. Rheum. Dis. 2023, 82 (Suppl. S1), 906–907. [Google Scholar]
- Kahan, B.D.; Grevel, J. Optimisation of cyclosporine therapy in renal transplantation by a pharmacokinetic strategy. Transplantation 1988, 46, 631–644. [Google Scholar] [CrossRef]
- Grevel, J.; Welsh, M.S.; Kahan, B.D. Cyclosporine monitoring in renal transplantation: Area under the curve monitoring is superior to trough-level monitoring. Ther. Drug Monit. 1989, 11, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Kumar, R.; Gupta, P. Utility of C-2 (Cyclosporine) monitoring in postrenal transplant patients: A study in the Indian population. Indian J. Nephrol. 2008, 18, 118. [Google Scholar] [CrossRef] [PubMed]
- Vari, C.E.; Tero-Vescan, A.; Imre, S.; Mutean, D.L. Therapeutic drug monitoring of cyclosporine in transplanted patients. Possibilities, controversy, causes for failure. Farmacia 2012, 60, 595–601. [Google Scholar]
- Uchida, K.; Tominaga, Y.; Haba, T.; Katayama, T.; Matsuoka, S.; Sato, T.; Goto, N.; Takeda, A.; Morozumi, K.; Takagi, H. Usefulness of monitoring of AUC0–4h during the induction period of immunosuppressive therapy with tacrolimus after renal transplantation. Transplant. Proc. 2002, 34, 1736–1737. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K.A.; Povlsen, J.V.; Madsen, S.; Madsen, M.; Hansen, H.E.; Pedersen, A.R.; Heinsvig, E.-M.; Poulsen, J. C2 (2-h) levels are not superior to trough levels as estimates of the area under the curve in tacrolimus-treated renal-transplant patients. Nephrol. Dial. Transplant. 2002, 17, 1487–1490. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Teng, Y.K.O.; Ginzler, E.M.; Arriens, C.; Caster, D.J.; Romero-Diaz, J.; Gibson, K.; Kaplan, J.; Lisk, L.; Navarra, S.; et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): A double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2070–2080. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Ginzler, E.M.; Gibson, K.; Satirapoj, B.; Santillán, A.E.Z.; Levchenko, O.; Navarra, S.; Atsumi, T.; Yasuda, S.; Chavez-Perez, N.N.; et al. Safety and efficacy of long-term voclosporin treatment for lupus nephritis in the phase 3 AURORA 2 clinical trial. Arthritis Rheumatol. 2024, 76, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, T.; Huizinga, R.B.; Noukens, J.; Lisk, L.J.; Solomons, N. Use of therapeutic drug monitoring does not add clinical value for voclosporin in patients with lupus nephritis. J. Am. Soc. Nephrol. 2020, 31, 594. [Google Scholar]
- Casteele, N.V.; Herfarth, H.; Katz, J.; Falck-Ytter, Y.; Singh, S. American Gastroenterological Association Institute technical review on the role of therapeutic drug monitoring in the management of inflammatory bowel diseases. Gastroenterology 2017, 153, 835–857.e6. [Google Scholar] [CrossRef]
- Gearry, R.B.; Barclay, M.L. Azathioprine and 6-mercaptopurine pharmacogenetics and metabolite monitoring in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2005, 20, 1149–1157. [Google Scholar] [CrossRef]
- Dean, L.; Kane, M. Mercaptopurine therapy and TPMT and NUDT15 genotype. In Medical Genetics Summaries; National Center for Biotechnology Information: Bethesda, MD, USA, 2020. [Google Scholar]
- Booth, R.A.; Ansari, M.; Loit, E.; Tricco, A.C.; Weeks, L.; Doucette, S.; Skidmore, B.; Sears, M.; Sy, R.; Karsh, J. Assessment of thiopurine S-methyltransferase activity in patients prescribed thiopurines: A systematic review. Ann. Intern. Med. 2011, 154, 814. [Google Scholar] [CrossRef]
- Ooi, C.J.; Hilmi, I.; Banerjee, R.; Chuah, S.W.; Ng, S.C.; Shu, W.; Makharia, G.K.; Pisespongsa, P.; Chen, M.H.; Ran, Z.H.; et al. Best practices on immunomodulators and biologic agents for ulcerative colitis and Crohn’s disease in Asia. Intest. Res. 2019, 17, 285–310. [Google Scholar] [CrossRef]
- Chao, K.; Huang, Y.; Zhu, X.; Tang, J.; Wang, X.; Lin, L.; Guo, H.; Zhang, C.; Li, M.; Yang, Q.; et al. Randomised clinical trial: Dose optimising strategy by NUDT15 genotyping reduces leucopenia during thiopurine treatment of Crohn’s disease. Aliment. Pharmacol. Ther. 2021, 54, 1124–1133. [Google Scholar] [CrossRef]
- Chang, J.Y.; Park, S.J.; Jung, E.S.; Jung, S.A.; Moon, C.M.; Chun, J.; Park, J.J.; Kim, E.S.; Park, Y.; Kim, T.-I.; et al. Genotype-based treatment with thiopurine reduces incidence of myelosuppression in patients with inflammatory bowel diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 2010–2018.e2. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.H.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Estevinho, M.M.; Afonso, J.; Rosa, I.; Lago, P.; Trindade, E.; Correia, L.; Dias, C.C.; Magro, F. A systematic review and meta-analysis of 6-thioguanine nucleotide levels and clinical remission in inflammatory bowel disease. J. Crohn’s Colitis 2017, 11, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- El-Azhary, R.A.; Farmer, S.A.; Drage, L.A.; Rogers, R.S.; McEvoy, M.T.; Davis, M.D.P.; Bridges, A.G.; Gibson, L.E. Thioguanine nucleotides and thiopurine methyltransferase in immunobullous diseases. Arch. Dermatol. 2009, 145, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Bradford, K.; Shih, D.Q. Optimizing 6-mercaptopurine and azathioprine therapy in the management of inflammatory bowel disease. World J. Gastroenterol. 2011, 17, 4166. [Google Scholar] [CrossRef] [PubMed]
- Askanase, A.; Wallace, D.J.; Weisman, M.H.; Tseng, C.; Bernstein, L.I.; Belmont, H.M.; Seidman, E.; Ishimori, M.; Izmirly, P.M.; Buyon, J.P. Use of pharmacogenetics, enzymatic phenotyping, and metabolite monitoring to guide treatment with azathioprine in patients with systemic lupus erythematosus. J. Rheumatol. 2009, 36, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Croyle, L.; Hoi, A.; Morand, E.F. Characteristics of azathioprine use and cessation in a longitudinal lupus cohort. Lupus Sci. Med. 2015, 2, e000105. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, C.; Lan, T.; Chen, H.; Chang, S.; Chen, Y.; Wang, J.-S.; Hung, W.-T.; Lan, J.-L.; Chen, D.-Y. Hydroxychloroquine reduces risk of incident diabetes mellitus in lupus patients in a dose-dependent manner: A population-based cohort study. Rheumatology 2015, 54, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.A.; Tun, K.M.; Atan, I.K.; Said, M.F.; Mustafar, R.; Zainuddin, A.A. New benefits of hydroxychloroquine in pregnant women with systemic lupus erythematosus: A retrospective study in a tertiary centre. Rev. Bras. De Ginecol. E Obs. 2020, 42, 705–711. [Google Scholar] [CrossRef]
- Appel, G.B.; Contreras, G.; Dooley, M.A.; Ginzler, E.M.; Isenberg, D.; Jayne, D.; Li, L.-S.; Mysler, E.; Sanchez-Guerrero, J.S.; Solomons, N.; et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J. Am. Soc. Nephrol. 2009, 20, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Houssiau, F.; D’Cruz, D.; Sangle, S.; Rémy, P.; Vasconcelos, C.; Petrović, R.; Fiehn, C.; Garrido, E.R.; Gilboe, I.-M.; Tektonidou, M.; et al. Azathioprine versus mycophenolate mofetil for long-term immunosuppression in lupus nephritis: Results from the MAINTAIN Nephritis Trial. Ann. Rheum. Dis. 2010, 69, 2083–2089. [Google Scholar] [CrossRef]
- Pisoni, C.N.; Sanchez, F.J.; Karim, Y.; Cuadrado, M.J.; D’Cruz, D.P.; Abbs, I.C.; Khamasta, M.A.; Hughes, G.R.V. Mycophenolate mofetil in systemic lupus erythematosus: Efficacy and tolerability in 86 patients. J. Rheumatol. 2005, 32, 1047–1052. [Google Scholar]
- Nannini, C.; Crowson, C.S.; Matteson, E.L.; Moder, K.G. Mycophenolate mofetil is effective in reducing disease flares in systemic lupus erythematosus patients: A retrospective study. Lupus 2009, 18, 394–399. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Drug | Hydroxychloroquine | Mycophenolate | Calcineurin Inhibitors | Azathioprine |
|---|---|---|---|---|
| Indication in SLE | - Anchor drug in SLE - Reduces lupus flares [6] and damage accrual - Prevents thrombosis [9] - Improves metabolic profile [94] - Reduces rates of preterm births, congenital heart block and pre-eclampsia [95] | - Induction and maintenance therapy in lupus nephritis [96,97] - Reduces glucocorticoid requirements and flares in extra-renal manifestations [98,99] | Induction and maintenance therapy in lupus nephritis as part of multitarget regimen [66,67] | Maintenance therapy for SLE |
| Key metabolisers | CYP3A4, 3A5, 2D6, 2C8 [21] | UDP-glucuronosyltransferases [47] | CYP3A4, 3A5 | Xanthine oxidase, TPMT, HPRT |
| Factors affecting blood concentration (besides dose and adherence) | - Age [25] - Gender [25] - CYP450 polymorphisms [26,27] - Renal function [23,30] | - Ethnicity [52] - Renal function [53] - Liver function [51] - Serum albumin levels [51] - Concomitant drugs (e.g., proton pump inhibitors, iron oxide) [51] | - CYP3A inhibitors or inducers including dietary polyphenols - CYP3A5 polymorphisms [70] | - Xanthine oxidase inhibitor - TPMT/NUDT15 polymorphisms [84] |
| Utility of monitoring | - Identify non-adherence, which predicts damage [39] and mortality - Predicts lupus flares and treatment failure [34,35] - Predicts thrombotic events [40] - Predicts HCQ retinopathy [41] | Determine appropriate dose to maximise efficacy and minimise toxicities | Determine appropriate dose to maximise efficacy and minimise toxicities | - 6-TGN levels can predict clinical response [89,90] - 6-MMP levels can predict hepatotoxicity [91] |
| Drug levels of clinical importance | - HCQ levels ≥ 750 ng/mL associated with 58% lower risk of active disease [36] - HCQ levels 750 to 1100 ng/mL associated with reduced odds of high lupus disease activity by 76 to 90% [38] - Mean HCQ levels > 1068 ng/mL, versus <648 ng/mL, reduce thrombotic events by 69% [40] - Mean HCQ levels > 1177 ng/mL associated with doubling of risk of retinopathy compared to 742 to 1176 ng/mL (7.9% versus 3.7%) [41] | - AUC0–12 of 30 to 60 mg × h/l associated with 68.8% complete renal remission of lupus nephritis at 12 months [64] - AUC threshold values of 30 to 45 mg × h/L and target trough threshold of 3 mg/L suggested by some authors [65] | - Tacrolimus trough levels of 5 to 7 ng/mL were maintained in the multitarget lupus nephritis trial [66] - Cyclosporine: no data | - 6-TGN levels of 235 to 450 pmol/8 × 108 RBCs associated with clinical remission in IBD patients [89] - 6-MMP levels > 5700 pmol/8 × 108 RBCs associated with hepatoxicity [91] - Optimal levels not yet established in SLE patients |
| Unknown gaps | - Pharmacokinetics, pharmacodynamics and optimal exposure of HCQ during pregnancy - Utility in predicting other adverse events besides retinopathy | Correlation between MPA trough level and MPA-AUC | Association of TDM with SLE outcomes | Target 6-TGN levels to predict therapeutic efficacy and toxicities in SLE patients |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, K.M.; Jiang, H.; Lo, E.A.G.; Hong, W.-Z.; Wong, E.T.-Y.; Chan, G.C.; Cho, J. Therapeutic Drug Monitoring in Patients with Systemic Lupus Erythematosus: Utility and Gaps. J. Clin. Med. 2024, 13, 451. https://doi.org/10.3390/jcm13020451
Chong KM, Jiang H, Lo EAG, Hong W-Z, Wong ET-Y, Chan GC, Cho J. Therapeutic Drug Monitoring in Patients with Systemic Lupus Erythematosus: Utility and Gaps. Journal of Clinical Medicine. 2024; 13(2):451. https://doi.org/10.3390/jcm13020451
Chicago/Turabian StyleChong, Kar Mun, He Jiang, Elaine Ah Gi Lo, Wei-Zhen Hong, Emmett Tsz-Yeung Wong, Gek Cher Chan, and Jiacai Cho. 2024. "Therapeutic Drug Monitoring in Patients with Systemic Lupus Erythematosus: Utility and Gaps" Journal of Clinical Medicine 13, no. 2: 451. https://doi.org/10.3390/jcm13020451
APA StyleChong, K. M., Jiang, H., Lo, E. A. G., Hong, W.-Z., Wong, E. T.-Y., Chan, G. C., & Cho, J. (2024). Therapeutic Drug Monitoring in Patients with Systemic Lupus Erythematosus: Utility and Gaps. Journal of Clinical Medicine, 13(2), 451. https://doi.org/10.3390/jcm13020451