Proposed Clinical Algorithm for Pleuroparenchymal Fibroelastosis (PPFE)

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
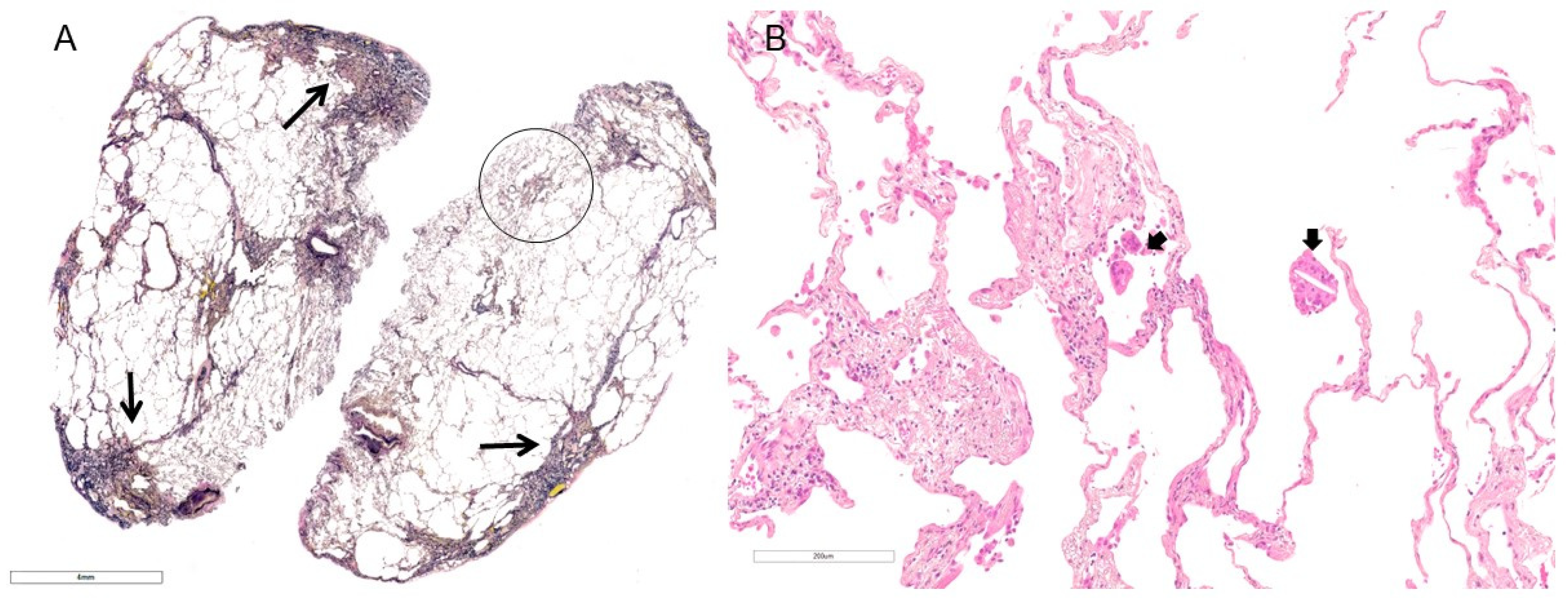
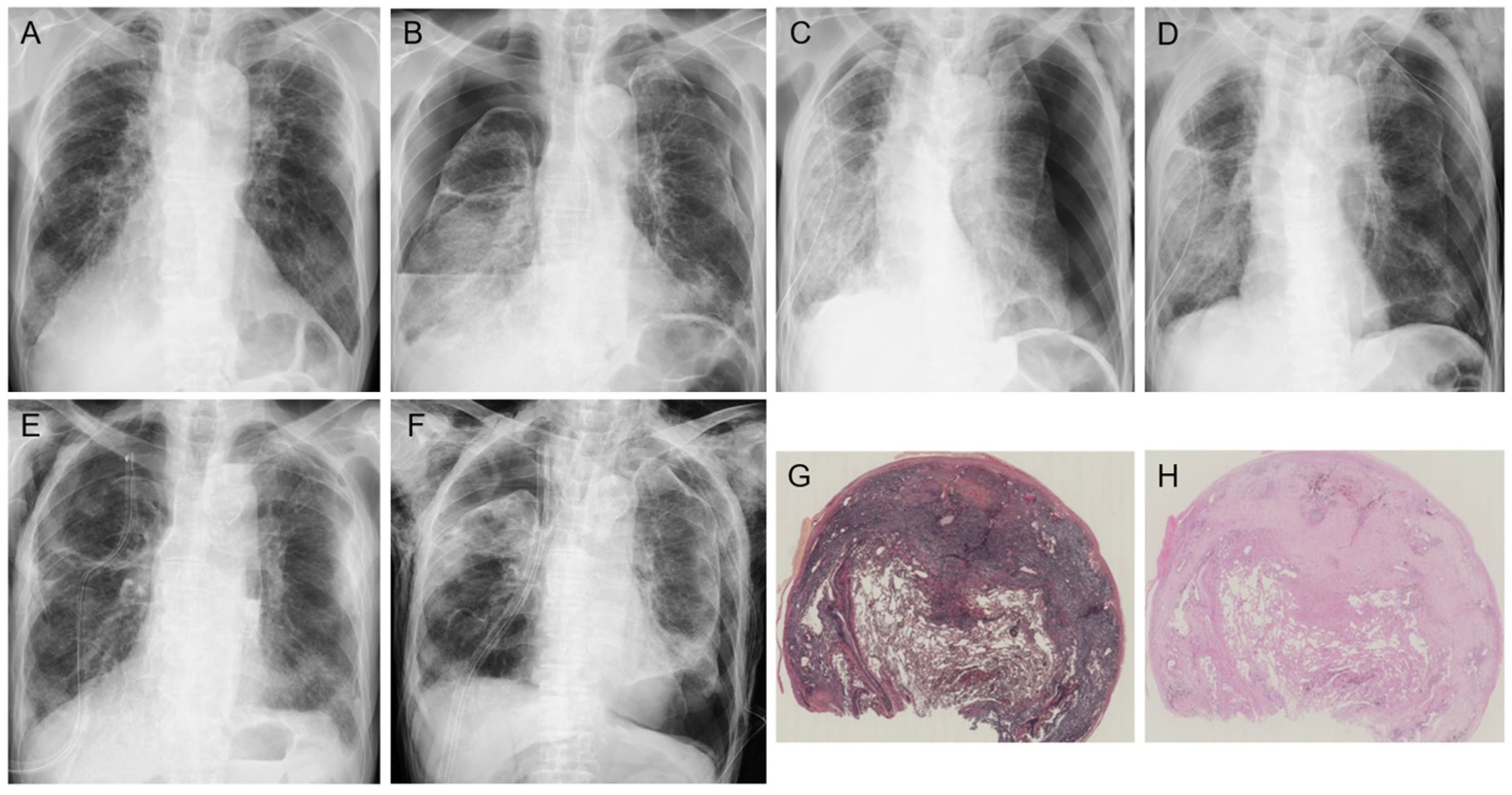
2. Clinical Features
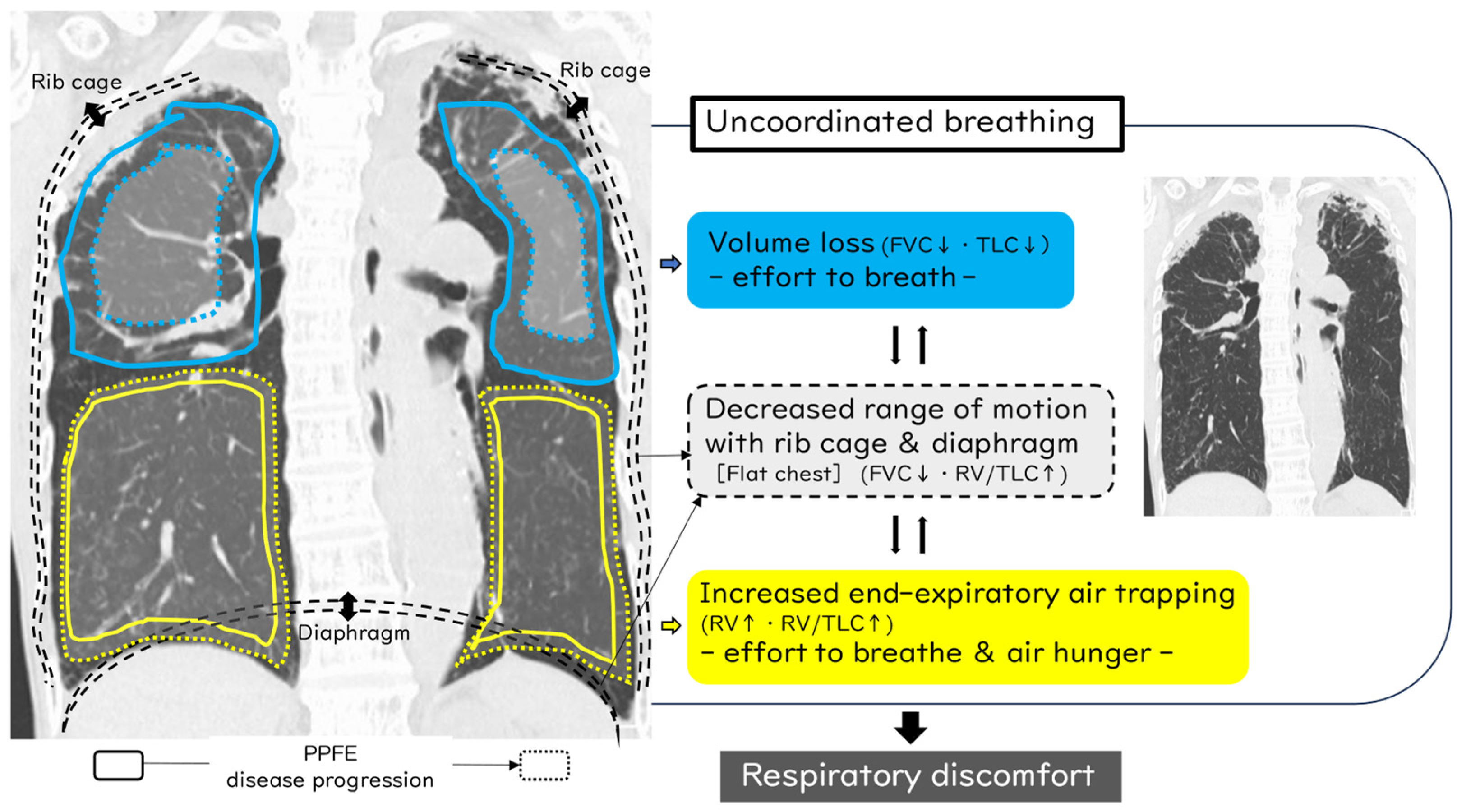
3. Important Points in the Mechanism of Dyspnea in PPFE Patients
4. Secondary PPFE
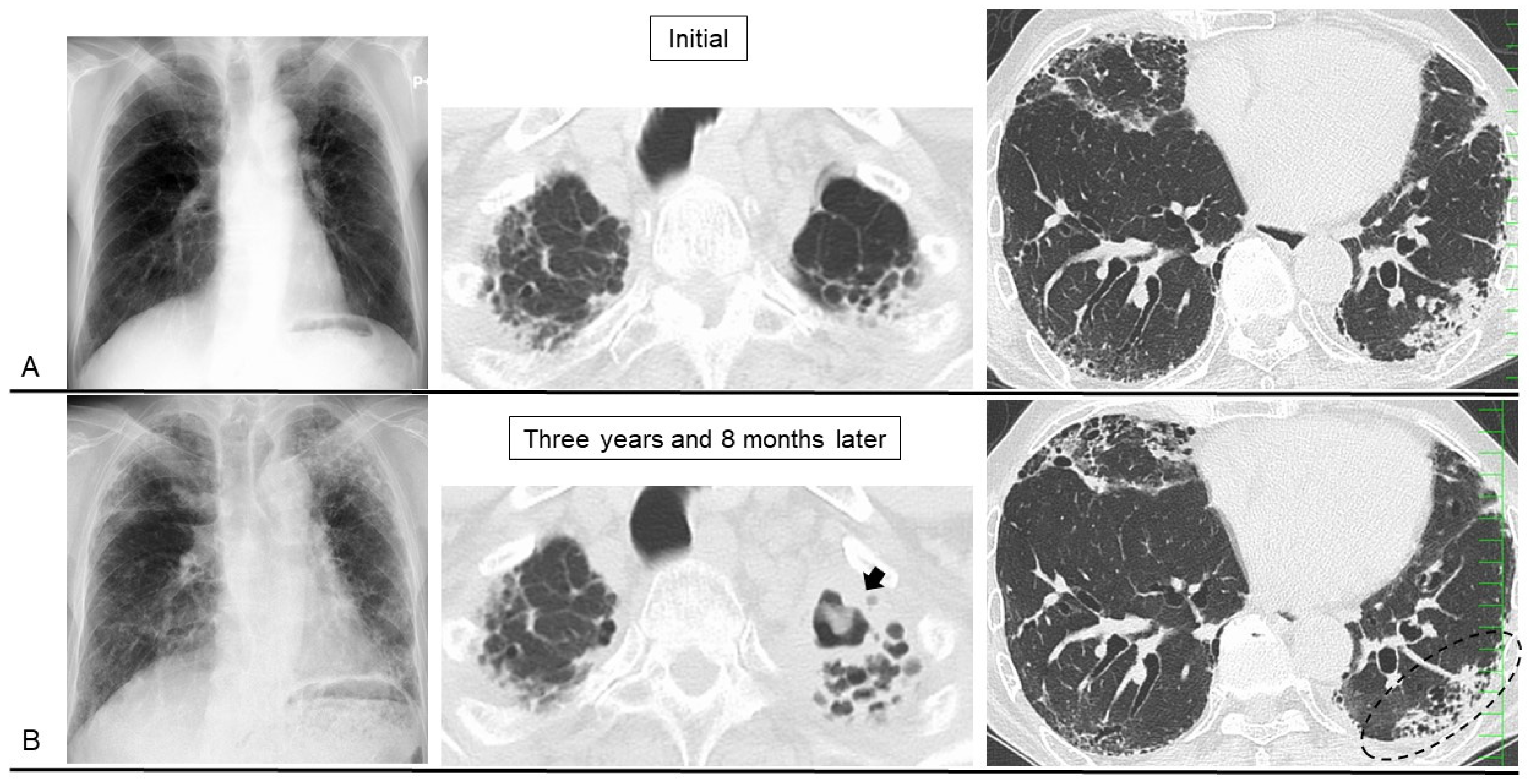
5. Disease Behavior and Monitoring Markers
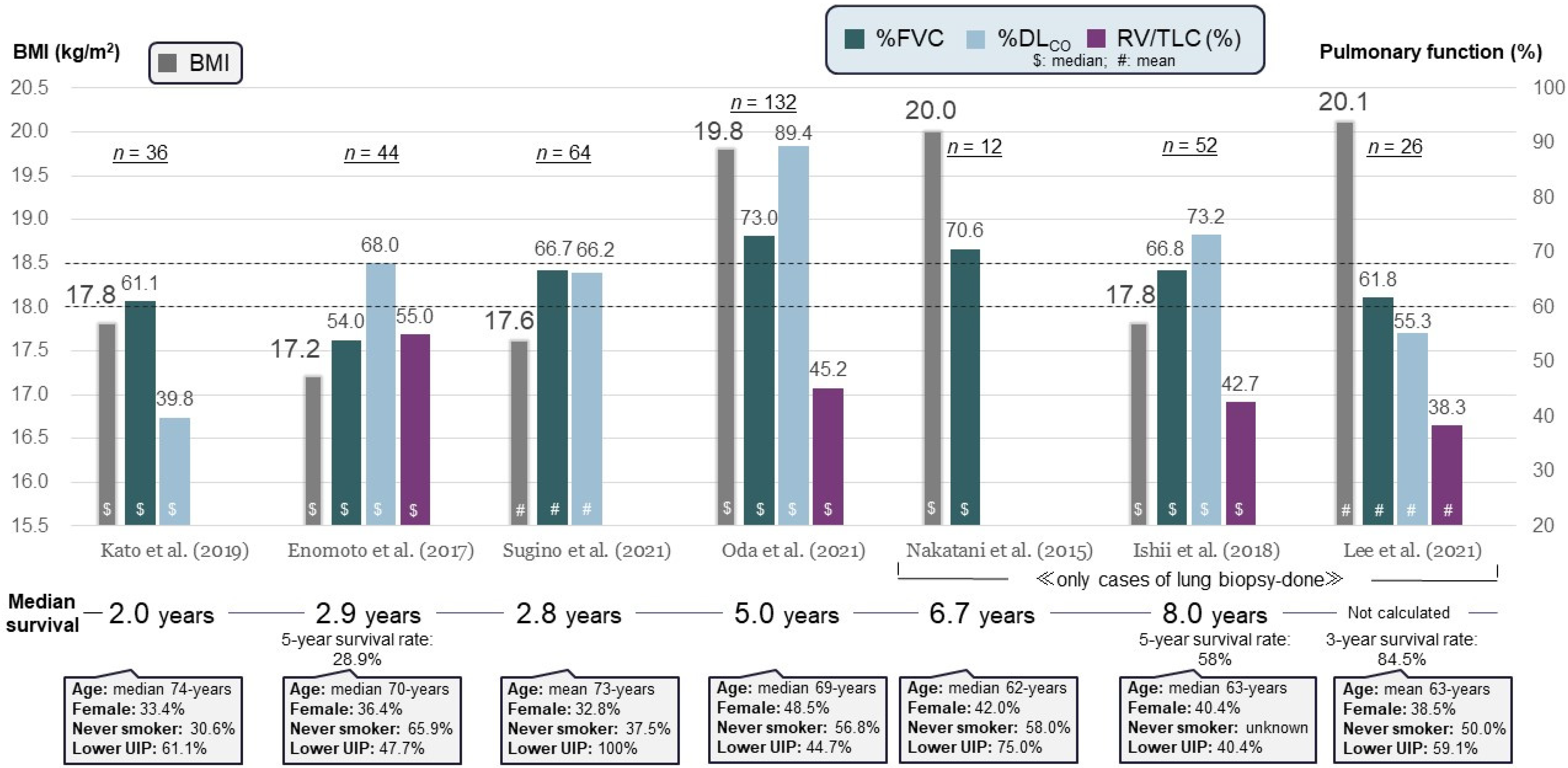
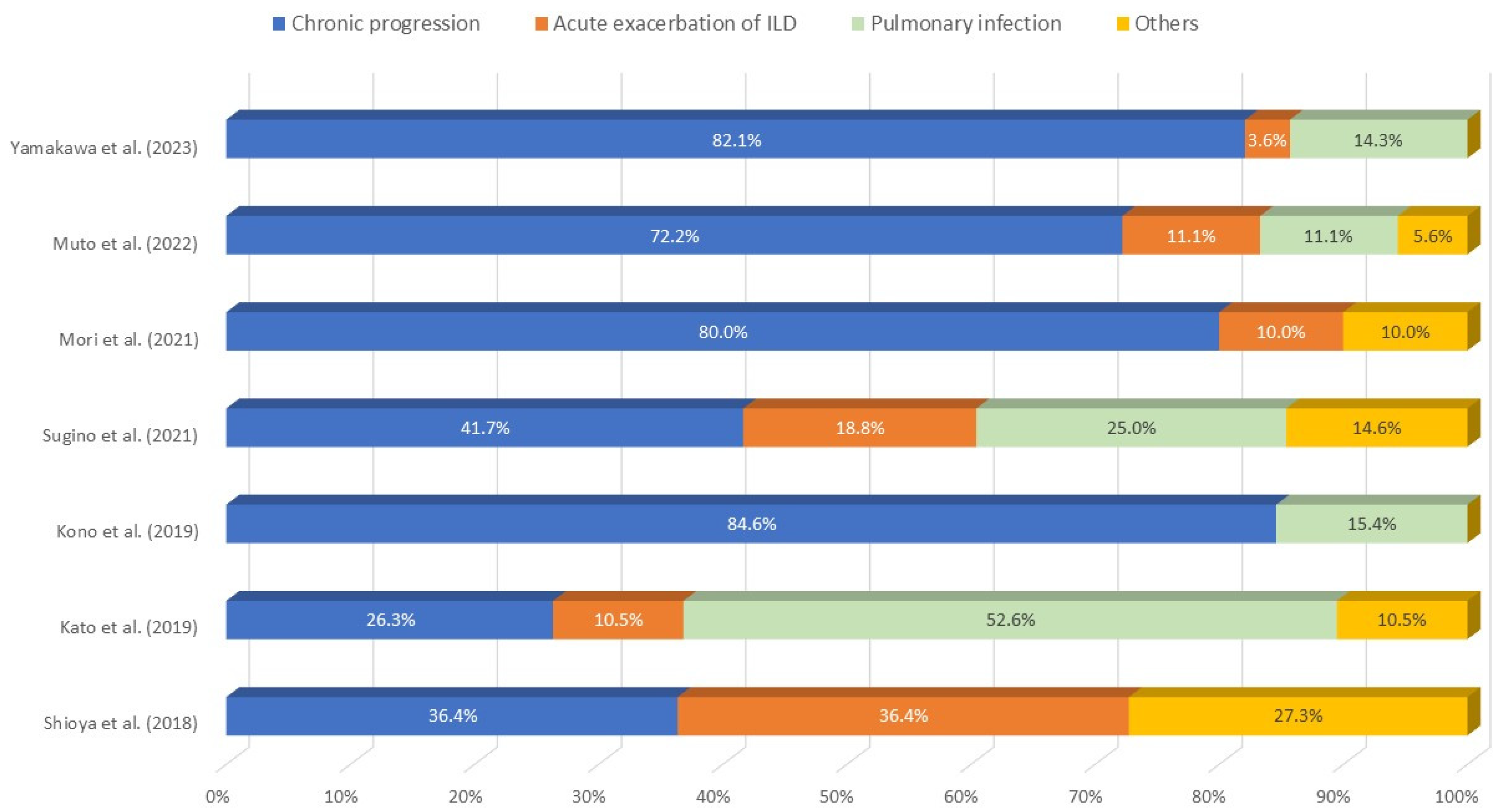
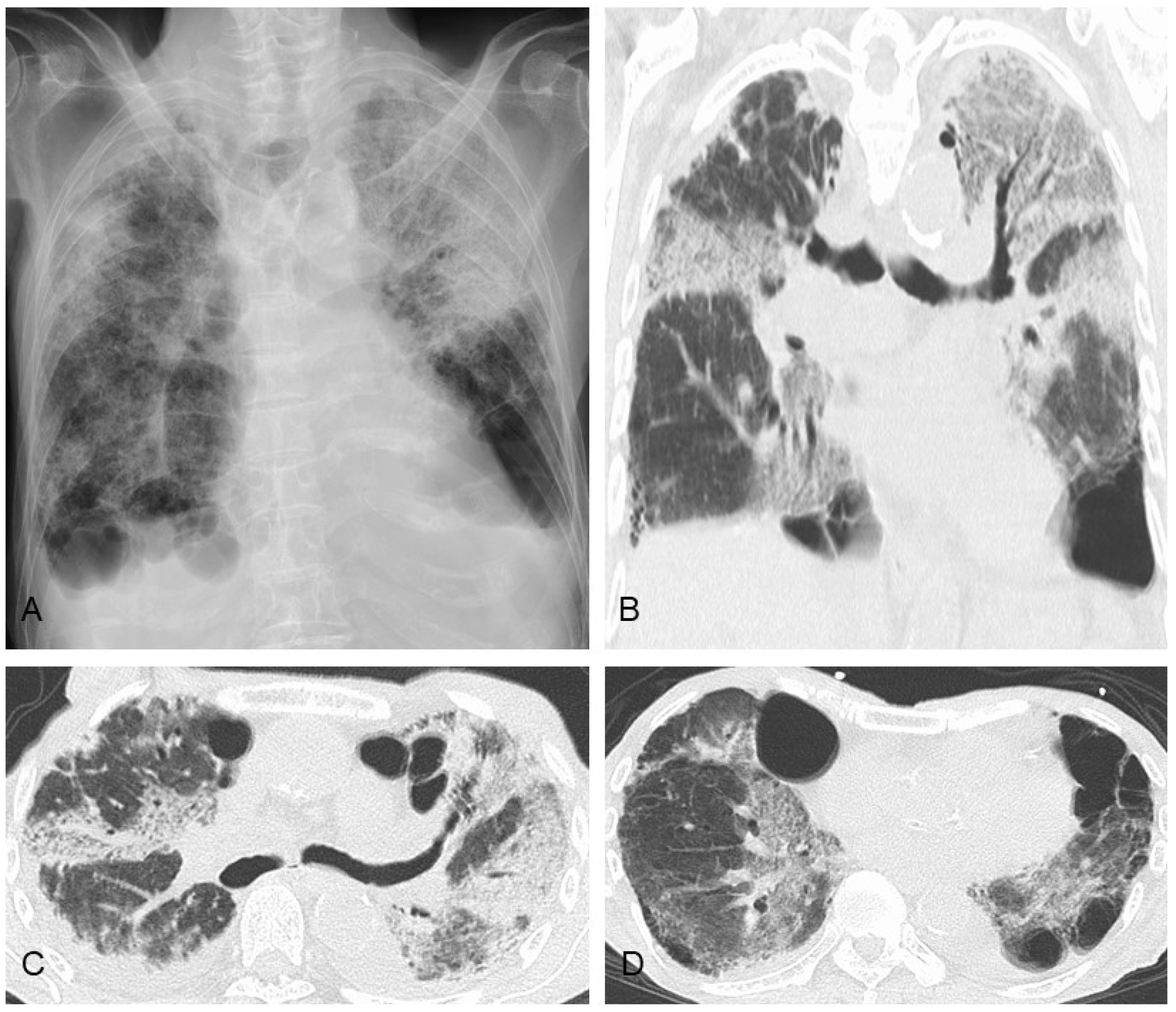
6. Prognostic Factors, Prognosis, and Causes of Death
7. Comorbidities
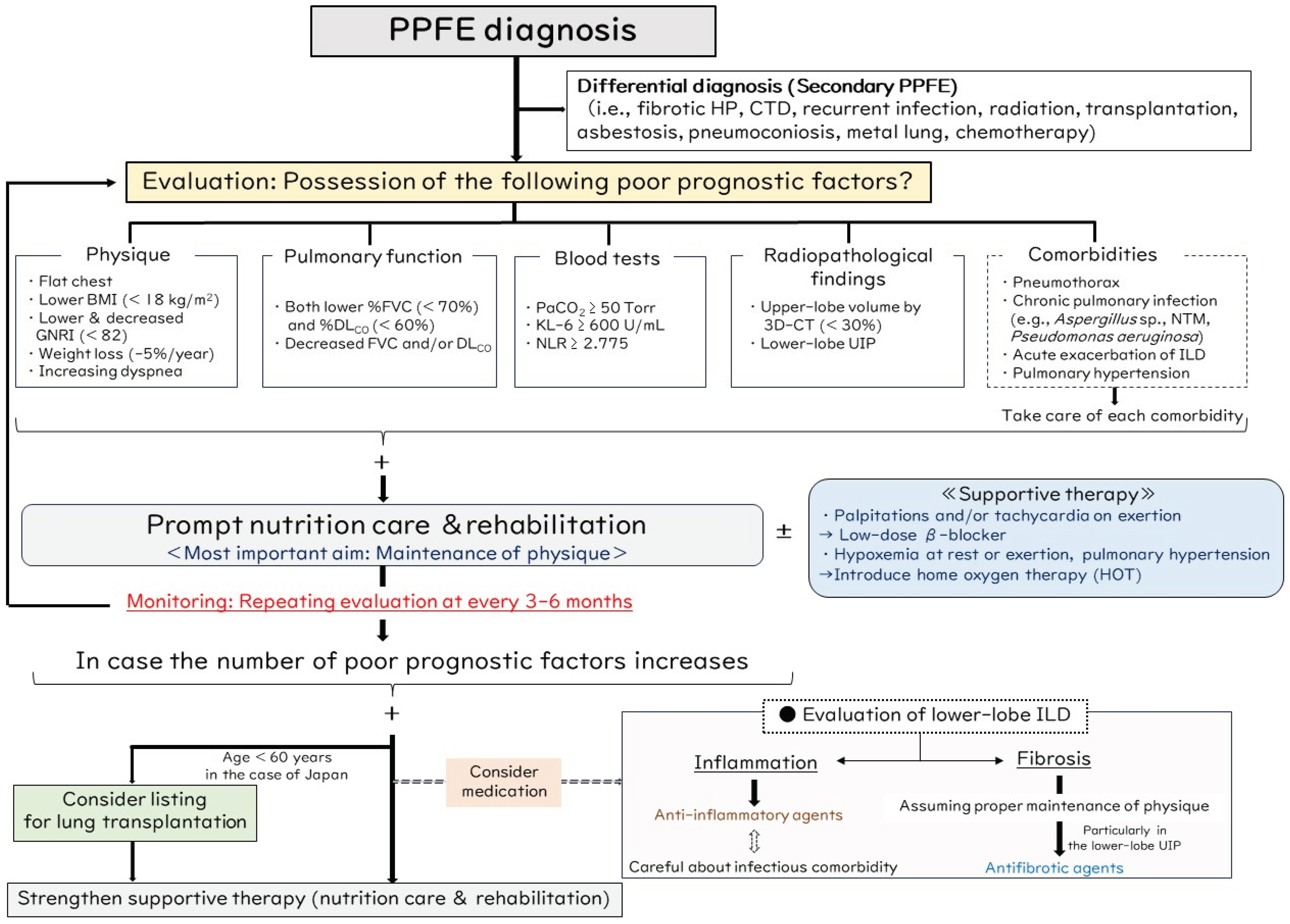
8. Proposed Management Algorithm for PPFE
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Amitani, R.; Niimi, A.; Kuse, F. Idiopathic pulmonary upper lobe fibrosis (IPUF). Kokyu 1992, 11, 693–699. [Google Scholar]
- Tetikkurt, C.; Ozturk, B.C.; Gungordu, N. Diagnosis of pleuroparenchymal fibroelastosis: A review. Monaldi Arch. Chest Dis. 2022, 93. [Google Scholar] [CrossRef]
- Watanabe, K. Pleuroparenchymal Fibroelastosis: Its Clinical Characteristics. Curr. Respir. Med. Rev. 2013, 9, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.L.; Tominaga, M.; Hansell, D.M.; von der Thusen, J.; Rassl, D.; Parfrey, H.; Guy, S.; Twentyman, O.; Rice, A.; Maher, T.M.; et al. Pleuroparenchymal fibroelastosis: A spectrum of histopathological and imaging phenotypes. Eur. Respir. J. 2012, 40, 377–385. [Google Scholar] [CrossRef]
- Watanabe, K.; Ishii, H.; Kiyomi, F.; Terasaki, Y.; Hebisawa, A.; Kawabata, Y.; Johkoh, T.; Sakai, F.; Kondoh, Y.; Inoue, Y.; et al. Criteria for the diagnosis of idiopathic pleuroparenchymal fibroelastosis: A proposal. Respir. Investig. 2019, 57, 312–320. [Google Scholar] [CrossRef]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sasaki, S.; Kurokawa, K.; Nakamura, T.; Yamada, T.; Sasano, H.; Arano, N.; Komura, M.; Ihara, H.; Nagashima, O.; et al. Usual interstitial pneumonia pattern in the lower lung lobes as a prognostic factor in idiopathic pleuroparenchymal fibroelastosis. Respiration 2019, 97, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Sugino, K.; Ono, H.; Shimizu, H.; Kurosawa, T.; Matsumoto, K.; Ando, M.; Mori, K.; Tsuboi, E.; Homma, S.; Kishi, K. Treatment with antifibrotic agents in idiopathic pleuroparenchymal fibroelastosis with usual interstitial pneumonia. ERJ Open Res. 2021, 7, 00196-2020. [Google Scholar] [CrossRef] [PubMed]
- Chua, F.; Desai, S.R.; Nicholson, A.G.; Deveraj, A.; Renzoni, E.; Rice, A.; Wells, A.U. Pleuroparenchymal fibroelastosis. A review of clinical, radiological, and pathological characteristics. Ann. Am. Thorac. Soc. 2019, 16, 1351–1359. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nagata, N.; Tsuruta, N.; Kitasato, Y.; Wakamatsu, K.; Yoshimi, M.; Ishii, H.; Hirota, T.; Hamada, N.; Fujita, M.; et al. Heterogeneous clinical features in patients with pulmonary fibrosis showing histology of pleuroparenchymal fibroelastosis. Respir. Investig. 2016, 54, 162–169. [Google Scholar] [CrossRef]
- Cottin, V.; Si-Mohamed, S.; Diesler, R.; Bonniaud, P.; Valenzuela, C. Pleuroparenchymal fibroelastosis. Curr. Opin. Pulm. Med. 2022, 28, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Cottin, V.; Beltramo, G. Pleuroparenchymal fibroelastosis: So many unmet needs. Eur. Respir. J. 2022, 60, 2201798. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Nakamura, Y.; Satake, Y.; Sumikawa, H.; Johkoh, T.; Colby, T.V.; Yasui, H.; Hozumi, H.; Karayama, M.; Suzuki, Y.; et al. Clinical diagnosis of idiopathic pleuroparenchymal fibroelastosis: A retrospective multicenter study. Respir. Med. 2017, 133, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Sekine, A.; Tabata, E.; Iwasawa, T.; Takemura, T.; Ogura, T. Comparison of Clinical characteristics and outcomes between idiopathic and secondary pleuroparenchymal fibroelastosis. J. Clin. Med. 2021, 10, 846. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T.; Arai, T.; Kitaichi, M.; Akira, M.; Tachibana, K.; Sugimoto, C.; Hirooka, A.; Tsuji, T.; Minomo, S.; Hayashi, S.; et al. Pleuroparenchymal fibroelastosis from a consecutive database: A rare disease entity? Eur. Respir. J. 2015, 45, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Watanabe, K.; Kushima, H.; Baba, T.; Watanabe, S.; Yamada, Y.; Arai, T.; Tsushima, K.; Kondoh, Y.; Nakamura, Y.; et al. Pleuroparenchymal fibroelastosis diagnosed by multidisciplinary discussions in Japan. Respir. Med. 2018, 141, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Chae, E.J.; Song, J.S.; Kim, M.; Song, J.W. Pleuroparenchymal fibroelastosis in Korean patients: Clinico-radiologic-pathologic features and 2-year follow-up. Korean J. Intern. Med. 2021, 36 (Suppl. S1), S132–S141. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Kinoshita, Y.; Kushima, H.; Ogura, T.; Watanabe, K. The upward shift of hilar structures and tracheal deviation in pleuroparenchymal fibroelastosis. Multidiscip. Respir. Med. 2019, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Kinoshita, Y.; Kushima, H.; Nagata, N.; Watanabe, K. The similarities and differences between pleuroparenchymal fibroelastosis and idiopathic pulmonary fibrosis. Chron. Respir. Dis. 2019, 16, 1479973119867945. [Google Scholar] [CrossRef]
- Kusagaya, H.; Nakamura, Y.; Kono, M.; Kaida, Y.; Kuroishi, S.; Enomoto, N.; Fujisawa, T.; Koshimizu, N.; Yokomura, K.; Inui, N.; et al. Idiopathic pleuroparenchymal fibroelastosis: Consideration of a clinicopathological entity in a series of Japanese patients. BMC Pulm. Med. 2012, 12, 72. [Google Scholar] [CrossRef]
- Watanabe, S.; Waseda, Y.; Takato, H.; Matsunuma, R.; Johkoh, T.; Egashira, R.; Kawabata, Y.; Ikeda, H.; Yasui, M.; Fujimura, M.; et al. Pleuroparenchymal fibroelastosis: Distinct pulmonary physiological features in nine patients. Respir. Investig. 2015, 53, 149–155. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Utsunomiya, T.; Koide, Y.; Wada, K.; Ueda, Y.; Yoshida, Y.; Kushima, H.; Ishii, H. Partial pressure of carbon dioxide levels reflect disease severity in idiopathic pleuroparenchymal fibroelastosis. Respir. Investig. 2023, 61, 379–386. [Google Scholar] [CrossRef]
- Collard, H.R.; King, T.E., Jr.; Bartelson, B.B.; Vourlekis, J.S.; Schwarz, M.I.; Brown, K.K. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 538–542. [Google Scholar] [CrossRef]
- Oda, T.; Ogura, T.; Kitamura, H.; Hagiwara, E.; Baba, T.; Enomoto, Y.; Iwasawa, T.; Okudela, K.; Takemura, T.; Sakai, F.; et al. Distinct characteristics of pleuroparenchymal fibroelastosis with usual interstitial pneumonia compared with idiopathic pulmonary fibrosis. Chest 2014, 146, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Fukada, A.; Suzuki, Y.; Mori, K.; Kono, M.; Hasegawa, H.; Hashimoto, D.; Yokomura, K.; Imokawa, S.; Tanaka, Y.; Inoue, Y.; et al. Idiopathic pleuroparenchymal fibroelastosis: Three-dimensional computed tomography assessment of upper-lobe lung volume. Eur. Respir. J. 2022, 60, 2200637. [Google Scholar] [CrossRef] [PubMed]
- Binks, A.P. Dyspnea. Handb. Clin. Neurol. 2022, 188, 309–338. [Google Scholar]
- Frankel, S.K.; Cool, C.D.; Lynch, D.A.; Brown, K.K. Idiopathic pleuroparenchymal fibroelastosis: Description of a novel clinicopathologic entity. Chest 2004, 126, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Mariani, F.; Gatti, B.; Rocca, A.; Bonifazi, F.; Cavazza, A.; Fanti, S.; Tomassetti, S.; Piciucchi, S.; Poletti, V.; Zompatori, M. Pleuroparenchymal fibroelastosis: The prevalence of secondary forms in hematopoietic stem cell and lung transplantation recipients. Diagn. Interv. Radiol. 2016, 22, 400–406. [Google Scholar] [CrossRef]
- von der Thüsen, J.H.; Hansell, D.M.; Tominaga, M.; Veys, P.A.; Ashworth, M.T.; Owens, C.M.; Nicholson, A.G. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod. Pathol. 2011, 24, 1633–1639. [Google Scholar] [CrossRef]
- Ofek, E.; Sato, M.; Saito, T.; Wagnetz, U.; Roberts, H.C.; Chaparro, C.; Waddell, T.K.; Singer, L.G.; Hutcheon, M.A.; Keshavjee, S.; et al. Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Mod. Pathol. 2013, 26, 350–356. [Google Scholar] [CrossRef]
- Enomoto, Y.; Nakamura, Y.; Colby, T.V.; Johkoh, T.; Sumikawa, H.; Nishimoto, K.; Yoshimura, K.; Matsushima, S.; Oyama, Y.; Hozumi, H.; et al. Radiologic pleuroparenchymal fibroelastosis-like lesion in connective tissue disease-related interstitial lung disease. PLoS ONE 2017, 12, e0180283. [Google Scholar] [CrossRef]
- Hassoun, D.; Dirou, S.; Arrigoni, P.P.; Durant, C.; Hamidou, M.; Néel, A.; Agard, C. Radiological pleuroparenchymal fibroelastosis associated to limited cutaneous systemic sclerosis: A case report. BMC Pulm. Med. 2018, 18, 73. [Google Scholar] [CrossRef]
- Bonifazi, M.; Sverzellati, N.; Negri, E.; Jacob, J.; Egashira, R.; Moser, J.; Piciucchi, S.; Mei, F.; De Lauretis, A.; Visca, D.; et al. Pleuroparenchymal fibroelastosis in systemic sclerosis: Prevalence and prognostic impact. Eur. Respir. J. 2020, 56, 1902135. [Google Scholar] [CrossRef]
- Xu, L.; Rassaei, N.; Caruso, C. Pleuroparenchymal fibroelastosis with long history of asbestos and silicon exposure. Int. J. Surg. Pathol. 2018, 26, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.D.; Gil, J.; Padilla, M.L. Idiopathic pleuroparenchymal fibroelastosis: An unrecognized or misdiagnosed entity? Mod. Pathol. 2008, 21, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Kikuchi, N.; Ishii, Y.; Kawabata, Y.; Moriyama, H.; Terada, M.; Suzuki, E.; Kobayashi, M.; Watanabe, K.; Hizawa, N. Upper lobe-dominant pulmonary fibrosis showing deposits of hard metal component in the fibrotic lesions. Intern. Med. 2010, 49, 2143–2145. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, Y.; Goto, H.; Nonaka, M.; Tachi, H.; Akiyama, T.; Arai, N.; Ishikawa, H.; Hyodo, K.; Nemoto, K.; Miura, Y.; et al. A case of airway aluminosis with likely secondary pleuroparenchymal fibroelastosis. Multidiscip. Respir. Med. 2019, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Nagata, N.; Kitasato, Y.; Wakamatsu, K.; Nabeshima, K.; Harada, T.; Hirota, T.; Shiraishi, M.; Fujita, M. Rapid decrease in forced vital capacity in patients with idiopathic pulmonary upper lobe fibrosis. Respir. Investig. 2012, 50, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Baroke, E.; Heussel, C.P.; Warth, A.; Eichinger, M.; Oltmanns, U.; Palmowski, K.; Herth, F.J.; Kreuter, M. Pleuroparenchymal fibroelastosis in association with carcinomas. Respirology 2016, 21, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Beynat-Mouterde, C.; Beltramo, G.; Lezmi, G.; Pernet, D.; Camus, C.; Fanton, A.; Foucher, P.; Cottin, V.; Bonniaud, P. Pleuroparenchymal fibroelastosis as a late complication of chemotherapy agents. Eur. Respir. J. 2014, 44, 523–527. [Google Scholar] [CrossRef]
- Khiroya, R.; Macaluso, C.; Montero, M.A.; Wells, A.U.; Chua, F.; Kokosi, M.; Maher, T.M.; Devaraj, A.; Rice, A.; Renzoni, E.A.; et al. Pleuroparenchymal fibroelastosis: A review of histopathologic features and the relationship between histologic parameters and survival. Am. J. Surg. Pathol. 2017, 41, 1683–1689. [Google Scholar] [CrossRef]
- Jacob, J.; Odink, A.; Brun, A.L.; Macaluso, C.; de Lauretis, A.; Kokosi, M.; Devaraj, A.; Desai, S.; Renzoni, E.; Wells, A.U. Functional associations of pleuroparenchymal fibroelastosis and emphysema with hypersensitivity pneumonitis. Respir. Med. 2018, 138, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, M.; Landini, N.; Bruni, C.; Sambataro, G.; Nardi, C.; Bargagli, E.; Tomassetti, S.; Occhipinti, M.; Bellando Randone, S.; Guiducci, S.; et al. Pleuroparenchymal fibroelastosis in rheumatic autoimmune diseases: A systematic literature review. Rheumatology 2020, 59, 3645–3656. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.K.; Chuah, K.L. Pleuroparenchymal Fibroelastosis of the Lung: A Review. Arch. Pathol. Lab. Med. 2016, 140, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Fernández Pérez, E.R.; Swigris, J.J.; Forssén, A.V.; Tourin, O.; Solomon, J.J.; Huie, T.J.; Olson, A.L.; Brown, K.K. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013, 144, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.; Prosch, H.; Vašáková, M. Diagnosis, course and management of hypersensitivity pneumonitis. Eur. Respir. Rev. 2022, 31, 210169. [Google Scholar] [CrossRef] [PubMed]
- Jee, A.S.; Sheehy, R.; Hopkins, P.; Corte, T.J.; Grainge, C.; Troy, L.K.; Symons, K.; Spencer, L.M.; Reynolds, P.N.; Chapman, S.; et al. Diagnosis and management of connective tissue disease-associated interstitial lung disease in Australia and New Zealand: A position statement from the Thoracic Society of Australia and New Zealand. Respirology 2021, 26, 23–51. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Makino, S.; Ogura, T.; Suda, T.; Tomioka, H.; Amano, H.; Anraku, M.; Enomoto, N.; Fujii, T.; Fujisawa, T.; et al. 2020 guide for the diagnosis and treatment of interstitial lung disease associated with connective tissue disease. Respir. Investig. 2021, 59, 709–740. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Stowasser, S.; Voss, F.; Bendstrup, E.; Kreuter, M.; Martinez, F.J.; Sime, P.J.; Stock, C. Decline in forced vital capacity as a surrogate for mortality in patients with pulmonary fibrosis. Respirology 2023, 28, 1147–1153. [Google Scholar] [CrossRef]
- Kono, M.; Nakamura, Y.; Enomoto, Y.; Yasui, H.; Hozumi, H.; Karayama, M.; Suzuki, Y.; Furuhashi, K.; Miki, Y.; Hashimoto, D.; et al. Pneumothorax in patients with idiopathic pleuroparenchymal fibroelastosis: Incidence, clinical features, and risk factors. Respiration 2021, 100, 19–26. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Utsunomiya, T.; Koide, Y.; Wada, K.; Ueda, Y.; Yoshida, Y.; Kushima, H.; Ishii, H. Changes in body weight reflect disease progression in pleuroparenchymal fibroelastosis. Respir. Med. Res. 2023, 83, 100980. [Google Scholar] [CrossRef]
- Harada, T.; Yoshida, Y.; Kitasato, Y.; Tsuruta, N.; Wakamatsu, K.; Hirota, T.; Tanaka, M.; Tashiro, N.; Ishii, H.; Shiraishi, M.; et al. The thoracic cage becomes flattened in the progression of pleuroparenchymal fibroelastosis. Eur. Respir. Rev. 2014, 23, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Sato, S.; Ohta, H.; Kusano, K.; Kawabe, R.; Oba, T.; Uzuka, C.; Sasaki, H.; Akasaka, K.; Amano, M.; et al. Body weight loss is a simple and useful indicator of prognosis and predictive tolerability in the first year of nintedanib therapy in patients with interstitial lung disease. Respir. Investig. 2024, 62, 551–557. [Google Scholar] [CrossRef]
- Mori, Y.; Yamano, Y.; Kataoka, K.; Yokoyama, T.; Matsuda, T.; Kimura, T.; Ogawa, T.; Watanabe, F.; Kondoh, Y. Pulmonary rehabilitation for idiopathic pleuroparenchymal fibroelastosis: A retrospective study on its efficacy, feasibility, and safety. Respir. Investig. 2021, 59, 849–858. [Google Scholar] [CrossRef]
- Muto, Y.; Sekine, A.; Hagiwara, E.; Komatsu, S.; Baba, T.; Oda, T.; Tabata, E.; Sakayori, M.; Fukui, K.; Iwasawa, T.; et al. Clinical characteristics of pulmonary hypertension in patients with pleuroparenchymal fibroelastosis. Respir. Investig. 2022, 60, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Kushima, H.; Kinoshita, Y.; Fujita, M.; Watanabe, K. The serum KL-6 levels in untreated idiopathic pulmonary fibrosis can naturally decline in association with disease progression. Clin. Respir. J. 2018, 12, 2411–2418. [Google Scholar] [CrossRef]
- Suzuki, Y.; Fukada, A.; Mori, K.; Kono, M.; Hasegawa, H.; Hashimoto, D.; Yokomura, K.; Imokawa, S.; Inoue, Y.; Yasui, H.; et al. Assessment of malnutrition-related risk in patients with idiopathic pleuroparenchymal fibroelastosis. ERJ Open Res. 2023, 9, 00749–02022. [Google Scholar] [CrossRef]
- Yabuuchi, Y.; Saito, T.; Hirano, H.; Nonaka, M.; Arai, N.; Hyodo, K.; Kanazawa, J.; Miura, Y.; Usui, S.; Tamura, K.; et al. Impact of sleep-related hypoventilation in patients with pleuroparenchymal fibroelastosis. Respir. Res. 2022, 23, 295. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kono, M.; Hasegawa, H.; Hashimoto, D.; Yokomura, K.; Imokawa, S.; Inoue, Y.; Hozumi, H.; Karayama, M.; Furuhashi, K.; et al. Neutrophil-lymphocyte ratio in patients with idiopathic pleuroparenchymal fibroelastosis. BMJ Open Respir. Res. 2023, 10, e001763. [Google Scholar] [CrossRef]
- Shioya, M.; Otsuka, M.; Yamada, G.; Umeda, Y.; Ikeda, K.; Nishikiori, H.; Kuronuma, K.; Chiba, H.; Takahashi, H. Poorer prognosis of idiopathic pleuroparenchymal fibroelastosis compared with idiopathic pulmonary fibrosis in advanced stage. Can. Respir. J. 2018, 2018, 6043053. [Google Scholar] [CrossRef]
- Yamakawa, H.; Sato, S.; Ohta, H.; Takatsuka, M.; Kusano, K.; Kawabe, R.; Oba, T.; Akasaka, K.; Amano, M.; Araya, J.; et al. Clinical significance of para-carinal air cysts in patients with pleuroparenchymal fibroelastosis: The relationship with pneumomediastinum and pneumothorax. Clin. Respir. J. 2023, 17, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Camus, P.; von der Thüsen, J.; Hansell, D.M.; Colby, T.V. Pleuroparenchymal fibroelastosis: One more walk on the wild side of drugs? Eur. Respir. J. 2014, 44, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.; Fujisawa, T.; Yoshimura, K.; Enomoto, Y.; Enomoto, N.; Nakamura, Y.; Inui, N.; Sumikawa, H.; Johkoh, T.; Colby, T.V.; et al. The prognostic significance of pneumothorax in patients with idiopathic pulmonary fibrosis. Respirology 2018, 23, 519–525. [Google Scholar] [CrossRef] [PubMed]
- von der Thüsen, J.H. Pleuroparenchymal fibroelastosis: Its pathological characteristics. Curr. Respir. Med. Rev. 2013, 9, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, Y.; Taniguchi, H.; Kondoh, Y.; Kataoka, K.; Hamada, N.; Hashiguchi, T.; Ichikado, K.; Kishaba, T.; Sato, S.; Udo, E.; et al. Pulmonary interstitial emphysema is a risk factor for poor prognosis and a cause of air leaks. Respir. Investig. 2019, 57, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Soto-Hurtado, E.J.; Peñuela-Ruíz, L.; Rivera-Sánchez, I.; Torres-Jiménez, J. Tracheal diverticulum: A review of the literature. Lung 2006, 184, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, R.; Bruzzi, G.; Manicardi, L.; Tabbì, L.; Fantini, R.; Castaniere, I.; Andrisani, D.; Gozzi, F.; Pellegrino, M.R.; Trentacosti, F.; et al. Risk factors for pulmonary air leak and clinical prognosis in patients with covid-19 related acute respiratory failure: A retrospective matched control study. Front. Med. 2022, 9, 848639. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Tsukahara, Y.; Sato, S.; Ohta, H.; Kida, G.; Nakamura, T.; Nishizawa, T.; Kawabe, R.; Oba, T.; Akasaka, K.; et al. Impact of progressive fibrosing interstitial lung disease (ILD) in ILD patients complicated with secondary spontaneous pneumothorax. Sarcoidosis Vasc. Diffus. Lung Dis. 2022, 38, e2021042. [Google Scholar]
- Yamakawa, H.; Nishizawa, T.; Ohta, H.; Tsukahara, Y.; Nakamura, T.; Sato, S.; Kawabe, R.; Oba, T.; Akasaka, K.; Amano, M.; et al. Patient background and prognosis of chronic pulmonary aspergillosis in fibrosing interstitial lung disease. Medicine 2022, 101, e29936. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tsujino, K.; Kuge, T.; Okabe, F.; Kawasaki, T.; Matsuki, T.; Kagawa, H.; Miki, M.; Miki, K.; Mori, M.; et al. Pleuroparenchymal fibroelastosis in Mycobacterium avium complex pulmonary disease: Clinical characteristics and prognostic impact. ERJ Open Res. 2021, 7, 00765–02020. [Google Scholar] [CrossRef]
- Holm, J.P.; Hilberg, O.; Noerskov-Lauritsen, N.; Bendstrup, E. Pseudomonas aeruginosa in patients without cystic fibrosis is strongly associated with chronic obstructive lung disease. Dan. Med. J. 2013, 60, A4636. [Google Scholar] [PubMed]
- Miyamoto, A.; Uruga, H.; Morokawa, N.; Moriguchi, S.; Takahashi, Y.; Ogawa, K.; Murase, K.; Hanada, S.; Takaya, H.; Kurosaki, A.; et al. Various bronchiolar lesions accompanied by idiopathic pleuroparenchymal fibroelastosis with a usual interstitial pneumonia pattern demonstrating acute exacerbation. Intern. Med. 2019, 58, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Makino, T.; Yagisawa, M.; Ishige, M.; Akimoto, Y.; Ito, K.; Anami, Y.; Kono, M. Acute exacerbation of pleuroparenchymal fibroelastosis with lower lobe usual interstitial pneumonia: An autopsy case. Respir. Med. Case Rep. 2023, 43, 101846. [Google Scholar] [CrossRef] [PubMed]
- Namba, M.; Masuda, T.; Takao, S.; Terada, H.; Yamaguchi, K.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; Nakashima, T.; Iwamoto, H.; et al. Extent of pulmonary fibrosis on high-resolution computed tomography is a prognostic factor in patients with pleuroparenchymal fibroelastosis. Respir. Investig. 2020, 58, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Bondue, B.; Pesci, A.; Miyazaki, Y.; Song, J.W.; Bhatt, N.Y.; Huggins, J.T.; Oldham, J.M.; Padilla, M.L.; Roman, J.; et al. Acute exacerbations of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180071. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Kondoh, Y.; Brown, K.K.; Johkoh, T.; Kataoka, K.; Fukuoka, J.; Kimura, T.; Matsuda, T.; Yokoyama, T.; Fukihara, J.; et al. Acute exacerbations of fibrotic interstitial lung diseases. Respirology 2020, 25, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Eastridge, C.E.; Hamman, J.L. Pneumothorax complicated by chronic steroid treatment. Am. J. Surg. 1973, 126, 784–787. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Shiiya, H.; Tian, D.; Sato, M.; Karasaki, T.; Kitano, K.; Nagayama, K.; Anraku, M.; Kaga, K.; Matsui, Y.; Nakajima, J. Differences between patients with idiopathic pleuroparenchymal fibroelastosis and those with other types of idiopathic interstitial pneumonia in candidates for lung transplants. Transplant. Proc. 2019, 51, 2014–2021. [Google Scholar] [CrossRef]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014--an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef]
- Hirama, T.; Akiba, M.; Watanabe, T.; Watanabe, Y.; Notsuda, H.; Oishi, H.; Niikawa, H.; Okada, Y. Waiting time and mortality rate on lung transplant candidates in Japan: A single-center retrospective cohort study. BMC Pulm. Med. 2021, 21, 390. [Google Scholar] [CrossRef]
- Shiiya, H.; Sato, M. Lung transplantation for pleuroparenchymal fibroelastosis. J. Clin. Med. 2021, 10, 957. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Ogura, T.; Kameda, H.; Kishaba, T.; Iwasawa, T.; Takemura, T.; Kuwano, K. Decision-making strategy for the treatment of rheumatoid arthritis-associated interstitial lung disease (RA-ILD). J. Clin. Med. 2021, 10, 3806. [Google Scholar] [CrossRef]
- Zamora-Legoff, J.A.; Krause, M.L.; Crowson, C.S.; Ryu, J.H.; Matteson, E.L. Risk of serious infection in patients with rheumatoid arthritis-associated interstitial lung disease. Clin. Rheumatol. 2016, 35, 2585–2589. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Watanabe, K.; Ishii, H.; Kushima, H.; Nabeshima, K. Lower-lobe predominant pleuroparenchymal fibroelastosis. Pathol. Int. 2019, 69, 536–540. [Google Scholar] [CrossRef]
- Nasser, M.; Si-Mohamed, S.; Turquier, S.; Traclet, J.; Ahmad, K.; Philit, F.; Bonniaud, P.; Chalabreysse, L.; Thivolet-Béjui, F.; Cottin, V. Nintedanib in idiopathic and secondary pleuroparenchymal fibroelastosis. Orphanet J. Rare Dis. 2021, 16, 419. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Miyamura, T.; Ikeda, T.; Ueda, Y.; Yoshida, Y.; Kushima, H.; Ishii, H. Limited efficacy of nintedanib for idiopathic pleuroparenchymal fibroelastosis. Respir. Investig. 2022, 60, 562–569. [Google Scholar] [CrossRef]
- Tomioka, H.; Iwabayashi, M.; Yokota, M.; Hashimoto, R.; Amimoto, H. Weight loss in nintedanib-treated patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2023, 80, 102213. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Bell, E.C.; Cox, N.S.; Goh, N.; Glaspole, I.; Westall, G.P.; Watson, A.; Holland, A.E. Oxygen therapy for interstitial lung disease: A systematic review. Eur. Respir. Rev. 2017, 26, 160080. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Ikeda, T.; Miyamura, T.; Ueda, Y.; Yoshida, Y.; Kushima, H.; Fujita, M.; Ogura, T.; Watanabe, K.; Ishii, H. A proposed prognostic prediction score for pleuroparenchymal fibroelastosis. Respir. Res. 2021, 22, 215. [Google Scholar] [CrossRef] [PubMed]
- Beiser, G.D.; Epstein, S.E.; Stampfer, M.; Goldstein, R.E.; Noland, S.P.; Levitsky, S. Impairment of cardiac function in patients with pectus excavatum, with improvement after operative correction. N. Engl. J. Med. 1972, 287, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Chan Wah Hak, Y.S.; Lim, Y.P.; Liew, R.; Hsu, L.F. Pectus excavatum: Uncommon electrical abnormalities caused by extrinsic right ventricular compression. J. Cardiovasc. Electrophysiol. 2014, 25, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, M.; Tachi, T.; Yamashita, T. Atrial tachycardia in a patient with severe pectus excavatum. Heart Rhythm. 2023, 20, 631–632. [Google Scholar] [CrossRef]
- Bandorski, D.; Bogossian, H.; Ghofrani, A.; Schmitt, J.; Höltgen, R. Tachycardia and pulmonary arterial hypertension. Herzschrittmacherther. Elektrophysiol. 2020, 31, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yamashiro, T.; Moriya, H.; Tsubakimoto, M.; Tsuchiya, N.; Nagatani, Y.; Matsuoka, S.; Murayama, S. Hyperinflated lungs compress the heart during expiration in COPD patients: A new finding on dynamic-ventilation computed tomography. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 3123–3131. [Google Scholar] [CrossRef] [PubMed]
- Holguin, F.; Folch, E.; Redd, S.C.; Mannino, D.M. Comorbidity and mortality in COPD-related hospitalizations in the United States, 1979 to 2001. Chest 2005, 128, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Asai, K.; Furuse, E.; Nakamura, S.; Murai, K.; Tsukada, Y.T.; Shimizu, W. Impact of beta-blocker selectivity on long-term outcomes in congestive heart failure patients with chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2015, 10, 515–523. [Google Scholar] [CrossRef]
- Page, R.L.; Joglar, J.A.; Caldwell, M.A.; Calkins, H.; Conti, J.B.; Deal, B.J.; Estes, N.A., 3rd; Field, M.E.; Goldberger, Z.D.; Hammill, S.C.; et al. 2015 ACC/AHA/HRS Guideline for the Management of Adult Patients with Supraventricular Tachycardia: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2015, 133, e506–e574. [Google Scholar] [CrossRef]
- Wołowiec, Ł.; Grześk, G.; Osiak, J.; Wijata, A.; Mędlewska, M.; Gaborek, P.; Banach, J.; Wołowiec, A.; Głowacka, M. Beta-blockers in cardiac arrhythmias-clinical pharmacologist’s point of view. Front. Pharmacol. 2023, 13, 1043714. [Google Scholar] [CrossRef]
- Fujisawa, T.; Akiyama, N.; Morita, T.; Koyauchi, T.; Matsuda, Y.; Mori, M.; Miyashita, M.; Tachikawa, R.; Tomii, K.; Tomioka, H.; et al. Palliative care for interstitial lung disease: A nationwide survey of pulmonary specialists. Respirology 2023, 28, 659–668. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author (Year) | No. of Patients | BMI (kg/m2) | %FVC | %DLCO | Poor Prognostic Factor | Median Survival (Years) | |
|---|---|---|---|---|---|---|---|
| + | − | ||||||
| Ishii et al. [56] (2018) | 52 | Median 17.8 | Median 66.8 | Median 73.2 | KL-6 ≥ 600 U/mL | 2 | 5.1 |
| Kato et al. [7] (2019) | 36 | Mean 17.8 | Mean 61.1 | Mean 39.8 | Lower-lobe UIP | 1 | 5.2 |
| Kono et al. [50] (2021) | 89 | Mean 17.0 | Mean 61.9 | Mean 84.9 | Pneumothorax | 1.5 | 2.8 |
| Yabuuchi et al. [58] (2022) | 52 | Median 17.5 | Median 52.2 | Median 60.7 | Sleep-related hypoventilation | 0.9 | NC |
| Muto et al. [55] (2022) * | 83 | Median 17.7 | Median 62.2 | Median 84.7 | Pulmonary hypertension | 1.3 | 4.2 |
| Fukada et al. [25] (2022) | 132 | Mean 16.8 | Mean 64.4 | Mean 90.7 | Standardized upper-lobe volume < 30% | 2.5 | 6.1 |
| Kinoshita et al. [22] (2023) | 47 | Mean 17.2 | Mean 61.9 | Mean 82.4 | PaCO2 ≥ 50 Torr | 0.97 | 1.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamakawa, H.; Oda, T.; Sugino, K.; Hirama, T.; Komatsu, M.; Katano, T.; Fukuda, T.; Takemura, T.; Kubota, Y.; Kishaba, T.; et al. Proposed Clinical Algorithm for Pleuroparenchymal Fibroelastosis (PPFE). J. Clin. Med. 2024, 13, 3675. https://doi.org/10.3390/jcm13133675
Yamakawa H, Oda T, Sugino K, Hirama T, Komatsu M, Katano T, Fukuda T, Takemura T, Kubota Y, Kishaba T, et al. Proposed Clinical Algorithm for Pleuroparenchymal Fibroelastosis (PPFE). Journal of Clinical Medicine. 2024; 13(13):3675. https://doi.org/10.3390/jcm13133675
Chicago/Turabian StyleYamakawa, Hideaki, Tsuneyuki Oda, Keishi Sugino, Takashi Hirama, Masamichi Komatsu, Takuma Katano, Taiki Fukuda, Tamiko Takemura, Yoshiaki Kubota, Tomoo Kishaba, and et al. 2024. "Proposed Clinical Algorithm for Pleuroparenchymal Fibroelastosis (PPFE)" Journal of Clinical Medicine 13, no. 13: 3675. https://doi.org/10.3390/jcm13133675
APA StyleYamakawa, H., Oda, T., Sugino, K., Hirama, T., Komatsu, M., Katano, T., Fukuda, T., Takemura, T., Kubota, Y., Kishaba, T., Norisue, Y., Araya, J., & Ogura, T. (2024). Proposed Clinical Algorithm for Pleuroparenchymal Fibroelastosis (PPFE). Journal of Clinical Medicine, 13(13), 3675. https://doi.org/10.3390/jcm13133675