

[18F]F-Poly(ADP-Ribose) Polymerase Inhibitor Radiotracers for Imaging PARP Expression and Their Potential Clinical Applications in Oncology

 , , , ,
, , , ,  ,
,  and
and 
Abstract
1. Introduction
2. [18F]F-Radiolabeled Molecular Probes for PET Imaging
2.1. Olaparib-Based [18F]F-Radiolabeled Molecular Probes
2.1.1. [18F]F-BO
2.1.2. [18F]F-PARPi-FL
2.1.3. [18F]F-PARPi
2.1.4. [18F]F-Olaparib
2.1.5. Other Olaparib-Based [18F]F PET Radiotracers ([18F]FPyPARP; [18F]F-20; [18F]F-9e; and [18F]F-AZD2461)
2.2. Rucaparib-Based [18F]F-Radiolabeled Molecular Probes
2.2.1. [18F]F-FluorThanatrace ([18F]FTT)
2.2.2. [18F]F-WC-DZ-F and [18F]F-Rucaparib
2.3. Talazoparib-Based and Other [18F]F-PARP Inhibitor Molecular Probes
[18F]F-Talazoparib
3. Discussion
4. Conclusions
5. Future Perspectives
- Assessing PARP expression status in lieu of biopsy in lesions. This could be in lesions that are inaccessible to biopsy or in suspected tumor heterogeneity, especially in patients with metastatic disease.
- At the time of initial diagnosis of primary or metastatic disease when considering PARP therapy.
- Detecting PARP expression when other imaging tests are equivocal or suspicious. This may also include patients with other immunohistochemical subtypes that may have BRCA-like mutations. BRCA1/2 mutations, PARP expression, and uptake of [18F]FTT have been seen even in patients with hormonal receptor expression. Should conventional therapies fail, the addition of PARP inhibitor therapy may be considered.
- PARP inhibitor therapeutic monitoring. This includes early assessment of adequate blockade of PARP.
- Differentiating therapy or inflammation-related findings from malignancy in which [18F]FDG may not be able to differentiate. This also includes better delineation of tumor from adjacent inflammation or areas with intense metabolic activity, in which [18F]FDG performs dismally.
- Assessing in vivo resistance mechanisms in patients unresponsive to PARP inhibitor therapy, especially P-glycoprotein-related resistance, despite confirmed PARP upregulation on immunohistochemistry.
- Establishing the optimal scan modality (static vs. dynamic), timing, and other technical aspects to better address the value of PARP inhibitor imaging in various clinical scenarios.
- Evaluation of the added value of PARP inhibitor PET/MRI.
- Development of less lipophilic PARP inhibitor tracers to facilitate the detection of PARP upregulation in hepatic and abdominal lesions. This would require the identification of a specific carrier which would allow the hydrophilic molecule to cross the cellular membrane since PARP is located within the nucleus.
- Correlation of PARP expression based on validated IHC scoring criteria and the semi-quantitative evaluation of PARP expression on PET-based imaging.
- Determining the prognostic and predictive role of PARP inhibitor PET/CT imaging using semi-quantitative parameters in patients being worked up for PARP inhibitor therapy.

{kind=link}
{kind=link}
| Scaffolding | Radiotracer | Preclinical/Clinical (P/C) | Pertinent Conclusion(s) |
|---|---|---|---|
| Olaparib | [18F]F-BO | P | Development of [18F]F-BO from Olaparib scaffolding with high-yield fluorination technique [45]. Uptake in PARP1-expressing tumor cell lines in in vivo mice models, and inhibition of uptake by cold Olaparib (AZ2D2281). In comparison with [18F]FDG, [18F]F-BO showed early changes post-PARP inhibition compared to [18F]FDG [46,47]. |
| [18F]F-PARPi-FL | P | Development of [18F]F-PARPi-FL [48]. Use in glioblastoma tumor models and xenografts. Specificity to PARP1 expression demonstrated after correlation with immunohistochemistry. Possible application in treatment planning and assessment of PARPi blockade [49,50]. | |
| [18F]F-PARPi | P/C | Development and improvement of the synthesis of [18F]F-PARPi from Olaparib by conjugating a 2H-phthalazin-1-one scaffold to 4-[18F]fluorobenzoic acid [51]. [18F]F-PARPi images tumor heterogeneity [52]. [18F]F-PARPi uptake is specific to tumor uptake [52,55,92]. Target engagement and potential for monitoring PARP1 blockage by PARPi [92]. Discriminating radiation injury from the recurrent tumor with [18F]PARPi: Comparison with [18F]FET [55]. Differentiating malignant from inflamed lymph nodes [56]. Phase I/2 study in head and neck tumors: Comparison with [18F]FDG [57]. Better performance of [18F]F-PARPi in detecting primary tongue tumors: Comparison with [18F]FDG [58]. | |
| [18F]F-Olaparib | P/C | Development and refinement of [18F]F-Olaparib production with high radiochemical yield [59,60,61]. [18F]F-Olaparib uptake proportional to PARP1 expression. Effective blockade seen post-PARP inhibition; can be used to assess therapeutic efficacy. Increased uptake as a DNA damage response post-irradiation. (In some cases PARPi may be more effective post-irradiation.) [18F]F-Olaparib uptake correlated to ER5 hypoxia marker; therefore, this may be effective in hypoxic tissue [59]. Tumor uptake of radiolabeled PARP inhibitors, such as [18F]F-Olaparib, is governed by more than PARP expression levels alone [62]. | |
| [18F]F-20 | P | Synthesis of [18F]F-20. Specific uptake in PARP-expressing tissue or cells. Problematic hepatobiliary excretion and in vivo defluorination leads to non-specific [18F]F bone uptake [63]. | |
| [18F]F-9e and [18F]F-AZD2461 | P | Synthesis of [18F]F-9e and [18F]F-AZD2461. Non-blood barrier permeants. [18F]F-AZD2461 uptake not affected by Olaparib blockade; therefore, cannot be used in cases of Olarapib blockade [64,65]. | |
| [18F]FPyPARP | P | Less lipophilicity with less hepatobiliary excretion. Favorable for theranostic translation [42]. | |
| Rucaparib | [18F]FTT | P/C | Development of [18F]FTT. Increased specificity for PARP1, competitive inhibition with Olaparib for PARP [67]. PARP expression and activity correlates to [18F]FTT uptake in vitro and in a xenograft model of breast and ovarian cancer [68,69]. [18F]FTT uptake a surrogate predictor of response to PARP inhibitor adjuvant therapy to radiation [70,72]. Higher SUVs in HRR than non-HRR but overlap was noted [72]. SUVs at 60 min a robust metric for noninvasively quantifying PARP1 expression in vivo [73]. [18F]FTT is a predictive and pharmacokinetic biomarker which can be used for patient selection for PARPi [74,75]. Evaluating in vivo PARP1 expression with [18F]FTT PET/CT in primary or recurrent breast cancer (NCT03083288; NCT03846167; NCT05226663 and NCT03604315). Serial imaging of the novel radiotracer [18F]FTT by PET/CT (NCT03604315). Summary of preclinical to clinical progress of [18F]FTT [71]. |
| [18F]F-rucaparib | P | Uptake correlated to PARP expression [77]. | |
| [18F]F-WC-DZ-F “aka” [18F]F-PARPZ | P | Uptake correlated to PARP expression. Increased uptake correlated to PARP expression as a DNA damage response post-[225Ac]Ac-PSMA-11 therapy [78]. | |
| Talazoparib | [18F]F-talazoparib | P | Development and uptake correlating to PARP expression [81]. |
| Other | [18F]F-SuPAR | P | Prediction of response. Development and uptake correlating to PARP expression [82]. |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative Stress and Its Role in Cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Oksenych, V.; Kainov, D.E. Dna Damage Response. Biomolecules 2021, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Huang, J. DNA Double-Strand Break Repair Pathway Choice: The Fork in the Road. Genome Instab. Dis. 2020, 1, 10–19. [Google Scholar] [CrossRef]
- Abbotts, R.; Wilson, D.M. Coordination of DNA Single Strand Break Repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, W.; Wang, Y. PARP-1 and Its Associated Nucleases in DNA Damage Response. DNA Repair 2019, 81, 102651. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Vyas, A.; Kassab, M.A.; Singh, A.K.; Yu, X. The Role of Poly ADP-Ribosylation in the First Wave of DNA Damage Response. Nucleic Acids Res. 2017, 45, 8129–8141. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The Underlying Mechanism for the PARP and BRCA Synthetic Lethality: Clearing up the Misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Afghahi, A.; Telli, M.L.; Kurian, A.W. Genetics of Triple-Negative Breast Cancer: Implications for Patient Care. Curr. Probl. Cancer 2016, 40, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Olaparib: First Global Approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Al-akhras, A.; Chehade, C.H.; Narang, A. PARP Inhibitors in Metastatic Castration-Resistant Prostate Cancer: Unraveling the Therapeutic Landscape. Life 2024, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Rucaparib: First Global Approval. Drugs 2017, 77, 585–592. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate Biomarkers of PARP Inhibitor Sensitivity in Ovarian Cancer beyond the BRCA Genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, L.; Garber, J.E. PARP Inhibitors in the Management of Breast Cancer: Current Data and Future Prospects. BMC Med. 2015, 13, 188. [Google Scholar] [CrossRef] [PubMed]
- Russnes, H.G.; Navin, N.; Hicks, J.; Borresen-Dale, A.L. Insight into the Heterogeneity of Breast Cancer through Next-Generation Sequencing. J. Clin. Investig. 2011, 121, 3810–3818. [Google Scholar] [CrossRef] [PubMed]
- Janku, F. Tumor Heterogeneity in the Clinic: Is It a Real Problem? Ther. Adv. Med Oncol. 2014, 6, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thürlimann, B.; Senn, H.J. Strategies for Subtypes-Dealing with the Diversity of Breast Cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Hahnen, E.; Hauke, J.; Engel, C.; Neidhardt, G.; Rhiem, K.; Schmutzler, R.K. Germline Mutations in Triple-Negative Breast Cancer. Breast Care 2017, 12, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Bou Zerdan, M.; Ghorayeb, T.; Saliba, F.; Allam, S.; Bou Zerdan, M.; Yaghi, M.; Bilani, N.; Jaafar, R.; Nahleh, Z. Triple Negative Breast Cancer: Updates on Classification and Treatment in 2021. Cancers 2022, 14, 1253. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.-D.; Liu, Y.-R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Derakhshan, F.; Reis-filho, J.S. Pathogenesis of Triple-Negative Breast Cancer. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.; Kobayashi, T.; Miyakami, Y.; Sumida, S.; Kakimoto, T.; Saijo, Y.; Uehara, H. Triple-Negative Breast Cancer and Basal-like Subtype: Pathology and Targeted Therapy. J. Med. Investig. 2021, 68, 213–219. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells Through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 4490. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Bates, S. Mechanisms of Resistance to PARP Inhibitors-Three and Counting. Cancer Discov. 2013, 3, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Washington, C.R.; Moore, K.N. Resistance to Poly (ADP-Ribose) Polymerase Inhibitors (PARPi): Mechanisms and Potential to Reverse. Curr. Oncol. Rep. 2022, 24, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Francica, P.; Rottenberg, S. Mechanisms of PARP Inhibitor Resistance in Cancer and Insights into the DNA Damage Response. Genome Med. 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yang, Y.; Jin, D.; Zhang, Z.; Shen, K.; Yang, J.; Chen, H.; Zhao, X.; Yang, L.; Lu, H. PARP Inhibitor Resistance in Breast and Gynecological Cancer: Resistance Mechanisms and Combination Therapy Strategies. Front. Pharmacol. 2022, 13, 967633. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Veskimäe, K.; Staff, S.; Grönholm, A.; Pesu, M.; Laaksonen, M.; Nykter, M.; Isola, J.; Mäenpää, J. Assessment of PARP Protein Expression in Epithelial Ovarian Cancer by ELISA Pharmacodynamic Assay and Immunohistochemistry. Tumor Biol. 2016, 37, 11991–11999. [Google Scholar] [CrossRef] [PubMed]
- Katal, S.; Eibschutz, L.S.; Saboury, B.; Gholamrezanezhad, A.; Alavi, A. Advantages and Applications of Total-Body PET Scanning. Diagnostics 2022, 12, 426. [Google Scholar] [CrossRef]
- Le Bars, D. Fluorine-18 and Medical Imaging: Radiopharmaceuticals for Positron Emission Tomography. J. Fluor. Chem. 2006, 127, 1488–1493. [Google Scholar] [CrossRef]
- Siraj, A.K.; Pratheeshkumar, P.; Parvathareddy, S.K.; Divya, S.P.; Al-Dayel, F.; Tulbah, A.; Ajarim, D.; Al-Kuraya, K.S. Overexpression of PARP Is an Independent Prognostic Marker for Poor Survival in Middle Eastern Breast Cancer and Its Inhibition Can Be Enhanced with Embelin Co-Treatment. Oncotarget 2018, 9, 37319–37332. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.A.; Mankoff, D.A.; Clark, A.S.; Fowler, A.M.; Linden, H.M.; Peterson, L.M.; Dehdashti, F.; Kurland, B.F.; Mortimer, J.; Mouabbi, J.; et al. Summary: Appropriate Use Criteria for Estrogen Receptor-Targeted PET Imaging with 16α-18F-Fluoro-17β-Fluoroestradiol. J. Nucl. Med. 2023, 64, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Ndlovu, H.; Lawal, I.; Mokoala, K.; Disenyane, D.; Nkambule, N.; Bassa, S.; Mzizi, Y.; Bida, M.; Sathekge, M. Imaging PARP Upregulation with [123 I]I-PARPi SPECT/CT in Small Cell Neuroendocrine Carcinoma. J. Nucl. Med. 2023, 65, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Stotz, S.; Kinzler, J.; Nies, A.T.; Schwab, M.; Maurer, A. Two Experts and a Newbie: [18F]PARPi vs [18F]FTT vs [18F]FPyPARP—A Comparison of PARP Imaging Agents. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Dziadkowiec, K.N.; Gasiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP Inhibitors: Review of Mechanisms of Action and BRCA1/2 Mutation Targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Keliher, E.J.; Reiner, T.; Turetsky, A.; Hilderbrand, S.A.; Weissleder, R. High-Yielding, Two-Step 18F Labeling Strategy for 18F-PARP1 Inhibitors. ChemMedChem 2011, 6, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Reiner, T.; Lacy, J.; Keliher, E.J.; Yang, K.S.; Ullal, A.; Kohler, R.H.; Vinegoni, C.; Weissleder, R. Imaging Therapeutic PARP Inhibition in Vivo through Bioorthogonally Developed Companion Imaging Agents. Neoplasia 2012, 14, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Reiner, T.; Keliher, E.J.; Earley, S.; Marinelli, B.; Weissleder, R. Synthesis and In Vivo Imaging of a 18 F-Labeled PARP1 Inhibitor Using a Chemically Orthogonal Scavenger-Assisted High-Performance Method. Angew. Chem. Int. Ed. 2011, 50, 1922–1925. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lin, T.P.; Li, D.; Leamer, L.; Shan, H.; Li, Z.; Gabbaï, F.P.; Conti, P.S. Lewis Acid-Assisted Isotopic 18F-19F Exchange in BODIPY Dyes: Facile Generation of Positron Emission Tomography/Fluorescence Dual Modality Agents for Tumor Imaging. Theranostics 2013, 3, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Irwin, C.P.; Portorreal, Y.; Brand, C.; Zhang, Y.; Desai, P.; Salinas, B.; Weber, W.A.; Reiner, T. PARPi-FL—A Fluorescent PARP1 Inhibitor for Glioblastoma Imaging. Neoplasia 2014, 16, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Carlucci, G.; Carney, B.; Brand, C.; Kossatz, S.; Irwin, C.P.; Carlin, S.D.; Keliher, E.J.; Weber, W.; Reiner, T. Dual-Modality Optical/PET Imaging of PARP1 in Glioblastoma. Mol. Imaging Biol. 2015, 17, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Carney, B.; Carlucci, G.; Salinas, B.; Di Gialleonardo, V.; Kossatz, S.; Vansteene, A.; Longo, V.A.; Bolaender, A.; Chiosis, G.; Keshari, K.R.; et al. Non-invasive PET Imaging of PARP1 Expression in Glioblastoma Models. Mol. Imaging Biol. 2016, 18, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; DemCrossed D Sign©trio De Souza França, P.; Pirovano, G.; Piotrowski, A.F.; Nicklin, P.J.; Riedl, C.C.; Schwartz, J.; Bale, T.A.; Donabedian, P.L.; Kossatz, S.; et al. Preclinical and First-in-Human-Brain-Cancer Applications of [18F]Poly (ADP-Ribose) Polymerase Inhibitor PET/MR. Neurooncol. Adv. 2020, 2, vdaa119. [Google Scholar] [CrossRef] [PubMed]
- Purandare, N.C.; Puranik, A.D.; Shah, S.; Agrawal, A.; Rangarajan, V. Post-Treatment Appearances, Pitfalls, and Patterns of Failure in Head and Neck Cancer on FDG PET/CT Imaging. Indian J. Nucl. Med. 2014, 29, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Gao, Q.; Han, A.; Zhu, H.; Yu, J. The Potential Mechanism, Recognition and Clinical Significance of Tumor Pseudoprogression after Immunotherapy. Cancer Biol. Med. 2019, 16, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Donabedian, P.L.; Kossatz, S.; Engelbach, J.A.; Jannetti, S.A.; Carney, B.; Young, R.J.; Weber, W.A.; Garbow, J.R.; Reiner, T. Discriminating Radiation Injury from Recurrent Tumor with [18F]PARPi and Amino Acid PET in Mouse Models. EJNMMI Res. 2018, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Salloum, D.; Carney, B.; Brand, C.; Kossatz, S.; Sadique, A.; Lewis, J.S.; Weber, W.A.; Wendel, H.G.; Reiner, T. Targeted PET Imaging Strategy to Differentiate Malignant from Inflamed Lymph Nodes in Diffuse Large B-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, E7441–E7449. [Google Scholar] [CrossRef] [PubMed]
- Schöder, H.; Demétrio, P.; Souza França, D.; Nakajima, R.; Burnazi, E.; Roberts, S.; Brand, C.; Grkovski, M.; Mauguen, A.; Dunphy, M.P.; et al. PARP1/2 Imaging with 18F-PARPi in Patients with Head and Neck Cancer. medRxiv 2019, 19009381. [Google Scholar]
- Schöder, H.; de Souza França, P.D.; Nakajima, R.; Burnazi, E.; Roberts, S.; Brand, C.; Grkovski, M.; Mauguen, A.; Dunphy, M.P.; Ghossein, R.A.; et al. Safety and Feasibility of PARP1/2 Imaging with 18F-PARPi in Patients with Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 3110–3116. [Google Scholar] [CrossRef] [PubMed]
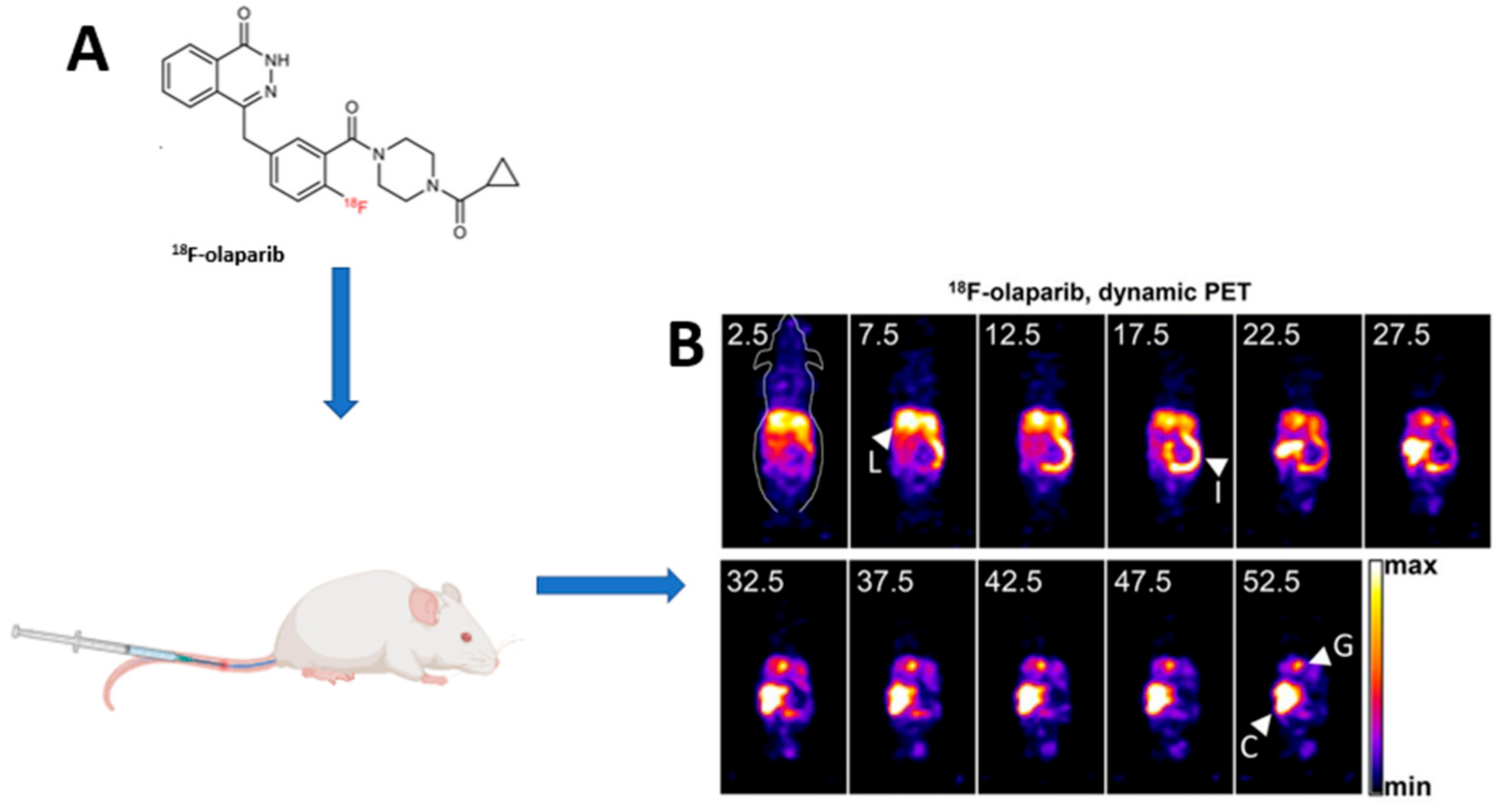
- Wilson, T.C.; Xavier, M.A.; Knight, J.; Verhoog, S.; Torres, J.B.; Mosley, M.; Hopkins, S.L.; Wallington, S.; Allen, P.D.; Kersemans, V.; et al. PET Imaging of PARP Expression Using 18F-Olaparib. J. Nucl. Med. 2019, 60, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Bowden, G.D.; Chailanggar, N.; Pichler, B.J.; Maurer, A. Scalable 18F Processing Conditions for Copper-Mediated Radiofluorination Chemistry Facilitate DoE Optimization Studies and Afford an Improved Synthesis of [18F]Olaparib. Org. Biomol. Chem. 2021, 19, 6995–7000. [Google Scholar] [CrossRef] [PubMed]
- Guibbal, F.; Isenegger, P.G.; Wilson, T.C.; Pacelli, A.; Mahaut, D.; Sap, J.B.I.; Taylor, N.J.; Verhoog, S.; Preshlock, S.; Hueting, R.; et al. Manual and Automated Cu-Mediated Radiosynthesis of the PARP Inhibitor [18F]Olaparib. Nat. Protoc. 2020, 15, 1525–1541. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Hopkins, S.L.; Guibbal, F.; Pacelli, A.; Baguña Torres, J.; Mosley, M.; Lau, D.; Isenegger, P.; Chen, Z.; Wilson, T.C.; et al. Correlation between Molar Activity, Injection Mass and Uptake of the PARP Targeting Radiotracer [18F]Olaparib in Mouse Models of Glioma. EJNMMI Res. 2022, 12, 67. [Google Scholar] [CrossRef] [PubMed]
- Zmuda, F.; Blair, A.; Liuzzi, M.C.; Malviya, G.; Chalmers, A.J.; Lewis, D.; Sutherland, A.; Pimlott, S.L. An 18 F-Labeled Poly(ADP-Ribose) Polymerase Positron Emission Tomography Imaging Agent. J. Med. Chem. 2018, 61, 4103–4114. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.W.; Puentes, L.N.; Schmitz, A.; Hsieh, C.J.; Weng, C.C.; Hou, C.; Li, S.; Kuo, Y.-M.; Padakanti, P.; Lee, H.; et al. Synthesis and Evaluation of an AZD2461 [18F]PET Probe in Non-Human Primates Reveals the PARP-1 Inhibitor to Be Non-Blood-Brain Barrier Penetrant. Bioorganic Chem. 2019, 83, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Guibbal, F.; Hopkins, S.L.; Pacelli, A.; Isenegger, P.G.; Mosley, M.; Torres, J.B.; Dias, G.M.; Mahaut, D.; Hueting, R.; Gouverneur, V.; et al. [18F]AZD2461, an Insight on Difference in PARP Binding Profiles for DNA Damage Response PET Imaging. Mol. Imaging Biol. 2020, 22, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ou, S.; Wei, H.; Qin, X.; Jiang, Q. Comparative Efficacy and Safety of Poly (ADP-Ribose) Polymerase Inhibitors in Patients with Ovarian Cancer: A Systematic Review and Network Meta-Analysis. Front. Oncol. 2022, 12, 815265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chu, W.; Xu, J.; Jones, L.A.; Peng, X.; Li, S.; Chen, D.L.; Mach, R.H. Synthesis, [18F] Radiolabeling, and Evaluation of Poly (ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors for In Vivo Imaging of PARP-1 Using Positron Emission Tomography. Bioorganic Med. Chem. 2014, 22, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, C.; Lieberman, B.; Xu, K.; Zeng, C.; Makvandi, M.; Li, S.; Hou, C.; Lee, H.; Greenberg, R.; Mankoff, D.; et al. Abstract P5-01-06: 18F-Radiolabeled PARP-1 Inhibitor Uptake as a Marker of PARP-1 Activity in Breast Cancer. Cancer Res. 2016, 76 (Suppl. S4), P5-01-06. [Google Scholar] [CrossRef]
- Makvandi, M.; Pantel, A.; Schwartz, L.; Schubert, E.; Xu, K.; Hsieh, C.J.; Hou, C.; Kim, H.; Weng, C.-C.; Winters, H.; et al. A PET Imaging Agent for Evaluating PARP-1 Expression in Ovarian Cancer. J. Clin. Investig. 2018, 128, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Effron, S.S.; Makvandi, M.; Lin, L.; Xu, K.; Li, S.; Lee, H.; Hou, C.; Pryma, D.A.; Koch, C.; Mach, R.H. PARP-1 Expression Quantified by [18F]FluorThanatrace: A Biomarker of Response to PARP Inhibition Adjuvant to Radiation Therapy. Cancer Biotherapy Radiopharm. 2017, 32, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Schwarz, S.W.; Schubert, E.K.; Chen, D.L.; Doot, R.K.; Makvandi, M.; Lin, L.L.; McDonald, E.S.; Mankoff, D.A.; Mach, R.H. The Development Of18F Fluorthanatrace: A PET Radiotracer for Imaging Poly (ADP-Ribose) Polymerase-1. Radiol. Imaging Cancer 2022, 4, e210070. [Google Scholar] [CrossRef] [PubMed]
- Dehdashti, F.; Reimers, M.A.; Shoghi, K.I.; Chen, D.L.; Luo, J.; Rogers, B.; Pachynski, R.K.; Sreekumar, S.; Weimholt, C.; Zhou, D. Pilot Study: PARP1 Imaging in Advanced Prostate Cancer. Mol. Imaging Biol. 2022, 24, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Young, A.J.; Pantel, A.R.; Viswanath, V.; Dominguez, T.L.; Makvandi, M.; Lee, H.; Li, S.; Schubert, E.K.; Pryma, D.A.; Farwell, M.D.; et al. Kinetic and Static Analysis of Poly-(Adenosine Diphosphate-Ribose) Polymerase-1-Targeted 18F-Fluorthanatrace PET Images of Ovarian Cancer. J. Nucl. Med. 2022, 63, 44–50. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.S.; Pantel, A.R.; Shah, P.D.; Farwell, M.D.; Clark, A.S.; Doot, R.K.; Pryma, D.A.; Carlin, S.D. In Vivo Visualization of PARP Inhibitor Pharmacodynamics. JCI Insight 2021, 6, 6–10. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.S.; Doot, R.K.; Pantel, A.R.; Farwell, M.D.; Mach, R.H.; Maxwell, K.N.; Mankoff, D.A. Positron Emission Tomography Imaging of Poly-(Adenosine Diphosphate-Ribose) Polymerase 1 Expression in Breast Cancer: A Nonrandomized Clinical Trial. JAMA Oncol. 2020, 6, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Destro, G.; Guibbal, F.; Chan, Y.; Cornelissen, B. Copper-Mediated Radiosynthesis of [18F] Rucaparib. Org. Lett. 2021, 23, 7290–7294. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Chen, Z.; Destro, G.; Veal, M.; Lau, D.; O’Neill, E.; Dias, G.; Mosley, M.; Kersemans, V.; Guibbal, F.; et al. Imaging PARP with [18F]Rucaparib in Pancreatic Cancer Models. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3668–3678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Abou, D.; Lu, P.; Hasson, A.M.; Villmer, A.; Benabdallah, N.; Jiang, W.; Ulmert, D.; Carlin, S.; Rogers, B.E.; et al. [18F]-Labeled PARP-1 PET Imaging of PSMA Targeted Alpha Particle Radiotherapy Response. Sci. Rep. 2022, 12, 13034. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, H.; Mpoy, C.; Afrin, S.; Rogers, B.E.; Garbow, J.R.; Katzenellenbogen, J.A.; Xu, J. Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as Parp-1-Targeting Agents. Biomedicines 2021, 9, 565. [Google Scholar] [CrossRef] [PubMed]
- Shuhendler, A.J.; Cui, L.; Chen, Z.; Shen, B.; Chen, M.; James, M.L.; Witney, T.H.; Bazalova-Carter, M.; Gambhir, S.S.; Chin, F.T.; et al. [18F]-SuPAR: A Radiofluorinated Probe for Noninvasive Imaging of DNA Damage-Dependent Poly(ADP-Ribose) Polymerase Activity. Bioconjugate Chem. 2019, 30, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, W.; Li, K.; Chen, K.; He, S.; Zhang, J.; Gu, B.; Xu, X.; Song, S. PET Imaging of PARP Expression Using 68Ga-Labelled Inhibitors. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 2606–2620. [Google Scholar] [CrossRef]
- Tu, Z.; Chu, W.; Zhang, J.; Dence, C.S.; Welch, M.J.; Mach, R.H. Synthesis and in Vivo Evaluation of [11C]PJ34, a Potential Radiotracer for Imaging the Role of PARP-1 in Necrosis. Nucl. Med. Biol. 2005, 32, 437–443. [Google Scholar] [CrossRef]
- Huang, T.; Hu, P.; Banizs, A.B.; He, J. Initial Evaluation of Cu-64 Labeled PARPi-DOTA PET Imaging in Mice with Mesothelioma. Bioorganic Med. Chem. Lett. 2017, 27, 3472–3476. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K. The Current Status of the Production and Supply of Gallium-68. Cancer Biother. Radiopharm. 2020, 35, 163–166. [Google Scholar] [CrossRef]
- Wenz, J.; Arndt, F.; Samnick, S. A New Concept for the Production of 11C-Labelled Radiotracers. EJNMMI Radiopharm. Chem. 2022, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Ferdani, R. Copper-64 Radiopharmaceuticals for PET Imaging of Cancer: Advances in Preclinical and Clinical Research. Cancer Biother. Radiopharm. 2009, 24, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.A.; Peil, J.; Vogg, A.T.J.; Bolm, C.; Terhorst, S.; Classen, A.; Bauwens, M.; Maurer, J.; Mottaghy, F.; Morgenroth, A. Auger Emitter Conjugated PARP Inhibitor for Therapy in Triple Negative Breast Cancers: A Comparative In-Vitro Study. Cancers 2022, 14, 230. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Pacelli, A.; Nader, M.; Kossatz, S. DNA Repair Enzyme Poly(ADP-Ribose) Polymerase 1/2 (PARP1/2)-Targeted Nuclear Imaging and Radiotherapy. Cancers 2022, 14, 1129. [Google Scholar] [CrossRef] [PubMed]
- Jannetti, S.A.; Carlucci, G.; Carney, B.; Kossatz, S.; Shenker, L.; Carter, L.M.; Salinas, B.; Brand, C.; Sadique, A.; Donabedian, P.L.; et al. PARP-1-Targeted Radiotherapy in Mouse Models of Glioblastoma. J. Nucl. Med. 2018, 59, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Laird, J.; Lok, B.H.; Carney, B.; Kossatz, S.; de Stanchina, E.; Reiner, T.; Poirier, J.T.; Rudin, C.M. Positron-Emission Tomographic Imaging of a Fluorine 18–Radiolabeled Poly(ADP-Ribose) Polymerase 1 Inhibitor Monitors the Therapeutic Efficacy of Talazoparib in SCLC Patient–Derived Xenografts. J. Thorac. Oncol. 2019, 14, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
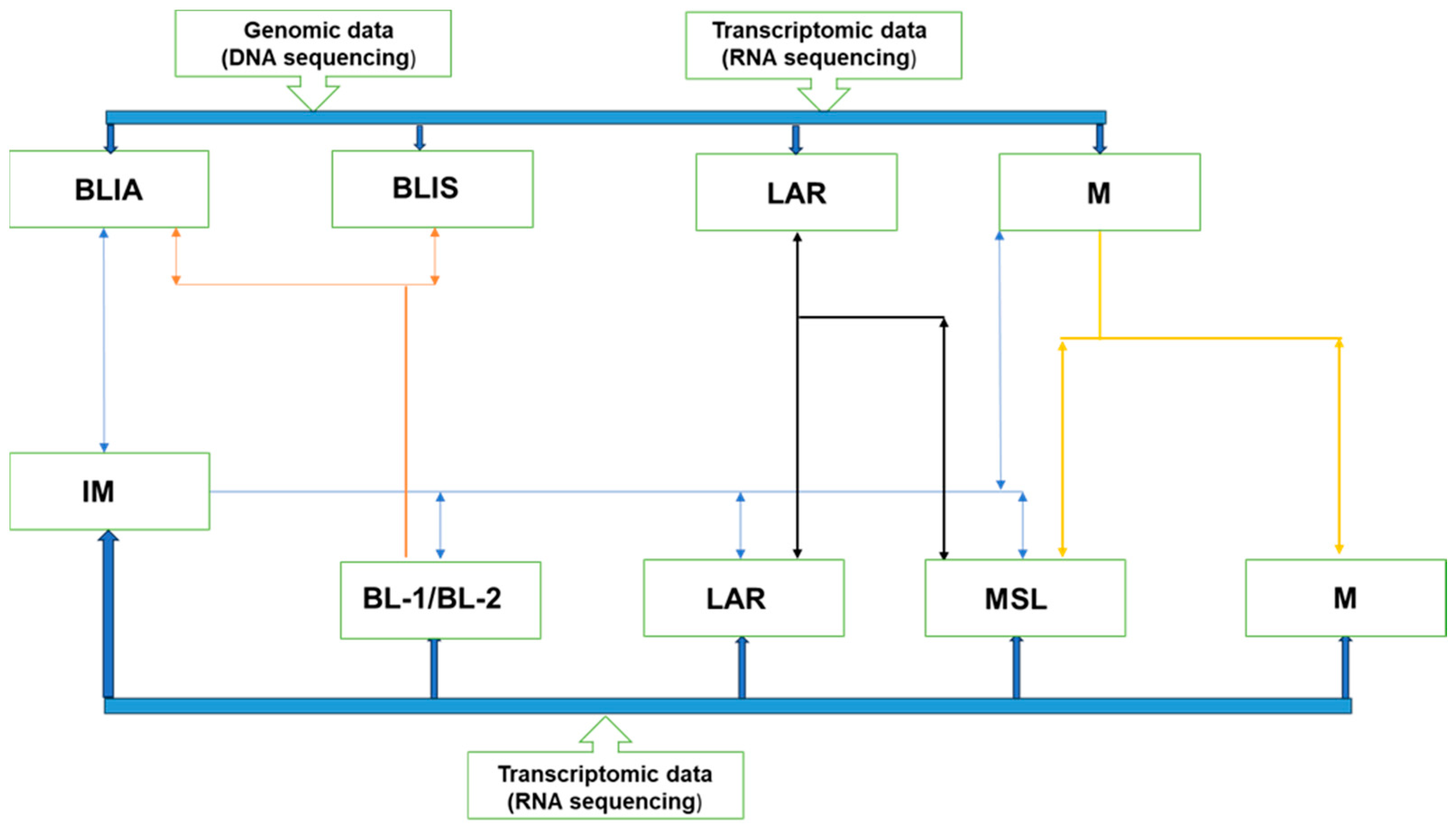
| Subtype | Molecular Features | Important Markers | Possible Therapies | |
|---|---|---|---|---|
| Basal-like (BL) | BL-1 | Elevated cell cycle and DDR gene expression. | ATR/BRCA, Ki-67 | Cisplatin and PARP inhibitors |
| BL-2 | Enriched in growth factor signaling, metabolic pathways, and myoepithelial signaling. | EGFR, IGF1R, NGF, MET, Wnt/b-catenin, EPHA2, TP63 | Growth factor inhibitors | |
| Immune enriched | IM and/or BSLIA | Genes involved in immune and cytokine signaling transduction pathways. | IL-12, IL-7, NFKB, TNF, JAK/STAT | Immune check point inhibitors |
| Mesenchymal | Mesenchymal | Gene expression for EMT, cell motility, and differentiation. | Wnt, ALK, TGF-β | |
| Mesenchymal-like | Increased growth factor signaling compared with (M), low proliferation, enrichment of genes associated with angiogenesis and stem cells, and low claudin expression. | EGFR, PDGF, ERK1/2, TGF-β, Wnt/β-catenin | EGFR, PDGF, ERK1/2, TGF-β inhibitors, growth factor inhibitors, Src inhibitors | |
| Luminal androgen receptor (LAR) | LAR | Increase in hormonally regulated pathways, AR signaling, and high rate of PIK3C-activating mutations. | AR, FOXA1, KRT18, XBP1, and ESR1 | AR antagonists, PI3K inhibitors, Hsp inhibitors, ER pathway inhibitors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndlovu, H.; Lawal, I.O.; Mdanda, S.; Kgatle, M.M.; Mokoala, K.M.G.; Al-Ibraheem, A.; Sathekge, M.M. [18F]F-Poly(ADP-Ribose) Polymerase Inhibitor Radiotracers for Imaging PARP Expression and Their Potential Clinical Applications in Oncology. J. Clin. Med. 2024, 13, 3426. https://doi.org/10.3390/jcm13123426
Ndlovu H, Lawal IO, Mdanda S, Kgatle MM, Mokoala KMG, Al-Ibraheem A, Sathekge MM. [18F]F-Poly(ADP-Ribose) Polymerase Inhibitor Radiotracers for Imaging PARP Expression and Their Potential Clinical Applications in Oncology. Journal of Clinical Medicine. 2024; 13(12):3426. https://doi.org/10.3390/jcm13123426
Chicago/Turabian StyleNdlovu, Honest, Ismaheel O. Lawal, Sipho Mdanda, Mankgopo M. Kgatle, Kgomotso M. G. Mokoala, Akram Al-Ibraheem, and Mike M. Sathekge. 2024. "[18F]F-Poly(ADP-Ribose) Polymerase Inhibitor Radiotracers for Imaging PARP Expression and Their Potential Clinical Applications in Oncology" Journal of Clinical Medicine 13, no. 12: 3426. https://doi.org/10.3390/jcm13123426
APA StyleNdlovu, H., Lawal, I. O., Mdanda, S., Kgatle, M. M., Mokoala, K. M. G., Al-Ibraheem, A., & Sathekge, M. M. (2024). [18F]F-Poly(ADP-Ribose) Polymerase Inhibitor Radiotracers for Imaging PARP Expression and Their Potential Clinical Applications in Oncology. Journal of Clinical Medicine, 13(12), 3426. https://doi.org/10.3390/jcm13123426