
Hypertensive Heart Disease: A Narrative Review Series—Part 1: Pathophysiology and Microstructural Changes



Abstract
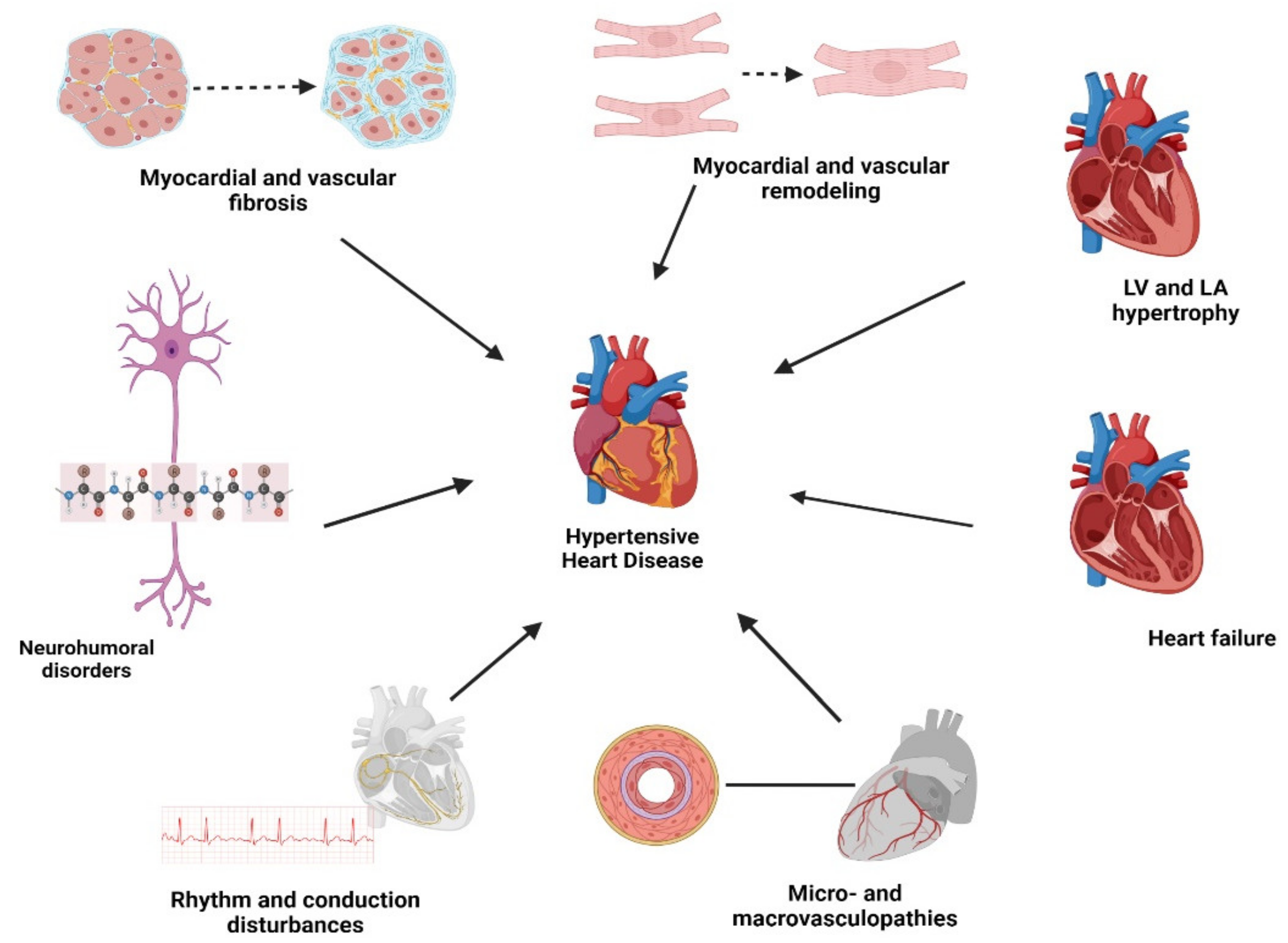
1. Introduction
2. Methods
Literature Search and Eligibility Criteria
3. Review
3.1. Myocardial Remodeling
3.2. Myocardial and Vascular Fibrosis
3.3. Extracellular Matrix (ECM)
3.4. Clinical Role of Fibrosis
3.5. Value of Circulating Biomarkers for Myocardial Fibrosis

{kind=link}
| Marker | Levels in Studied HHD/HTN/HHF | Role in Collagen Metabolism | Effect on Fibrosis | Study: HHD/HTN/HHF | Author, Year |
|---|---|---|---|---|---|
| Aldosterone | Increased | Profibrotic biomarker | Stimulates myocardial fibroblasts to synthesize and secrete procollagen into the ECM [58] | CHF with elevated plasma levels of natriuretic peptides, and LV EF ≤ 40% [48] HFpEF [58] HTN [52] | MR. Zile et al., 2019 [48] Paulus W.J., 2021 [58] Harvey A. et al., 2016 [52] |
| Angiotensin II ( Ang II) | Increased | Profibrotic biomarker | Can stimulate myofibroblasts through ANG II type 1, inhibits fibroblast apoptosis, stimulates fibroblast migration, and inducts TGF-β1. This promotes fibroblast proliferative, synthesis, and secretion activities of procollagen I and III [30,44,51] | HHD [44] HTN [52] | Schimmel K. et al., 2022 [30] Frangogiannis N.G., 2021 [51] Berk B.C. et al., 2007 [44] Harvey A. et al., 2016 [52] |
| Endothelin-1 (ET-1) | Increased | Profibrotic biomarker | Endothelin-1 secreted also by fibroblasts, cardiomyocytes, and macrophages and can bind to ET cardiac fibroblasts receptors, contributing to the development of myocardial fibrosis. | HTN [52,59] | Schimmel K. et al., 2022 [30] Fang T. et al., 2019 [59] Harvey A. et al., 2016 [52] |
| C-reactive protein (CRP) | Increased | Profibrotic biomarker | CRP directly induces collagen matrix and α-SMA expression in cardiac fibroblasts in vitro or promotes this fibrosis response in the presence of Ang II in vivo and in vitro [60] | with HFpEF [58,61] HTN [60] | Sanders-van Wijk S. et al., 2015 [61] Paulus W.J. et al., 2021 [58] Zhang R. et al., 2010 [60] |
| Soluble suppression of tumorigenicity-2 protein (sST2) | Increased | Profibrotic biomarker | Stimulate myocardial fibroblasts synthesize and secrete procollagen into the ECM [58] | CHF with elevated plasma levels of natriuretic peptides, and LV EF ≤ 40% [48] HFpEF [58] | MR. Zile et al., 2019 [48] Paulus W.J. et al., 2021 [58] |
| Interleukin 6 (IL6) | Increased | Profibrotic biomarker | The soluble IL-6 receptor in combination with IL-6 is essential in increasing collagen content regulated by isolated cardiac fibroblasts, and also plays a role in mediating a phenotypic conversion to myofibroblasts [49] | with HFpEF [58,61] HTN [59] HTN [49,58] | Sanders-van Wijk S. et al., 2015 [61] Paulus W.J. et al., 2021 [58] Fang T. et al., 2019 [59] Meléndez G.C. et al., 2010 [49] |
| Transforming growth factor-β1 (TGF-β1) | Increased | Profibrotic biomarker | A fibrogenic cytokine that upregulates the expression of the genes encoding fibrillar collagen type I and type III [44], stimulating both myofibroblast formation and collagen production [44]. Activation of vascular TGF-β1 increases the fibronectin, collagen, and plasminogen activator inhibitor-1 (PAI-1) synthesis that stimulates TIMP [52] | HHD [44] HTN [52] | Berk B.C. et al., 2007 [44] Harvey A. et al., 2016 [52] |
| Growth differentiation factor 15 (GDF-15) | Increased | Profibrotic biomarker | A member of the TGF-β cytokine superfamily, one of the factors controlling a transformation of cardiac fibroblasts to myofibroblasts [62] | HFpEF [58,61] HF [62] | Sanders-van Wijk S. et al., 2015 [61] Paulus W.J. et al., 2021 [58] Rochette L. et al., 2021 [62] |
| Fibroblast growth factor-21 (FGF21) | Increased | Antifibrotic biomarker | Able to reverse the myofibroblast phenotype in vitro and in vivo, indicating direct protective effects against cardiac fibrosis development [23] | HHD [23] | Ferrer-Curriu G. et al., 2019 [23] |
| Galectin-3 (Gal-3) | Increased | Profibrotic biomarker | Stimulates myocardial fibroblasts to synthesize and secrete procollagen into the ECM (extracellular matrix) [58] and reflects the general extent of fibrosis and the severity of HFpEF [63] | CHF with elevated plasma levels of natriuretic peptides, and LV EF ≤ 40% [48] HFpEF [58] HTN with HF [28] HTN [52] | MR. Zile et al., 2019 [48] Paulus W.J., 2021 [58] Mavrogeni S., et al., 2017 [28] Harvey A. et al., 2016 [52] |
| Plasminogen activator inhibitor-1 (PAI-1) | Increased | Profibrotic biomarker | Inhibits fibrinolysis, reduces plasmin generation that leads to accumulation of ECM proteins and tissue fibrosis by preventing tissue proteolytic activity and reducing collagen degradation [52] | HTN [52] | Harvey A. et al., 2016 [52] |
| Matrix metalloproteinase-1 (MMP-1) | Increased [45] Decreased [44] | Biomarker of collagen degradation | Related to the loss of the physiological mysial collagen scaffold and accumulation of pathologic non-mysial collagen, [45] initiates the ECM degradation process by cleaving the α-chains of type I and type II [44] collagens and type III [27] collagen | HHD with HF [45] HHD with LVH [44] | López B. et al., 2015 [45] Berk B.C. et al., 2007 [44] |
| Matrix metalloproteinase-2 (MMP-2) | Decreased [48,64] Increased [44] | Biomarker of collagen degradation | MMP-2 degrades basement membrane proteins, fibrillar collagen peptides, and newly synthesized type I, II, and III collagens [27,64]. Stimulation of TGF-β1 signaling; increases vascular smooth muscle cell production of collagens I, II, and III; increases fibronectin secretion that lead to collagen accumulation in the vascular wall [52] | CHF with elevated plasma levels of natriuretic peptides, and LV EF ≤ 40% [48] HTN with LVH [64] HTN [52] | MR. Zile et al., 2019 [48] Ahmed SH et al., 2006 [64] Harvey A. et al., 2016 [52] |
| Matrix metalloproteinase-9 (MMP-9) | Decreased [48] Increased [52,64] | Profibrotic biomarker | MMP-9 has significant effects on transforming growth factor-β and other “profibrotic” proteins and profibrotic pathways [64]. Increases in MMP-9 would be expected to increase ECM accumulation [52,64] | CHF with elevated plasma levels of natriuretic peptides, and LV EF ≤ 40% [48] HTN with LVH [64] HTN [52] | MR. Zile et al., 2019 [48] Ahmed S.H. et al., 2006 [64] Harvey A. et al., 2016 [52] |
| Matrix metalloproteinase-13 (MMP-13) | Decreased | Biomarker of collagen degradation | Collagenolytic enzyme. The reduction would be expected to cause reduced fibrillar collagen turnover, reduced degradation, and increased ECM accumulation [64] | HTN with LVH [64] HTN with LVH and CHF [64] HHD [44] | Ahmed S.H. et al., 2006 [64] Berk B.C. et al., 2007 [44] |
| Matrix metalloproteinase-1/Tissue inhibitor of matrix metalloproteinase-1 (MMP-1/TIMP-1) ratio | Increased | Reflects ECM proteolytic activity | Characterizes the balance of degradation and synthesis of ECM. | HTN with HF [19,65] HTN with LVH [44] | Drazner M.H., 2011 [19] Berk B.C. et al., 2007 [44] López B., et al., 2006 [65] |
| Tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) | Increased | Profibrotic biomarker | Increase in TIMPs inhibits MMPs enzymatic activity, which would facilitate ECM accumulation and decreased collagen degradation [64] | CHF with elevated plasma levels of natriuretic peptides, and LV EF ≤ 40% [48] HTN with LVH and CHF [64] HHD with LVH [44] | MR. Zile et al., 2019 [48] Ahmed S.H. et al., 2006 [64] Berk B.C. et al., 2007 [44] |
| Serum amino-terminal propeptide of procollagen type III (PIIINP) | Increased | Profibrotic biomarker | Plasma/serum concentrations reflect collagen synthesis rate [58] | CHF with elevated plasma levels of natriuretic peptides, and LV EF ≤ 40% [48] HFpEF [58] HHD with HF [45] | MR Zile et al., 2019 [48] Paulus W.J., 2021 [58] López B. et al., 2015 [45] |
| Procollagen type I N-terminal propeptide (PINP) | Increased | Profibrotic biomarker | Plasma/serum concentrations reflect collagen synthesis rate [58] | CHF with elevated plasma levels of natriuretic peptides, and LV EF ≤ 40% [48] HFpEF [58] | MR. Zile et al., 2019 [48] Paulus W.J., 2021 [58] |
| Serum carboxy-terminal propeptide of procollagen type I (PICP) | Increased | Profibrotic biomarker | Plasma/serum concentrations reflect collagen synthesis rate [58], acting as a marker of extracellular collagen type I synthesis [66,67] | HFpEF [58] HHD with/without HF [66,67] | Paulus W.J., 2021 [58] Querejeta R. et al., 2000 [66], 2004 [67] |
| Serum C-terminal telopeptide of collagen type I (CITP) | Decreased | Biomarker of collagen degradation | Serum concentrations related to the intensity of the degradation of collagen type I fibrils [58,66] | HHD and HF [21] HTN [66] | Querejeta. R. et al., 2000 [66] González, A., 2018 [21] |
| Carboxy-terminal propeptide of procollagen type III (PIIICP) | Increased | Profibrotic biomarker | Plasma/serum concentrations reflect collagen synthesis rate [58] | HFpEF [58] | Paulus, W.J., 2021 [58] |
3.6. Value of Imaging to Detection of Myocardial Fibrosis
3.7. Myocardial Fibrosis as Therapeutic Target
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- NCD Risk Factor Collaboration. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: A pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 2021, 398, 957–980. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, 1269–1324. [Google Scholar] [CrossRef]
- Al Ghorani, H.; Kulenthiran, S.; Lauder, L.; Bohm, M.; Mahfoud, F. Hypertension trials update. J. Hum. Hypertens. 2021, 35, 398–409. [Google Scholar] [CrossRef]
- Vischer, A.S.; Socrates, T.; Winterhalder, C.; Eckstein, J.; Mayr, M.; Burkard, T. Impact of single-visit American versus European office blood pressure measurement procedure on individual blood pressure classification: A cross-sectional study. Clin. Res. Cardiol. 2019, 108, 990–999. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart. J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef]
- The Criteria Committee of the New York Heart Association. Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels; Little, Brown Company: Boston, MA, USA, 1979; p. 12. [Google Scholar]
- Frohlich, E. Evaluation and management of the patients with essential hypertension. In Cardiology; Parmley, W.W., Chatterjee, K., Eds.; Lippincott: Philadelphia, PA, USA, 1989; Volume 23. [Google Scholar]
- Nwabuo, C.C.; Vasan, R.S. Pathophysiology of Hypertensive Heart Disease: Beyond Left Ventricular Hypertrophy. Curr. Hypertens. Rep. 2020, 22, 11. [Google Scholar] [CrossRef]
- Kuroda, K. Hypertensive cardiomyopathy: A clinical approach and literature review. World J. Hypertens. 2015, 5, 41–52. [Google Scholar] [CrossRef]
- Albakri, A. Hypertensive heart failure: A review of clinical status and meta-analyses of prognostic value of echocardiography and antihypertensive medication. Integr. Mol. Med. 2018, 5, 1–16. [Google Scholar] [CrossRef]
- Alegría-Ezquerra, E.; González-Juanatey, J.R.; González-Maqueda, I. Hypertensive Heart Disease: A Proposed Clinical Classification. Rev. Española De Cardiol. (Engl. Ed.) 2006, 59, 398–399. [Google Scholar] [CrossRef]
- Frohlich, E.D.; Apstein, C.; Chobanian, A.V.; Devereux, R.B.; Dustan, H.P.; Dzau, V.; Fauad-Tarazi, F.; Horan, M.J.; Marcus, M.; Massie, B.; et al. The heart in hypertension. N. Engl. J. Med. 1992, 327, 998–1008. [Google Scholar] [CrossRef]
- Izzo, J.L., Jr.; Gradman, A.H. Mechanisms and management of hypertensive heart disease: From left ventricular hypertrophy to heart failure. Med. Clin. North Am. 2004, 88, 1257–1271. [Google Scholar] [CrossRef]
- Prisant, L.M. Hypertensive heart disease. J. Clin. Hypertens. 2005, 7, 231–238. [Google Scholar] [CrossRef]
- Gonzalez, A.; Lopez, B.; Diez, J. New directions in the assessment and treatment of hypertensive heart disease. Curr. Opin. Nephrol. Hypertens. 2005, 14, 428–434. [Google Scholar] [CrossRef]
- Gradman, A.H.; Alfayoumi, F. From left ventricular hypertrophy to congestive heart failure: Management of hypertensive heart disease. Prog. Cardiovasc. Dis. 2006, 48, 326–341. [Google Scholar] [CrossRef]
- Gonzalez-Maqueda, I.; Alegria-Ezquerra, E.; Gonzalez-Juanatey, J.R. Hypertensive heart disease: A new clinical classification (VIA). E-J. ESC Counc. Cardiol. Pract. 2009, 7, 20. [Google Scholar]
- Drazner, M.H. The progression of hypertensive heart disease. Circulation 2011, 123, 327–334. [Google Scholar] [CrossRef]
- Díez, J. Hypertensive heart disease. Hypertension 2013, 152–166. [Google Scholar] [CrossRef]
- Gonzalez, A.; Ravassa, S.; Lopez, B.; Moreno, M.U.; Beaumont, J.; San Jose, G.; Querejeta, R.; Bayes-Genis, A.; Diez, J. Myocardial Remodeling in Hypertension. Hypertension 2018, 72, 549–558. [Google Scholar] [CrossRef]
- Kalogeropoulos, A.P.; Goulbourne, C.; Butler, J. Diagnosis and Prevention of Hypertensive Heart Failure. Heart Fail. Clin. 2019, 15, 435–445. [Google Scholar] [CrossRef]
- Ferrer-Curriu, G.; Redondo-Angulo, I.; Guitart-Mampel, M.; Ruperez, C.; Mas-Stachurska, A.; Sitges, M.; Garrabou, G.; Villarroya, F.; Fernandez-Sola, J.; Planavila, A. Fibroblast growth factor-21 protects against fibrosis in hypertensive heart disease. J. Pathol. 2019, 248, 30–40. [Google Scholar] [CrossRef]
- Tackling, G.; Borhade, M.B. Hypertensive Heart Disease; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Lu, Y.; Lan, T. Global, regional, and national burden of hypertensive heart disease during 1990-2019: An analysis of the global burden of disease study 2019. BMC Public Health 2022, 22, 841. [Google Scholar] [CrossRef]
- Iriarte, M.; Murga, N.; Sagastagoitia, D.; Morillas, M.; Boveda, J.; Molinero, E.; Etxebeste, J.; Salcedo, A.; Rodriguez, E.; Ormaetxe, J.M.; et al. Classification of hypertensive cardiomyopathy. Eur. Heart J. 1993, 14, 95–101. [Google Scholar]
- Gyongyosi, M.; Winkler, J.; Ramos, I.; Do, Q.T.; Firat, H.; McDonald, K.; Gonzalez, A.; Thum, T.; Diez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef]
- Mavrogeni, S.; Katsi, V.; Vartela, V.; Noutsias, M.; Markousis-Mavrogenis, G.; Kolovou, G.; Manolis, A. The emerging role of Cardiovascular Magnetic Resonance in the evaluation of hypertensive heart disease. BMC Cardiovasc. Disord. 2017, 17, 132. [Google Scholar] [CrossRef]
- Kuruvilla, S.; Janardhanan, R.; Antkowiak, P.; Keeley, E.C.; Adenaw, N.; Brooks, J.; Epstein, F.H.; Kramer, C.M.; Salerno, M. Increased extracellular volume and altered mechanics are associated with LVH in hypertensive heart disease, not hypertension alone. JACC Cardiovasc. Imaging 2015, 8, 172–180. [Google Scholar] [CrossRef]
- Schimmel, K.; Ichimura, K.; Reddy, S.; Haddad, F.; Spiekerkoetter, E. Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front. Cardiovasc. Med. 2022, 9, 886553. [Google Scholar] [CrossRef]
- Ojji, D.; Libhaber, E.; Lamont, K.; Thienemann, F.; Sliwa, K. Circulating biomarkers in the early detection of hypertensive heart disease: Usefulness in the developing world. Cardiovasc. Diagn. Ther. 2020, 10, 296–304. [Google Scholar] [CrossRef]
- Bozkurt, B.; Coats, A.J.S.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar] [CrossRef]
- Gonzalez, A.; Schelbert, E.B.; Diez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef]
- Zheng, Q.; Loo, G.; Le, T.-T.; Shi, L.; Chan, E.S.-Y.; Chin, C.W.L. Prognosis associated with geometric patterns of left ventricular remodeling: Systematic review and network meta-analysis. F1000Research 2019, 8, 1130. [Google Scholar] [CrossRef]
- Saliba, L.J.; Maffett, S. Hypertensive Heart Disease and Obesity: A Review. Heart Fail. Clin. 2019, 15, 509–517. [Google Scholar] [CrossRef]
- Schumann, C.L.; Jaeger, N.R.; Kramer, C.M. Recent Advances in Imaging of Hypertensive Heart Disease. Curr. Hypertens. Rep. 2019, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, F.; Yalcin, H.; Abraham, M.R.; Abraham, T.P. Ultimate phases of hypertensive heart disease and stressed heart morphology by conventional and novel cardiac imaging. Am. J. Cardiovasc. Dis. 2021, 11, 628–634. [Google Scholar] [PubMed]
- Cameli, M.; Mandoli, G.E.; Lisi, E.; Ibrahim, A.; Incampo, E.; Buccoliero, G.; Rizzo, C.; Devito, F.; Ciccone, M.M.; Mondillo, S. Left atrial, ventricular and atrio-ventricular strain in patients with subclinical heart dysfunction. Int. J. Cardiovasc. Imaging 2019, 35, 249–258. [Google Scholar] [CrossRef]
- Medi, C.; Kalman, J.M.; Spence, S.J.; Teh, A.W.; Lee, G.; Bader, I.; Kaye, D.M.; Kistler, P.M. Atrial electrical and structural changes associated with longstanding hypertension in humans: Implications for the substrate for atrial fibrillation. J. Cardiovasc. Electrophysiol. 2011, 22, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.H.; Shenasa, H.A.; Shenasa, M. Hypertension, Prehypertension, Hypertensive Heart Disease, and Atrial Fibrillation. Card. Electrophysiol. Clin. 2021, 13, 37–45. [Google Scholar] [CrossRef]
- Slivnick, J.; Lampert, B.C. Hypertension and Heart Failure. Heart Fail. Clin. 2019, 15, 531–541. [Google Scholar] [CrossRef]
- Cuspidi, C.; Facchetti, R.; Bombelli, M.; Tadic, M.; Sala, C.; Grassi, G.; Mancia, G. High Normal Blood Pressure and Left Ventricular Hypertrophy Echocardiographic Findings from the PAMELA Population. Hypertension 2019, 73, 612–619. [Google Scholar] [CrossRef]
- Cuspidi, C.; Sala, C.; Tadic, M.; Gherbesi, E.; Grassi, G.; Mancia, G. Pre-hypertension and subclinical cardiac damage: A meta-analysis of echocardiographic studies. Int. J. Cardiol. 2018, 270, 302–308. [Google Scholar] [CrossRef]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Invest. 2007, 117, 568–575. [Google Scholar] [CrossRef]
- Lopez, B.; Gonzalez, A.; Ravassa, S.; Beaumont, J.; Moreno, M.U.; San Jose, G.; Querejeta, R.; Diez, J. Circulating Biomarkers of Myocardial Fibrosis: The Need for a Reappraisal. J. Am. Coll. Cardiol. 2015, 65, 2449–2456. [Google Scholar] [CrossRef]
- Krishnan, A.; Chilton, E.; Raman, J.; Saxena, P.; McFarlane, C.; Trollope, A.F.; Kinobe, R.; Chilton, L. Are Interactions between Epicardial Adipose Tissue, Cardiac Fibroblasts and Cardiac Myocytes Instrumental in Atrial Fibrosis and Atrial Fibrillation? Cells 2021, 10, 2501. [Google Scholar] [CrossRef]
- Gluba, A.; Bielecka-Dabrowa, A.; Mikhailidis, D.P.; Wong, N.D.; Franklin, S.S.; Rysz, J.; Banach, M. An update on biomarkers of heart failure in hypertensive patients. J. Hypertens. 2012, 30, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; O’Meara, E.; Claggett, B.; Prescott, M.F.; Solomon, S.D.; Swedberg, K.; Packer, M.; McMurray, J.J.V.; Shi, V.; Lefkowitz, M.; et al. Effects of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients with HFrEF. J. Am. Coll. Cardiol. 2019, 73, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.; Ravassa, S.; Gonzalez, A.; Zubillaga, E.; Bonavila, C.; Berges, M.; Echegaray, K.; Beaumont, J.; Moreno, M.U.; San Jose, G.; et al. Myocardial Collagen Cross-Linking Is Associated with Heart Failure Hospitalization in Patients with Hypertensive Heart Failure. J. Am. Coll. Cardiol. 2016, 67, 251–260. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef]
- Conrad, C.H.; Brooks, W.W.; Hayes, J.A.; Sen, S.; Robinson, K.G.; Bing, O.H. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation 1995, 91, 161–170. [Google Scholar] [CrossRef]
- Rain, S.; Andersen, S.; Najafi, A.; Gammelgaard Schultz, J.; da Silva Goncalves Bos, D.; Handoko, M.L.; Bogaard, H.J.; Vonk-Noordegraaf, A.; Andersen, A.; van der Velden, J.; et al. Right Ventricular Myocardial Stiffness in Experimental Pulmonary Arterial Hypertension: Relative Contribution of Fibrosis and Myofibril Stiffness. Circ. Heart Fail. 2016, 9, e002636. [Google Scholar] [CrossRef] [PubMed]
- Tadic, M.; Cuspidi, C. Right Ventricle in Arterial Hypertension: Did We Forget Something? J. Clin. Med. 2022, 11, 6257. [Google Scholar] [CrossRef]
- Kosinski, A.; Piwko, G.M.; Kaminski, R.; Nowicka, E.; Kaczynska, A.; Zajaczkowski, M.; Czerwiec, K.; Gleinert-Rozek, M.; Karnecki, K.; Gos, T. Arterial hypertension and remodelling of the right ventricle. Folia Morphol. 2022, 81, 336–342. [Google Scholar] [CrossRef]
- Ravassa, S.; Lopez, B.; Querejeta, R.; Echegaray, K.; San Jose, G.; Moreno, M.U.; Beaumont, F.J.; Gonzalez, A.; Diez, J. Phenotyping of myocardial fibrosis in hypertensive patients with heart failure. Influence on clinical outcome. J. Hypertens. 2017, 35, 853–861. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure with Preserved Ejection Fraction Paradigm Revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef]
- Fang, T.; Guo, B.; Xue, L.; Wang, L. Atorvastatin Prevents Myocardial Fibrosis in Spontaneous Hypertension via Interleukin-6 (IL-6)/Signal Transducer and Activator of Transcription 3 (STAT3)/Endothelin-1 (ET-1) Pathway. Med. Sci. Monit. 2019, 25, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, Y.Y.; Huang, X.R.; Wu, Y.; Chung, A.C.; Wu, E.X.; Szalai, A.J.; Wong, B.C.; Lau, C.P.; Lan, H.Y. C-reactive protein promotes cardiac fibrosis and inflammation in angiotensin II-induced hypertensive cardiac disease. Hypertension 2010, 55, 953–960. [Google Scholar] [CrossRef]
- Sanders-van Wijk, S.; van Empel, V.; Davarzani, N.; Maeder, M.T.; Handschin, R.; Pfisterer, M.E.; Brunner-La Rocca, H.P.; TIME-CHF Investigators. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur. J. Heart Fail. 2015, 17, 1006–1014. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Zeller, M.; Cottin, Y.; Vergely, C. GDF15 and Cardiac Cells: Current Concepts and New Insights. Int. J. Mol. Sci. 2021, 22, 8889. [Google Scholar] [CrossRef]
- Michalska-Kasiczak, M.; Bielecka-Dabrowa, A.; von Haehling, S.; Anker, S.D.; Rysz, J.; Banach, M. Biomarkers, myocardial fibrosis and co-morbidities in heart failure with preserved ejection fraction: An overview. Arch. Med. Sci 2018, 14, 890–909. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.H.; Clark, L.L.; Pennington, W.R.; Webb, C.S.; Bonnema, D.D.; Leonardi, A.H.; McClure, C.D.; Spinale, F.G.; Zile, M.R. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: Relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation 2006, 113, 2089–2096. [Google Scholar] [CrossRef]
- Lopez, B.; Gonzalez, A.; Querejeta, R.; Larman, M.; Diez, J. Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J. Am. Coll. Cardiol. 2006, 48, 89–96. [Google Scholar] [CrossRef]
- Querejeta, R.; Varo, N.; Lopez, B.; Larman, M.; Artinano, E.; Etayo, J.C.; Martinez Ubago, J.L.; Gutierrez-Stampa, M.; Emparanza, J.I.; Gil, M.J.; et al. Serum carboxy-terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive heart disease. Circulation 2000, 101, 1729–1735. [Google Scholar] [CrossRef]
- Querejeta, R.; Lopez, B.; Gonzalez, A.; Sanchez, E.; Larman, M.; Martinez Ubago, J.L.; Diez, J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: Relation to myocardial fibrosis. Circulation 2004, 110, 1263–1268. [Google Scholar] [CrossRef]
- Crnkovic, S.; Egemnazarov, B.; Damico, R.; Marsh, L.M.; Nagy, B.M.; Douschan, P.; Atsina, K.; Kolb, T.M.; Mathai, S.C.; Hooper, J.E.; et al. Disconnect between Fibrotic Response and Right Ventricular Dysfunction. Am. J. Respir. Crit. Care Med. 2019, 199, 1550–1560. [Google Scholar] [CrossRef]
- Friedberg, M.K.; Cho, M.Y.; Li, J.; Assad, R.S.; Sun, M.; Rohailla, S.; Honjo, O.; Apitz, C.; Redington, A.N. Adverse biventricular remodeling in isolated right ventricular hypertension is mediated by increased transforming growth factor-beta1 signaling and is abrogated by angiotensin receptor blockade. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1019–1028. [Google Scholar] [CrossRef]
- Aurigemma, G.P.; Salerno, M. A Novel Index of Remodeling in Hypertensive Heart Disease. Circ. Cardiovasc. Imaging 2017, 10, e006975. [Google Scholar] [CrossRef]
- Rodrigues, J.C.; Amadu, A.M.; Dastidar, A.G.; Szantho, G.V.; Lyen, S.M.; Godsave, C.; Ratcliffe, L.E.; Burchell, A.E.; Hart, E.C.; Hamilton, M.C.; et al. Comprehensive characterisation of hypertensive heart disease left ventricular phenotypes. Heart 2016, 102, 1671–1679. [Google Scholar] [CrossRef]
- Rodrigues, J.C.; Amadu, A.M.; Ghosh Dastidar, A.; McIntyre, B.; Szantho, G.V.; Lyen, S.; Godsave, C.; Ratcliffe, L.E.; Burchell, A.E.; Hart, E.C.; et al. ECG strain pattern in hypertension is associated with myocardial cellular expansion and diffuse interstitial fibrosis: A multi-parametric cardiac magnetic resonance study. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 441–450. [Google Scholar] [CrossRef]
- Cavallo, A.U.; Troisi, J.; Muscogiuri, E.; Cavallo, P.; Rajagopalan, S.; Citro, R.; Bossone, E.; McVeigh, N.; Forte, V.; Di Donna, C.; et al. Cardiac Computed Tomography Radiomics-Based Approach for the Detection of Left Ventricular Remodeling in Patients with Arterial Hypertension. Diagnostics 2022, 12, 322. [Google Scholar] [CrossRef]
- Zou, Y.; Akazawa, H.; Qin, Y.; Sano, M.; Takano, H.; Minamino, T.; Makita, N.; Iwanaga, K.; Zhu, W.; Kudoh, S.; et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 2004, 6, 499–506. [Google Scholar] [CrossRef]
- Susic, D.; Varagic, J.; Ahn, J.; Matavelli, L.; Frohlich, E.D. Long-term mineralocorticoid receptor blockade reduces fibrosis and improves cardiac performance and coronary hemodynamics in elderly SHR. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H175–H179. [Google Scholar] [CrossRef]
- Morfino, P.; Aimo, A.; Castiglione, V.; Galvez-Monton, C.; Emdin, M.; Bayes-Genis, A. Treatment of cardiac fibrosis: From neuro-hormonal inhibitors to CAR-T cell therapy. Heart Fail. Rev. 2022, 28, 555–569. [Google Scholar] [CrossRef]
- Zannad, F.; Alla, F.; Dousset, B.; Perez, A.; Pitt, B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: Insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation 2000, 102, 2700–2706. [Google Scholar] [CrossRef]
- Graham, H.K.; Trafford, A.W. Spatial disruption and enhanced degradation of collagen with the transition from compensated ventricular hypertrophy to symptomatic congestive heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1364–H1372. [Google Scholar] [CrossRef]
- Diez, J.; Querejeta, R.; Lopez, B.; Gonzalez, A.; Larman, M.; Martinez Ubago, J.L. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation 2002, 105, 2512–2517. [Google Scholar] [CrossRef]
- Chang, S.A.; Kim, Y.J.; Lee, H.W.; Kim, D.H.; Kim, H.K.; Chang, H.J.; Sohn, D.W.; Oh, B.H.; Park, Y.B. Effect of rosuvastatin on cardiac remodeling, function, and progression to heart failure in hypertensive heart with established left ventricular hypertrophy. Hypertension 2009, 54, 591–597. [Google Scholar] [CrossRef]
- Abulhul, E.; McDonald, K.; Martos, R.; Phelan, D.; Spiers, J.P.; Hennessy, M.; Baugh, J.; Watson, C.; O’Loughlin, C.; Ledwidge, M. Long-term statin therapy in patients with systolic heart failure and normal cholesterol: Effects on elevated serum markers of collagen turnover, inflammation, and B-type natriuretic peptide. Clin. Ther. 2012, 34, 91–100. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Wu, Y.W.; Lee, J.K.; Lin, Y.M.; Lin, Y.T.; Kao, H.L.; Hung, C.S.; Lin, H.J.; Lin, Y.H. Effects of 12 weeks of atorvastatin therapy on myocardial fibrosis and circulating fibrosis biomarkers in statin-naive patients with hypertension with atherosclerosis. J. Investig. Med. 2016, 64, 1194–1199. [Google Scholar] [CrossRef]
- Ashton, E.; Windebank, E.; Skiba, M.; Reid, C.; Schneider, H.; Rosenfeldt, F.; Tonkin, A.; Krum, H. Why did high-dose rosuvastatin not improve cardiac remodeling in chronic heart failure? Mechanistic insights from the UNIVERSE study. Int. J. Cardiol. 2011, 146, 404–407. [Google Scholar] [CrossRef]
- Lewis, G.A.; Dodd, S.; Clayton, D.; Bedson, E.; Eccleson, H.; Schelbert, E.B.; Naish, J.H.; Jimenez, B.D.; Williams, S.G.; Cunnington, C.; et al. Pirfenidone in heart failure with preserved ejection fraction: A randomized phase 2 trial. Nat. Med. 2021, 27, 1477–1482. [Google Scholar] [CrossRef]

| Author (s) | Year | Definition |
|---|---|---|
| The Criteria Committee of the New York Heart Association | 1979 | “An anatomofunctional alteration characterized by left ventricular hypertrophy (LVH) and cardiac failure in patients with systemic hypertension” [7,12] |
| Frohlich E. et al. | 1989, 1992 | “HHD is the cardiac response to the afterload imposed on the left ventricle by the progressive increase in arterial pressure and peripheral vascular resistance as a consequence of hypertensive vascular disease” [8,13] |
| Joseph L. Izzo Jr. et al. | 2004 | “HHD is a spectrum of abnormalities that represents the accumulation of a lifetime of functional and structural adaptations to increased blood pressure load. LVH, increasing vascular and ventricular stiffness, and diastolic dysfunction are prominent intermediate features of this syndrome that operate in parallel with ischemic heart disease and ultimately cause heart failure if inadequately treated” [14] |
| L. Michael Prisant | 2005 | “HHD is the target organ response of systemic arterial hypertension. It is more than left ventricular hypertrophy or heart failure, it also includes ischemic heart disease, aortic root disease, left atrial enlargement, and arrhythmias” [15]. |
| A. González et al. | 2005 | “HHD can be defined as the response of the heart to the stress imposed on the left ventricle by the progressively increasing arterial pressure and is characterized by complex changes in myocardial composition that are responsible for the structural remodeling of the myocardium” [16]. |
| E. Alegría-Ezquerra et al. | 2006 | “The term “hypertensive heart disease” to encompass the complex and variable set of effects that cause the chronic increase in blood pressure in the heart of a patient with hypertension and includes the presence of anatomic or biochemical signs of left ventricular hypertrophy or ventricular dysfunction, be either diastolic or systolic, of myocardial ischemia and rhythm abnormalities” [12]. |
| Alan H. Gradman et al. | 2006 | “Conceptually, HHD may be thought of as those disease manifestations that can be directly related to the cardiac anatomical changes, which accompany chronic elevation of systolic and diastolic blood pressure” [17]. |
| I. Gonzalez-Maqueda et al. | 2009 | “HHD is a complex and variable syndrome that usually, but not necessarily, includes clinical manifestations derived from: left ventricular hypertrophy and dysfunction, be it diastolic or systolic; myocardial ischemia; and rhythm abnormalities, all of them caused by the effects on the heart of chronically elevated blood pressure” [18]. |
| Mark H. Drazner | 2011 | “HHD is a constellation of abnormalities that includes left ventricular hypertrophy, systolic and diastolic dysfunction, and their clinical manifestations including arrhythmias and symptomatic heart failure” [19]. |
| Javier Díez | 2013 | “From a pathophysiological viewpoint, HHD can be defined as the cardiomyopathy that results from the response of the myocardium to the biomechanical stress imposed on the left ventricle by the progressively increasing blood pressure. Clinically, HHD is characterized by the presence of left ventricular hypertrophy in the absence of a cause other than arterial hypertension” [20]. |
| González A. et al. | 2018 | “HHD is defined by the presence of left ventricular hypertrophy or LV systolic and diastolic dysfunction and their clinical manifestations, such as arrhythmias and symptomatic heart failure, appearing in patients with hypertension” [21] |
| A.P. Kalogeropoulos et al. | 2019 | “Prolonged exposure of the heart to elevated blood pressure causes a variety of changes in the myocardial structure, coronary vasculature, and conduction system of the heart, collectively known as hypertensive heart disease” [22]. |
| Ferrer-Curriu, G. et al. | 2019 | “Functional or structural heart changes, leading to diastolic dysfunction, progressive left-ventricular hypertrophy, interstitial fibrosis, systolic dysfunction and chronic heart failure in subjects with defined hypertension develop a disease commonly termed hypertensive heart disease” [23]. |
| Chike C. Nwabuo, Ramachandran S. Vasan | 2020 | “HHD is characterized by micro- and macroscopic myocardial alterations, structural phenotypic adaptations, and functional changes that include cardiac fibrosis, and the remodeling of the atria and ventricles and the arterial system” [9] |
| Tackling, G.; Borhade, M.B. | 2022 | “HHD refers to a constellation of changes in the left ventricle, left atrium, and coronary arteries as a result of chronic blood pressure elevation, which increases the workload on the heart inducing structural and functional changes” [24]. |
| Lu, Y.; Lan, T. | 2022 | “HHD includes left ventricular hypertrophy, systolic and diastolic dysfunction, and a broader spectrum of cardiac and vascular adaptations” [25]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemtsova, V.; Vischer, A.S.; Burkard, T. Hypertensive Heart Disease: A Narrative Review Series—Part 1: Pathophysiology and Microstructural Changes. J. Clin. Med. 2023, 12, 2606. https://doi.org/10.3390/jcm12072606
Nemtsova V, Vischer AS, Burkard T. Hypertensive Heart Disease: A Narrative Review Series—Part 1: Pathophysiology and Microstructural Changes. Journal of Clinical Medicine. 2023; 12(7):2606. https://doi.org/10.3390/jcm12072606
Chicago/Turabian StyleNemtsova, Valeriya, Annina S. Vischer, and Thilo Burkard. 2023. "Hypertensive Heart Disease: A Narrative Review Series—Part 1: Pathophysiology and Microstructural Changes" Journal of Clinical Medicine 12, no. 7: 2606. https://doi.org/10.3390/jcm12072606
APA StyleNemtsova, V., Vischer, A. S., & Burkard, T. (2023). Hypertensive Heart Disease: A Narrative Review Series—Part 1: Pathophysiology and Microstructural Changes. Journal of Clinical Medicine, 12(7), 2606. https://doi.org/10.3390/jcm12072606