
CAR T-Cell Therapy in Children with Solid Tumors

 and
and 
Abstract
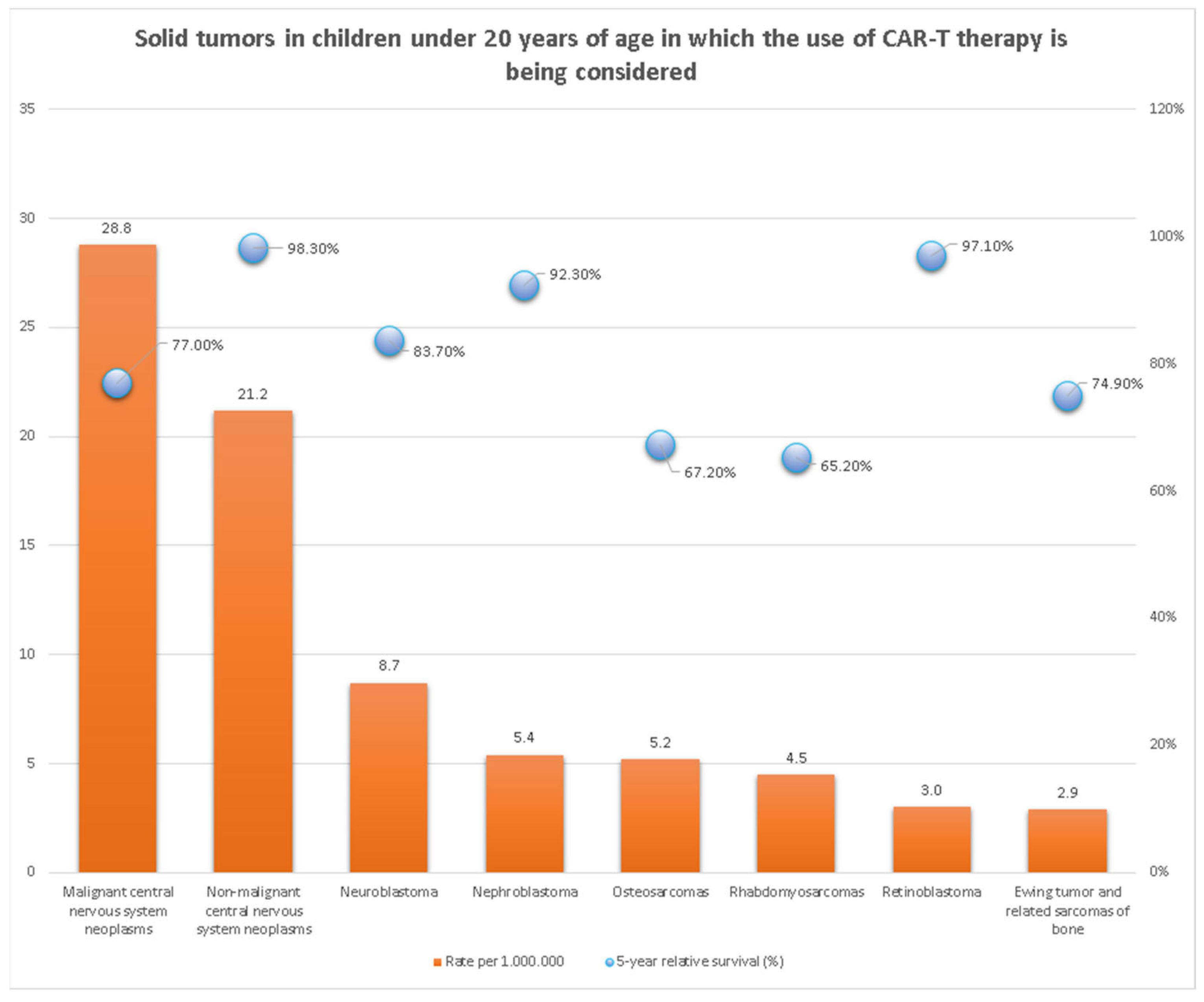
1. Introduction
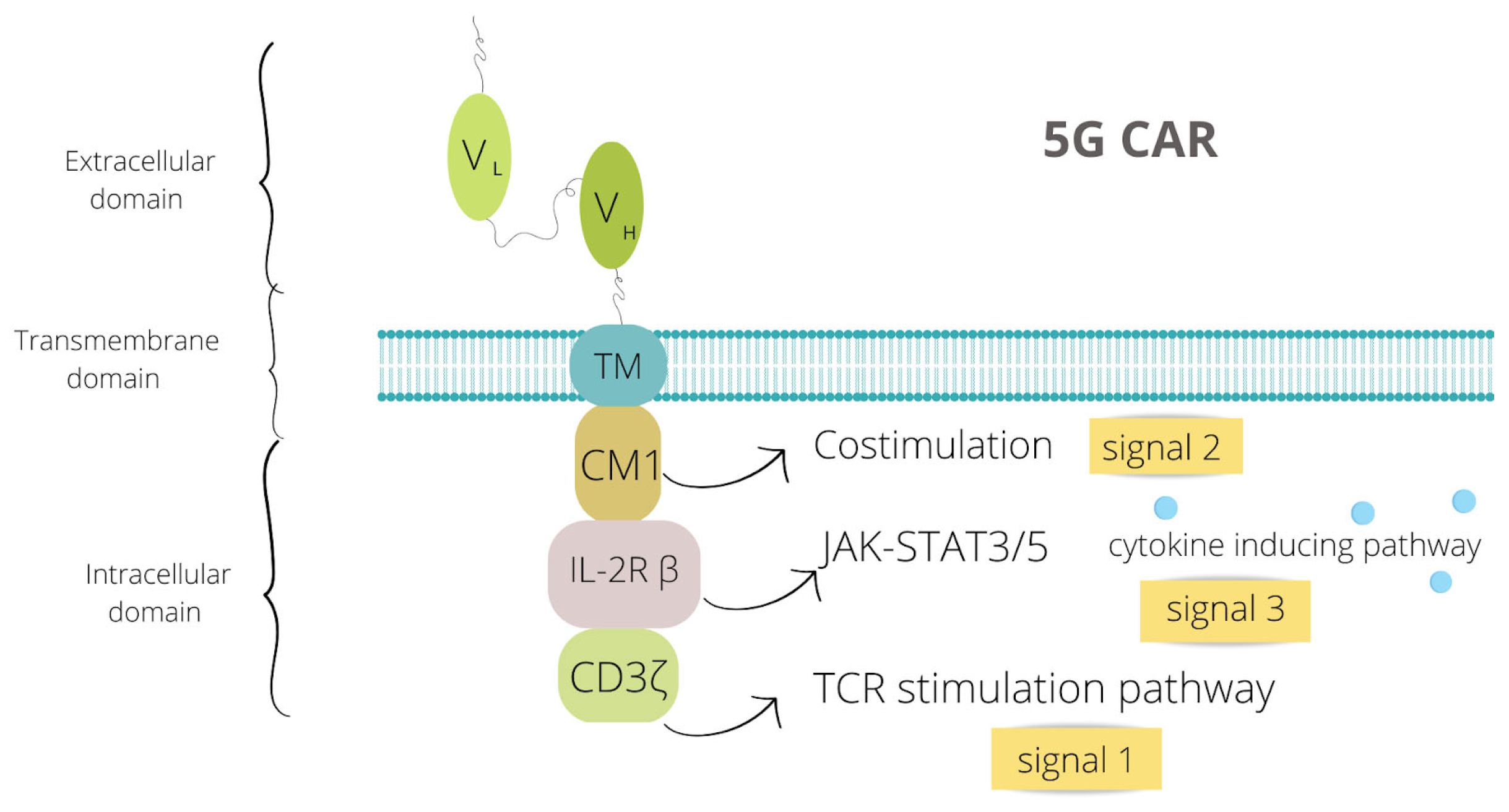
2. CAR Structure
3. Pediatric Solid Tumors—Preclinical and Clinical Reports
3.1. Brain Tumors
3.2. Neuroblastoma
3.2.1. Disialogangloside- GD2
3.2.2. CD171/L1-CAM—The L1-Cell Adhesion Molecule (CD171)
3.3. Wilms Tumor
3.4. Osteosarcoma
3.4.1. HER-2, GD2, B7-H3—Preclinical Researches and Clinical Trials
3.4.2. IL-11Ra, IGF1R, ROR1, and EphA2—Preclinical Researches
3.5. Rhabdomyosarcoma
3.6. Retinoblastoma
3.7. Ewing Sarcoma
3.7.1. VEGFR2, IGF1R, ROR1 and EphA2—Only Preclinical Research
3.7.2. GD2 and B7-H3—Clinical Trials Started
3.8. The Summary of Clinical Studies on Solid Tumors in Children
4. Limitations of Car-T Therapy
4.1. Antigen Escape
4.2. Immunosuppressive Tumor Microenvironment (TME)
4.3. Restricted Trafficking and Limited Tumor Infiltration
4.4. On-Target Off-Tumor Effects (OTOT)
4.5. Toxicity of the CAR-T Cell Therapy

{kind=link}
{kind=link}
| Limitations to Overcome | Main Potential Solution | References |
|---|---|---|
| Antigen escape | Multiple target therapy | [102,104,105] |
| Immunosuppressive TME | Usage of immune checkpoint inhibitors and oncolytic viruses | [108,109,110,111,112] |
| Restricted trafficking and limited infiltration | Local delivery of CAR-T | [14,22,25] |
| On-target off-tumor effects | Modification of the scFv and other domains | [117] |
| Toxicity of the therapy | Administration of corticosteroids and cytokine inhibitors | [120] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Allen-Rhoades, W.; Whittle, S.B.; Rainusso, N. Pediatric Solid Tumors of Infancy: An Overview. Pediatr. Rev. 2018, 39, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 12, 3855–3864. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Yang, J.F.; Deng, B.P.; Zhao, X.J.; Zhang, X.; Lin, Y.H.; Wu, Y.N.; Deng, Z.L.; Zhang, Y.L.; Liu, S.H.; et al. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia 2017, 31, 2587–2593. [Google Scholar] [CrossRef]
- National Cancer Institute. Available online: https://nccrexplorer.ccdi.cancer.gov/ (accessed on 19 February 2023).
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw Dejenie, T.; Tiruneh G/Medhin, M.; Dessie Terefe, G.; Tadele Admasu, F.; Wale Tesega, W.; Chekol Abebe, E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum. Vaccines Immunother. 2022, 18, 2114254. [Google Scholar] [CrossRef]
- Wells, E.M.; Packer, R.J. Pediatric brain tumors. Continuum 2015, 21, 373–396. [Google Scholar] [CrossRef]
- Antonucci, L.; Canciani, G.; Mastronuzzi, A.; Carai, A.; Del Baldo, G.; Del Bufalo, F. CAR-T Therapy for Pediatric High-Grade Gliomas: Peculiarities, Current Investigations and Future Strategies. Front. Immunol. 2022, 13, 867154. [Google Scholar] [CrossRef] [PubMed]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Odé, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro-Oncology 2021, 23, 999–1011. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. J. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Rousso-Noori, L.; Mastandrea, I.; Talmor, S.; Waks, T.; Globerson Levin, A.; Haugas, M.; Teesalu, T.; Alvarez-Vallina, L.; Eshhar, Z.; Friedmann-Morvinski, D. P32-specific CAR T cells with dual antitumor and antiangiogenic therapeutic potential in gliomas. Nat. Commun. 2021, 12, 3615. [Google Scholar] [CrossRef]
- Foster, J.B.; Griffin, C.; Rokita, J.L.; Stern, A.; Brimley, C.; Rathi, K.; Lane, M.V.; Buongervino, S.N.; Smith, T.; Madsen, P.J.; et al. Development of GPC2-directed chimeric antigen receptors using mRNA for pediatric brain tumors. J. Immunother. Cancer. 2022, 10, e004450. [Google Scholar] [CrossRef] [PubMed]
- de Billy, E.; Pellegrino, M.; Orlando, D.; Pericoli, G.; Ferretti, R.; Businaro, P.; Ajmone-Cat, M.A.; Rossi, S.; Petrilli, L.L.; Maestro, N.; et al. Dual IGF1R/IR inhibitors in combination with GD2-CAR T-cells display a potent anti-tumor activity in diffuse midline glioma H3K27M-mutant. Neuro-Oncology 2022, 24, 1150–1163. [Google Scholar] [CrossRef]
- Li, G.; Wang, H.; Wu, H.; Chen, J. B7-H3-targeted CAR-T cell therapy for solid tumors. Int. Rev. Immunol. 2022, 41, 625–637. [Google Scholar] [CrossRef]
- Kontos, F.; Michelakos, T.; Kurokawa, T.; Sadagopan, A.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. B7-H3: An Attractive Target for Antibody-based Immunotherapy. Clin. Cancer Res. 2021, 27, 1227–1235. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. J. Nat. Med. 2002, 26, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Wilson, A.L.; Huang, W.; Seidel, K.; Brown, C.; Gustafson, J.A.; Yokoyama, J.K.; Johnson, A.J.; Baxter, B.A.; Koning, R.W.; et al. Intraventricular B7-H3 CAR T Cells for Diffuse Intrinsic Pontine Glioma: Preliminary First-in-Human Bioactivity and Safety. Cancer Discov. 2023, 13, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Li, D.P.; Zhang, N.; Zhang, J.Y.; Zhang, D.; Liu, T.T.; Yang, J.; Ge, M. Spinal Cord Diffuse Midline Glioma with Histone H3 K27M Mutation in a Pediatric Patient. Front. Surg. 2021, 8, 616334. [Google Scholar] [CrossRef] [PubMed]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. J. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Künkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. J. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Mulcahy Levy, J.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT03638167 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT05298995 (accessed on 19 February 2023).
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef]
- Rossig, C.; Bollard, C.M.; Nuchtern, J.G.; Merchant, D.A.; Brenner, M.K. Targeting of GD2-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int. J. Cancer 2021, 94, 228–236. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. J. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J. Cancer Res. Clin. Oncol. 2022, 148, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT01460901 (accessed on 19 February 2023).
- Tumino, N.; Weber, G.; Besi, F.; Del Bufalo, F.; Bertaina, V.; Paci, P.; Quatrini, L.; Antonucci, L.; Sinibaldi, M.; Quintarelli, C.; et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J. Hematol. Oncol. 2021, 14, 191. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tashiro, H.; Omer, B.; Lapteva, N.; Ando, J.; Ngo, M.; Mehta, B.; Dotti, G.; Kinchington, P.R.; Leen, A.M.; et al. Vaccination Targeting Native Receptors to Enhance the Function and Proliferation of Chimeric Antigen Receptor (CAR)-Modified T Cells. Clin. Cancer Res. 2017, 23, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04539366 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03721068 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02107963 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03635632 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02919046 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04637503 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03294954 (accessed on 19 February 2023).
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT02311621 (accessed on 19 February 2023).
- Stokes, C.L.; Stokes, W.A.; Kalapurakal, J.A.; Paulino, A.C.; Cost, N.G.; Cost, C.R.; Garrington, T.P.; Greffe, B.S.; Roach, J.P.; Bruny, J.L.; et al. Timing of Radiation Therapy in Pediatric Wilms Tumor: A Report from the National Cancer Database. Int. J. Radiat. Oncol. Biol. Psys. 2018, 101, 453–461. [Google Scholar] [CrossRef]
- Capurro, M.I.; Xiang, Y.Y.; Lobe, C.; Filmus, J. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 2005, 65, 6245–6254. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Zitomersky, N.L.; Eskenazi, A.E.; Voigt, R.W.; Strauch, E.D.; Sun, C.C.; Huber, R.; Meltzer, S.J.; Schlessinger, D. Glypican-3 expression in Wilms tumor and hepatoblastoma. J. Pediatr. Hematol. Oncol. 2001, 23, 496–499. [Google Scholar] [CrossRef]
- Saikali, Z.; Sinnett, D. Expression of glypican 3 (GPC3) in embryonal tumors. Int. J. Cancer 2000, 89, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04377932 (accessed on 19 February 2023).
- Ravanpay, A.C.; Gust, J.; Johnson, A.J.; Rolczynski, L.S.; Cecchini, M.; Chang, C.A.; Hoglund, V.J.; Mukherjee, R.; Vitanza, N.A.; Orentas, R.J.; et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget 2019, 10, 7080–7095. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT03618381 (accessed on 19 February 2023).
- Steinberger, P.; Majdic, O.; Derdak, S.V.; Pfistershammer, K.; Kirchberger, S.; Klauser, C.; Zlabinger, G.; Pickl, W.F.; Stöckl, J.; Knapp, W. Molecular characterization of human 4Ig-B7-H3, a member of the B7 family with four Ig-like domains. J. Immunol. 2004, 172, 2352–2359. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04483778 (accessed on 19 February 2023).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Cheung, N.V. GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J. Hematol. Oncol. 2020, 13, 172. [Google Scholar] [CrossRef]
- Huang, G.; Yu, L.; Cooper, L.J.; Hollomon, M.; Huls, H.; Kleinerman, E.S. Genetically modified T cells targeting interleukin-11 receptor α-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012, 72, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Liu, B.; Liu, K.; Zhu, G.; Dai, Z.; Xie, Y. Overexpression of fibroblast activation protein and its clinical implications in patients with osteosarcoma. J. Surg. Oncol. 2013, 108, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.; Middlemiss, S.; Saletta, F.; Gottschalk, S.; McCowage, G.B.; Kramer, B. Chimeric Antigen Receptor-modified T cells targeting EphA2 for the immunotherapy of paediatric bone tumours. Cancer Gene Ther. 2021, 28, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Manara, M.C.; Hattinger, C.M.; Benini, S.; Perdichizzi, S.; Pasello, M.; Bacci, G.; Zanella, L.; Bertoni, F.; Picci, P.; et al. Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing’s sarcoma. Eur. J. Cancer 2005, 41, 1349–1361. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef]
- Rainusso, N.; Brawley, V.S.; Ghazi, A.; Hicks, M.J.; Gottschalk, S.; Rosen, J.M.; Ahmed, N. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 2012, 19, 212–217. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Linkowski, M.; Tarim, J.; Piperdi, S.; Sowers, R.; Geller, D.; Gill, J.; Gorlick, R. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer 2014, 120, 548–554. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef]
- Chulanetra, M.; Morchang, A.; Sayour, E.; Eldjerou, L.; Milner, R.; Lagmay, J.; Cascio, M.; Stover, B.; Slayton, W.; Chaicumpa, W.; et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 2020, 10, 674–687. [Google Scholar] [PubMed]
- Murty, S.; Labanieh, L.; Murty, T.; Gowrishankar, G.; Haywood, T.; Alam, I.S.; Beinat, C.; Robinson, E.; Aalipour, A.; Klysz, D.D.; et al. PET Reporter Gene Imaging and Ganciclovir-Mediated Ablation of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2020, 80, 4731–4740. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04897321 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04864821 (accessed on 19 February 2023).
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.clinicaltrials.gov/ct2/show/NCT05312411 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04433221 (accessed on 19 February 2023).
- Linch, M.; Miah, A.B.; Thway, K.; Judson, I.R.; Benson, C. Systemic treatment of soft-tissue sarcoma-gold standard and novel therapies. Nat. Rev. Clin. Oncol. 2014, 11, 187–202. [Google Scholar] [CrossRef]
- Shah, M.H.; Lorigan, P.; O’Brien, M.E.; Fossella, F.V.; Moore, K.N.; Bhatia, S.; Kirby, M.; Woll, P.J. Phase I study of IMGN901, a CD56-targeting antibody-drug conjugate, in patients with CD56-positive solid tumors. Investig. New Drugs 2016, 34, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Howley, S.; Stack, D.; Morris, T.; McDermott, M.; Capra, M.; Betts, D.; O’Sullivan, M.J. Ectomesenchymoma with t(1;12)(p32;p13) evolving from embryonal rhabdomyosarcoma shows no rearrangement of ETV6. Hum. Pathol. 2012, 43, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, F.; Collini, P.; Aiello, A.; Barzanò, E.; Gambirasio, F.; Podda, M.; Meazza, C.; Ferrari, A.; Luksch, R. Flow cytometric phenotype of rhabdomyosarcoma bone marrow metastatic cells and its implication in differential diagnosis with neuroblastoma. Anticancer Res. 2008, 28, 1565–1569. [Google Scholar] [PubMed]
- Jiang, C.; Zhao, W.; Qin, M.; Jin, M.; Chang, L.; Ma, X. CD56-chimeric antigen receptor T-cell therapy for refractory/recurrent rhabdomyosarcoma: A 3.5-year follow-up case report. Medicine 2019, 98, e17572. [Google Scholar] [CrossRef]
- Hegde, M.; Joseph, S.K.; Pashankar, F.; DeRenzo, C.; Sanber, K.; Navai, S.; Byrd, T.T.; Hicks, J.; Xu, M.L.; Gerken, C. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020, 11, 3549. [Google Scholar] [CrossRef] [PubMed]
- Manrique, M.; Akinbolue, D.; Madigan, W.P.; Bregman, J. Update on the Treatment of Retinoblastoma. NeoReviews 2021, 22, e423–e437. [Google Scholar] [CrossRef] [PubMed]
- Andersch, L.; Radke, J.; Klaus, A.; Schwiebert, S.; Winkler, A.; Schumann, E.; Grunewald, L.; Zirngibl, F.; Flemmig, C.; Jensen, M.C.; et al. CD171- and GD2-specific CAR-T cells potently target retinoblastoma cells in preclinical in vitro testing. BMC Cancer 2019, 19, 895. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, Y.; Ahn, S.; Zheng, M.; Landoni, E.; Dotti, G.; Savoldo, B.; Han, Z. GD2-specific CAR T cells encapsulated in an injectable hydrogel control retinoblastoma and preserve vision. Nat. Cancer 2020, 1, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Sujjitjoon, J.; Sayour, E.; Tsao, S.T.; Uiprasertkul, M.; Sanpakit, K.; Buaboonnam, J.; Yenchitsomanus, P.T.; Atchaneeyasakul, L.O.; Chang, L.J. GD2-specific chimeric antigen receptor-modified T cells targeting retinoblastoma—Assessing tumor and T cell interaction. Transl. Oncol. 2021, 14, 100971. [Google Scholar] [CrossRef]
- Lin, Z.; Wu, Z.; Luo, W. A Novel Treatment for Ewing’s Sarcoma: Chimeric Antigen Receptor-T Cell Therapy. Front. Immunol. 2021, 12, 707211. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Zhou, Z.; Bolontrade, M.F.; Reddy, K.; Duan, X.; Guan, H.; Yu, L.; Hicklin, D.J.; Kleinerman, E.S. Suppression of Ewing’s sarcoma tumor growth, tumor vessel formation, and vasculogenesis following anti vascular endothelial growth factor receptor-2 therapy. Clin. Cancer Res. 2007, 13, 4867–4873. [Google Scholar] [CrossRef]
- Kreuter, M.; Paulussen, M.; Boeckeler, J.; Gerss, J.; Buerger, H.; Liebscher, C.; Kessler, T.; Jurgens, H.; Berdel, W.E.; Mesters, R.M. Clinical significance of Vascular Endothelial Growth Factor—A expression in Ewing’s sarcoma. Eur. J. Cancer 2006, 42, 1904–1911. [Google Scholar] [CrossRef]
- Englisch, A.; Altvater, B.; Kailayangiri, S.; Hartmann, W.; Rossig, C. VEGFR2 as a target for CAR T cell therapy of Ewing sarcoma. Pediatr. Blood Cancer 2020, 67, e28313. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Bui, M.; Ahmed, A.A. IGF1R immunohistochemistry in Ewing’s sarcoma as predictor of response to targeted therapy. Int. J. Health Sci. 2020, 14, 17–21. [Google Scholar]
- Golinelli, G.; Grisendi, G.; Dall’Ora, M.; Casari, G.; Spano, C.; Talami, R.; Banchelli, F.; Prapa, M.; Chiavelli, C.; Rossignoli, F.; et al. Anti-GD2 CAR MSCs against metastatic Ewing’s sarcoma. Transl. Oncol. 2022, 15, 101240. [Google Scholar] [CrossRef]
- Available online: https://www.clinicaltrials.gov/ct2/show/NCT03356782 (accessed on 19 February 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04099797?cond=NCT04099797&draw=2&rank=1 (accessed on 10 March 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04196413 (accessed on 10 March 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02442297 (accessed on 10 March 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03500991 (accessed on 10 March 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04185038 (accessed on 10 March 2023).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03373097 (accessed on 19 February 2023).
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2021, 131, e152477. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT02208362 (accessed on 19 February 2023).
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 2018, 20, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Gu, J.; Xu, H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 2018, 17, 7. [Google Scholar] [CrossRef]
- Cocco, C.; Morandi, F.; Airoldi, I. Immune Checkpoints in Pediatric Solid Tumors: Targetable Pathways for Advanced Therapeutic Purposes. Cells 2021, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.L.; Meyran, D.; Fleuren, E.D.G.; Mayoh, C.; Zhu, J.; Omer, N.; Ziegler, D.S.; Haber, M.; Darcy, P.K.; Trapani, J.A.; et al. Chimeric Antigen Receptor T cell Therapy and the Immunosuppressive Tumor Microenvironment in Pediatric Sarcoma. Cancers 2021, 13, 4704. [Google Scholar] [CrossRef]
- Park, J.A.; Cheung, N.V. Limitations and opportunities for immune checkpoint inhibitors in pediatric malignancies. Cancer Treat. Rev. 2017, 58, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Author Correction: Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 536. [Google Scholar] [CrossRef]
- Chastkofsky, M.I.; Pituch, K.C.; Katagi, H.; Zannikou, M.; Ilut, L.; Xiao, T.; Han, Y.; Sonabend, A.M.; Curiel, D.T.; Bonner, E.R.; et al. Mesenchymal Stem Cells Successfully Deliver Oncolytic Virotherapy to Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2021, 27, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Olsen, H.E.; Lynn, G.M.; Valdes, P.A.; Cerecedo Lopez, C.D.; Ishizuka, A.S.; Arnaout, O.; Bi, W.L.; Peruzzi, P.P.; Chiocca, E.A.; Friedman, G.K.; et al. Therapeutic cancer vaccines for pediatric malignancies: Advances, challenges, and emerging technologies. Neuro-Oncol. Adv. 2021, 3, vdab027. [Google Scholar] [CrossRef]
- Njonkou, R.; Jackson, C.M.; Woodworth, G.F.; Hersh, D.S. Pediatric glioblastoma: Mechanisms of immune evasion and potential therapeutic opportunities. Cancer Immunol. Immunother. 2022, 71, 1813–1822. [Google Scholar] [CrossRef]
- Michaelides, S.; Obeck, H.; Kechur, D.; Endres, S.; Kobold, S. Migratory Engineering of T Cells for Cancer Therapy. Vaccines 2022, 10, 1845. [Google Scholar] [CrossRef]
- Steentoft, C.; Migliorini, D.; King, T.R.; Mandel, U.; June, C.H.; Posey, A.D. Glycan-directed CAR-T cells. Glycobiology 2018, 28, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Chen, R.; Huang, Y.; Meng, X.; Chen, J.; Liao, C.; Tang, Y.; Zhou, C.; Gao, X.; Sun, J. Tuning the ignition of CAR: Optimizing the affinity of scFv to improve CAR-T therapy. Cell. Mol. Life Sci. 2021, 79, 14. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Y.; Huang, Z.; Wang, X.; Jin, Z.; Li, J.; Limsakul, P.; Zhu, L.; Allen, M.; Pan, Y.; et al. Control of the activity of CAR-T cells within tumours via focused ultrasound. Nat. Biomed. Eng. 2021, 5, 1336–1347. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.W.; Bhattarai, N. CAR-T Cell Therapy: Mechanism, Management, and Mitigation of Inflammatory Toxicities. Front. Immunol. 2021, 12, 693016. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Teachey, D.T.; Pequignot, E.; Frey, N.; Porter, D.; Maude, S.L.; Grupp, S.A.; June, C.H.; Melenhorst, J.J.; Lacey, S.F. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J. Immunol. 2016, 434, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]


| Tumor | Number of Patients Treated with CAR-T | Treatment Effects | References |
|---|---|---|---|
| Diffuse Intrinsic Pontine Glioma | 3 | 6 months after treatment: 2 pts- increased tumor bulk with infiltration in the right brachium pontis and dentate nuclei 1 pt- mild decrease in tumor size | [22] |
| H3K27M-mutated diffuse midline gliomas | 4 | 3 pts- initially: exhibited clinical and radiographic improvement, 2 pts died in 7 and 10 months after first cell infusion, 1 pt survived after the data cutt-off 1 pt- death in 3 months after first cell infusion | [25] |
| Neuroblastoma | 11 | 6 weeks after treatment: 4 pts- no evidence of disease 2 pts- stable disease 2 pts- tumor necrosis 2 pts- progressive disease 1 pt- partial response | [32] |
| Neuroblastoma | 19 | 6 weeks after treatment: 8 pts- no evidence of disease 4 pts- progressive disease 2 pts- complete response 2 pts- tumor necrosis 2 pts- stable disease 1 pt- partial response | [33] |
| Neuroblastoma | 11 | 6 weeks after treatment: 6 pts- progressive disease 5 pts- stable disease | [34] |
| Neuroblastoma | 12 | 6 pts- no in vivo expansion of CAR T-cells was detected, no immune activation or antitumor activity was seen 3 pts- disease progression 2 pts- mixed response 1 pt- near complete clearance bone marrow infiltration | [35] |
| Neuroblastoma | 10 | 6 months after treatment: 6 pts- stable disease 4 pts- progressive disease | [36] |
| Neuroblastoma | 3 | All died of the disease within a year | [37] |
| Neuroblastoma | 6 | 56 days after treatment: 5 pts- progressive disease 1 pt- partial response | [47] |
| Osteosarcoma (16 pts) Ewing sarcoma (1 pt) Primitive neuroectodermal tumor (1 pt) Desmoplastic small round cell tumor (1 pt) | 19 | 6 weeks after treatment: 13 pts- progressive disease 4 pts- stable disease 2 pts- not evaluable | [66] |
| Rhabdomyosarcoma | 1 | Complete remission | [80] |
| Rhabdomyosarcoma | 1 | In remission for 20 month at the time of the report | [81] |
| Solid Tumor | Receptor | Phase of Research | Responsible Party | References |
|---|---|---|---|---|
| Brain Tumors | GD2 | Phase 1 | Franco Locatelli, Bambino Gesù Hospital and Research Institute | [29] |
| Phase 1 | Bilal Omer, Baylor College of Medicine | [94] | ||
| Phase 1 | Crystal Mackall, MD, Stanford University | [95] | ||
| HER2 | Phase 1 | Nabil Ahmed, Baylor College of Medicine | [96] | |
| Phase 1 | Rebecca Gardner, Seattle Children’s Hospital | [97] | ||
| B7H3 | Phase 1 | Rebecca Gardner, Seattle Children’s Hospital | [98] | |
| Neuroblastoma | GD2, PSMA and CD276 | Phase 2 | Shenzhen Geno-Immune Medical Institute | [45] |
| Osteosarcoma | CD276 | Early Phase 1 | PersonGen BioTherapeutics (Suzhou) Co., Ltd. | [72] |
| FITC-E2 | Phase 1 | Rebecca Gardner, Seattle Children’s Hospital | [74] | |
| Neuroblastoma Osteosarcoma | GD2 | Phase 1 | National Cancer Institute (NCI) | [40] |
| Phase 1 | UNC Lineberger Comprehensive Cancer Center | [41] | ||
| Neuroblastoma Osteosarcoma Ewing Sarcoma Rhabdomyosarcoma | GD2 | Phase 1 | Bilal Omer, Baylor College of Medicine | [43] |
| Neuroblastoma Osteosarcoma Ewing Sarcoma | Phase 2 | Franco Locatelli, Bambino Gesù Hospital and Research Institute | [99] | |
| Neuroblastoma Osteosarcoma Rhabdomyosarcoma Ewing Sarcoma Wilms Tumor | B7H3 | Phase 1 | Rebecca Gardner, Seattle Children’s Hospital | [57] |
| Phase 1 | St. Jude Children’s Research Hospital | [71] | ||
| EGFR806 | Phase 1 | Rebecca Gardner, Seattle Children’s Hospital | [55] | |
| Rhabdomyosarcoma Wilms Tumor | GPC3 | Phase 1 | Andras Heczey, Baylor College of Medicine | [53] |
| Osteosarcoma Ewing Sarcoma | GD2, PSMA, Her2, CD276 and other markers | Phase 2 | Shenzhen Geno-Immune Medical Institute | [75] |
| CD133, GD2, Muc1, CD117 and other markers | Phase 2 | Lung-Ji Chang, Shenzhen Geno-Immune Medical Institute | [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulczycka, M.; Derlatka, K.; Tasior, J.; Lejman, M.; Zawitkowska, J. CAR T-Cell Therapy in Children with Solid Tumors. J. Clin. Med. 2023, 12, 2326. https://doi.org/10.3390/jcm12062326
Kulczycka M, Derlatka K, Tasior J, Lejman M, Zawitkowska J. CAR T-Cell Therapy in Children with Solid Tumors. Journal of Clinical Medicine. 2023; 12(6):2326. https://doi.org/10.3390/jcm12062326
Chicago/Turabian StyleKulczycka, Marika, Kamila Derlatka, Justyna Tasior, Monika Lejman, and Joanna Zawitkowska. 2023. "CAR T-Cell Therapy in Children with Solid Tumors" Journal of Clinical Medicine 12, no. 6: 2326. https://doi.org/10.3390/jcm12062326
APA StyleKulczycka, M., Derlatka, K., Tasior, J., Lejman, M., & Zawitkowska, J. (2023). CAR T-Cell Therapy in Children with Solid Tumors. Journal of Clinical Medicine, 12(6), 2326. https://doi.org/10.3390/jcm12062326