
Preimplantation Genetic Testing (PGT) and Prenatal Diagnosis of Schaaf-Yang Syndrome: A Report of Three Families and a Research on Genotype–Phenotype Correlations
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. DNA Extraction and Variant Detection
2.3. In Vitro Fertilization (IVF)
2.4. Identity Testing and Haplotype Analysis
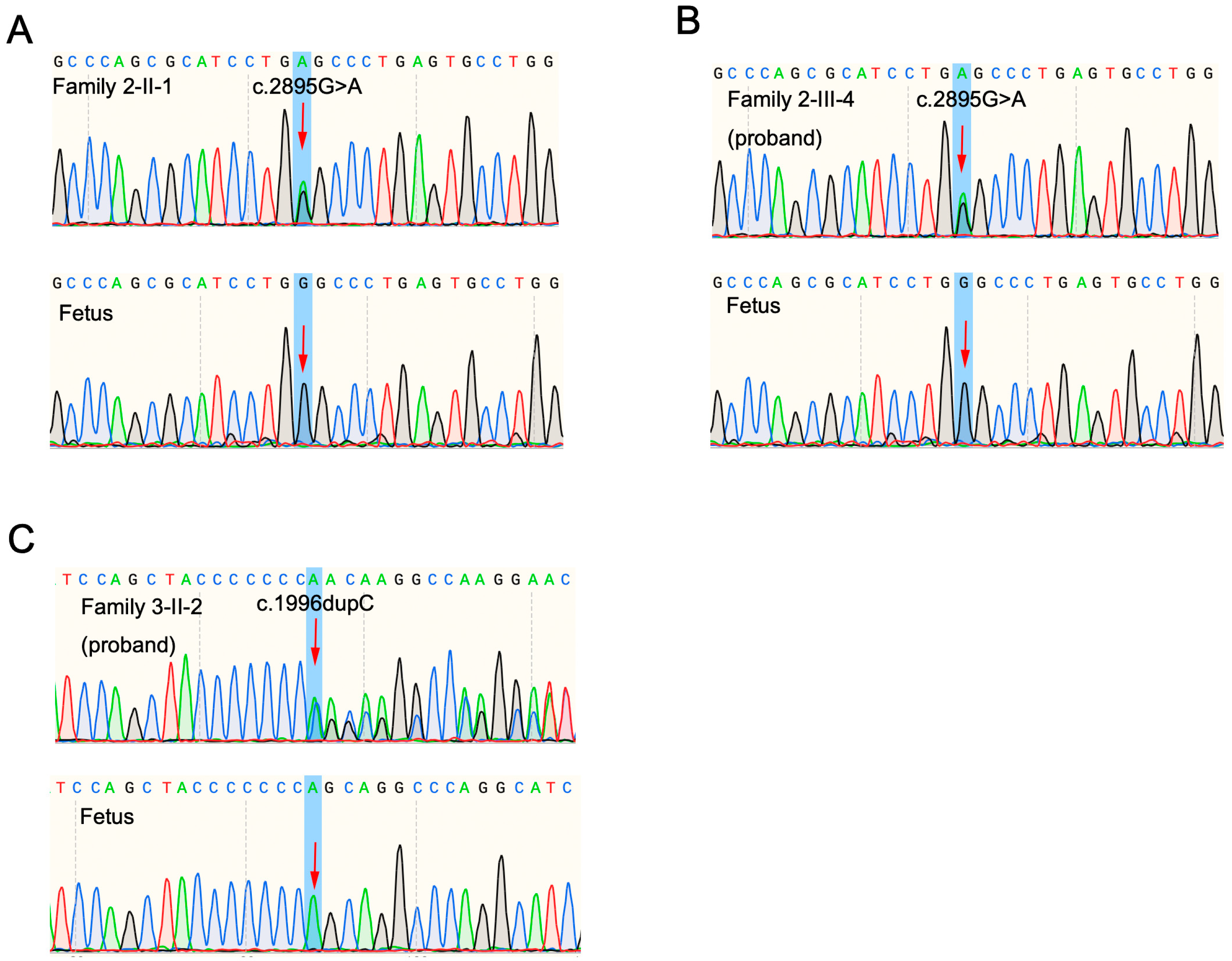
2.5. Prenatal Diagnosis
2.6. Literature Review and Genotype–Clinical Phenotype Correlation Analysis
2.7. Statistical Analysis
3. Results
3.1. History of Three Families
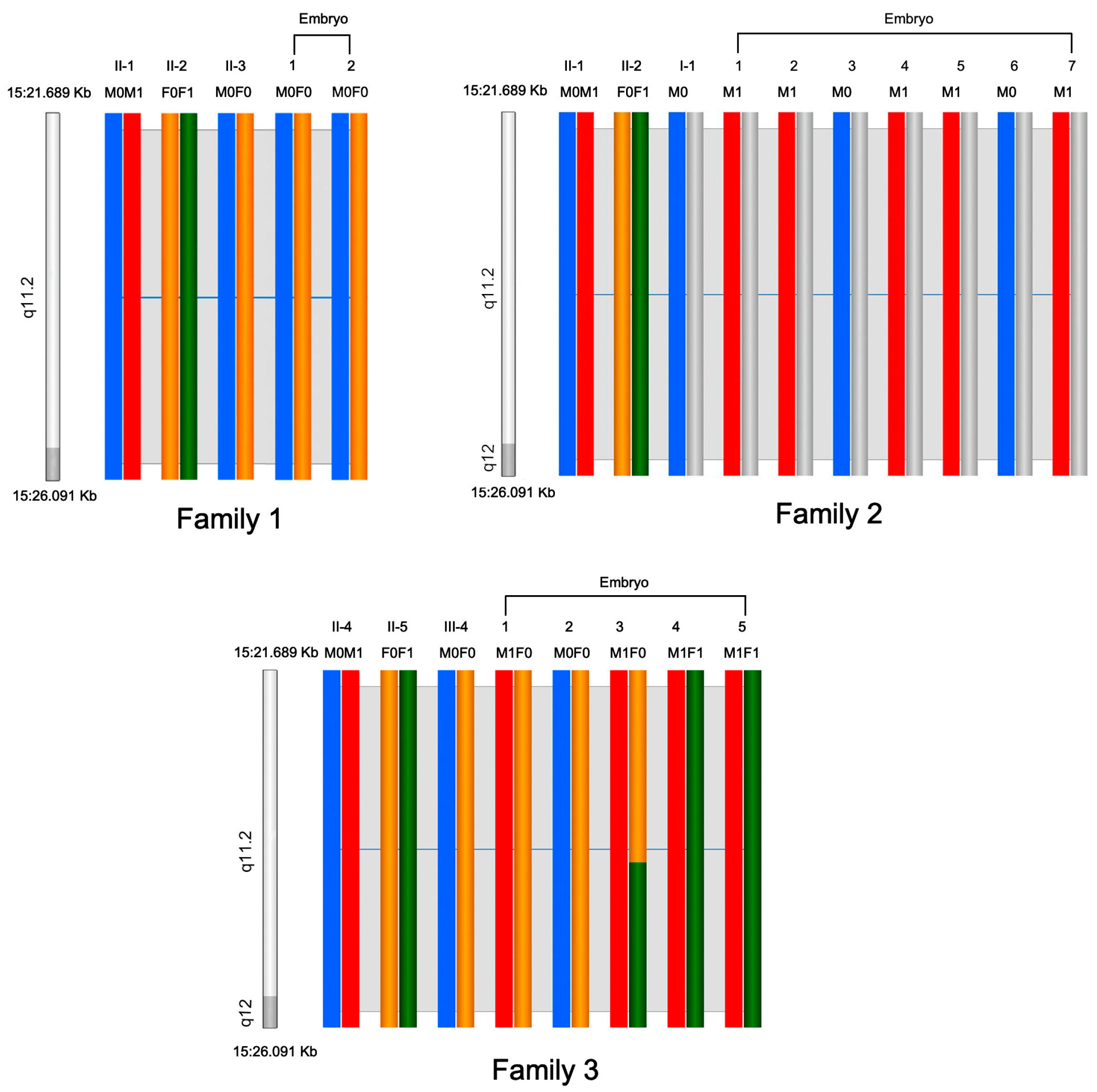
3.1.1. Family 1
3.1.2. Family 2
3.1.3. Family 3
3.2. Preimplantation Genetic Testing and Prenatal Diagnosis
3.3. Literature Review
3.4. MAGEL2 Gene Variant Spectrum
3.5. Clinical Phenotype Profile
3.6. MAGEL2 Genotype–Clinical Phenotype Correlation Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age | Variant Location | Respiratory Distress | Hypotonia | Hypogonadism | Other Clinical Phenotypes |
|---|---|---|---|---|---|---|---|
| Family 1-III-1 | M | 7 d | c.1996dupC | Neonatal anoxia | + | + | Camptodactyly |
| Family 1-III-2 | M | 15 d | c.1996dupC | Neonatal anoxia | + | + | Camptodactyly, patent foramen ovale |
| Family 1-III-3 | F | 110 d | c.1996dupC | Recurrent apnea, respiratory failure | + | − | Camptodactyly, simian line |
| Family 1-III-4 | M | 17 d | c.1996dupC | Neonatal anoxia | + | + | Camptodactyly, simian line, small mandible, high palate arch, small tongue, low ear position |
| Family 1-III-5 | F | 9 d | c.1996dupC | Neonatal anoxia | − | − | Camptodactyly, high palate arch, low ear position, bilateral polydactyly |
| Family 2-III-4 | M | 11 y | c.2895G>A | Neonatal anoxia | − | + | Feeding difficulties, intellectual disability, obesity, bulimia |
| Family 2-III-1/2/3/5 | M | 1–14 d | c.2895G>A | Neonatal anoxia, atelectasis | + | + | - |
| Family 3-III-4 | F | 32 d | c.1996dupC | Neonatal pneumonia | − | − | Neonatal sepsis, neonatal encephalopathy, intracranial hemorrhage, camptodactyly, thyroid-stimulating hormone, atrial septal defect, and patent foramen ovale |


References
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Gao, L.; Wang, X.; Cai, Y.; Shu, Y.; Zou, X.; Qin, Y.; Hu, C.; Dai, Y.; Zhu, M.; et al. Dysregulated adipose tissue expansion and impaired adipogenesis in Prader-Willi syndrome children before obesity-onset. Metabolism 2022, 136, 155295. [Google Scholar] [CrossRef]
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408. [Google Scholar] [CrossRef]
- Fountain, M.D.; Aten, E.; Cho, M.T.; Juusola, J.; Walkiewicz, M.A.; Ray, J.W.; Xia, F.; Yang, Y.; Graham, B.H.; Bacino, C.A.; et al. The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet. Med. 2017, 19, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Mejlachowicz, D.; Nolent, F.; Maluenda, J.; Ranjatoelina-Randrianaivo, H.; Giuliano, F.; Gut, I.; Sternberg, D.; Laquerrière, A.; Melki, J. Truncating Mutations of MAGEL2, a Gene within the Prader-Willi Locus, Are Responsible for Severe Arthrogryposis. Am. J. Hum. Genet. 2015, 97, 616–620. [Google Scholar] [CrossRef]
- Negishi, Y.; Ieda, D.; Hori, I.; Nozaki, Y.; Yamagata, T.; Komaki, H.; Tohyama, J.; Nagasaki, K.; Tada, H.; Saitoh, S. Schaaf-Yang syndrome shows a Prader-Willi syndrome-like phenotype during infancy. Orphanet J. Rare Dis. 2019, 14, 277. [Google Scholar] [CrossRef] [PubMed]
- Jobling, R.; Stavropoulos, D.J.; Marshall, C.R.; Cytrynbaum, C.; Axford, M.M.; Londero, V.; Moalem, S.; Orr, J.; Rossignol, F.; Lopes, F.D.; et al. Chitayat-Hall and Schaaf-Yang syndromes: A common aetiology: Expanding the phenotype of MAGEL2-related disorders. J. Med. Genet. 2018, 55, 316–321. [Google Scholar] [CrossRef]
- Patak, J.; Gilfert, J.; Byler, M.; Neerukonda, V.; Thiffault, I.; Cross, L.; Amudhavalli, S.; Pacio-Miguez, M.; Palomares-Bralo, M.; Garcia-Minaur, S.; et al. MAGEL2-related disorders: A study and case series. Clin. Genet. 2019, 96, 493–505. [Google Scholar] [CrossRef]
- Chen, X.; Ma, X.; Zou, C. Phenotypic spectrum and genetic analysis in the fatal cases of Schaaf-Yang syndrome: Two case reports and literature review. Medicine 2020, 99, e20574. [Google Scholar] [CrossRef]
- Tong, W.; Wang, Y.; Lu, Y.; Ye, T.; Song, C.; Xu, Y.; Li, M.; Ding, J.; Duan, Y.; Zhang, L.; et al. Whole-exome Sequencing Helps the Diagnosis and Treatment in Children with Neurodevelopmental Delay Accompanied Unexplained Dyspnea. Sci. Rep. 2018, 8, 5214. [Google Scholar] [CrossRef]
- Ahn, H.; Seo, G.H.; Oh, A.; Lee, Y.; Keum, C.; Heo, S.H.; Kim, T.; Choi, J.; Kim, G.H.; Ko, T.S.; et al. Diagnosis of Schaaf-Yang syndrome in Korean children with developmental delay and hypotonia. Medicine 2020, 99, e23864. [Google Scholar] [CrossRef]
- Carlson, L.M.; Vora, N.L. Prenatal Diagnosis: Screening and Diagnostic Tools. Obstet. Gynecol. Clin. N. Am. 2017, 44, 245–256. [Google Scholar] [CrossRef]
- Dokras, A.; Barlow, D. Preimplantation genetic testing for monogenic diseases-bench to bedside reflections. Fertil. Steril. 2022, 118, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Vermeesch, J.R.; Voet, T.; Devriendt, K. Prenatal and pre-implantation genetic diagnosis. Nat. Rev. Genet. 2016, 17, 643–656. [Google Scholar] [CrossRef]
- Besser, A.G.; Blakemore, J.K.; Grifo, J.A.; Mounts, E.L. Transfer of embryos with positive results following preimplantation genetic testing for monogenic disorders (PGT-M): Experience of two high-volume fertility clinics. J. Assist. Reprod. Genet. 2019, 36, 1949–1955. [Google Scholar] [CrossRef]
- Gibbons, W.E. Preimplantation diagnosis for genetic susceptibility. N. Engl. J. Med. 2006, 355, 2048. [Google Scholar] [CrossRef] [PubMed]
- Beaudet, A.L. Preimplantation genetic screens. Science 2015, 349, 1423. [Google Scholar] [CrossRef] [PubMed]
- Dolan, S.M.; Goldwaser, T.H.; Jindal, S.K. Preimplantation Genetic Diagnosis for Mendelian Conditions. JAMA 2017, 318, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Treff, N.R.; Fedick, A.; Tao, X.; Devkota, B.; Taylor, D.; Scott, R.T., Jr. Evaluation of targeted next-generation sequencing-based preimplantation genetic diagnosis of monogenic disease. Fertil. Steril. 2013, 99, 1377–1384.e1376. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Alur-Gupta, S.; Gallop, R.; Dokras, A. Utilization of preimplantation genetic testing for monogenic disorders. Fertil. Steril. 2020, 114, 854–860. [Google Scholar] [CrossRef]
- Chen, S.; Fei, H.; Zhang, J.; Chen, Y.; Huang, H.; Lu, D.; Xu, C. Classification and Interpretation for 11 FBN1 Variants Responsible for Marfan Syndrome and Pre-implantation Genetic Testing (PGT) for Two Families Successfully Blocked Transmission of the Pathogenic Mutations. Front. Mol. Biosci. 2021, 8, 749842. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Marbach, F.; Elgizouli, M.; Rech, M.; Beygo, J.; Erger, F.; Velmans, C.; Stumpel, C.; Stegmann, A.P.A.; Beck-Wödl, S.; Gillessen-Kaesbach, G.; et al. The adult phenotype of Schaaf-Yang syndrome. Orphanet J. Rare Dis. 2020, 15, 294. [Google Scholar] [CrossRef] [PubMed]
- Halloun, R.; Habib, C.; Ekhilevitch, N.; Weiss, R.; Tiosano, D.; Cohen, M. Expanding the spectrum of endocrinopathies identified in Schaaf-Yang syndrome—A case report and review of the literature. Eur. J. Med. Genet. 2021, 64, 104252. [Google Scholar] [CrossRef]
- Kleinendorst, L.; Pi Castán, G.; Caro-Llopis, A.; Boon, E.M.J.; van Haelst, M.M. The role of obesity in the fatal outcome of Schaaf-Yang syndrome: Early onset morbid obesity in a patient with a MAGEL2 mutation. Am. J. Med. Genet. Part A 2018, 176, 2456–2459. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Yokoyama, K.; Yokoyama, A.; Nukata, T.; Ikeda, Y.; Hara, S. Longitudinal analysis of electroencephalography pattern changes in an infant with Schaaf-Yang syndrome and a novel mutation in melanoma antigen L2 (MAGEL2). Mol. Genet. Genomic. Med. 2022, 10, e1932. [Google Scholar] [CrossRef]
- Duan, Y.; Liu, L.; Zhang, X.; Jiang, X.; Xu, J.; Guan, Q. Phenotypic spectrum and mechanism analysis of Schaff Yang syndrome: A case report on new mutation of MAGEL2 gene. Medicine 2021, 100, e26309. [Google Scholar] [CrossRef] [PubMed]
- Gregory, L.C.; Shah, P.; Sanner, J.R.F.; Arancibia, M.; Hurst, J.; Jones, W.D.; Spoudeas, H.; Le Quesne Stabej, P.; Williams, H.J.; Ocaka, L.A.; et al. Mutations in MAGEL2 and L1CAM Are Associated with Congenital Hypopituitarism and Arthrogryposis. J. Clin. Endocrinol. Metab. 2019, 104, 5737–5750. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Nie, Y.; Yan, Z.; Zhu, X.; Wang, Y.; Guan, S.; Kuo, Y.; Zhang, W.; Zhi, X.; Wei, Y.; et al. Genetic testing and PGD for unexplained recurrent fetal malformations with MAGEL2 gene mutation. Sci. China Life Sci. 2019, 62, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.; Xavier, M.; Lourenço, C.; Melo, M.; Godinho, C. Schaaf-Yang Syndrome: A Real Challenge for Prenatal Diagnosis. Cureus 2021, 13, e20414. [Google Scholar] [CrossRef]
- Fountain, M.D.; Schaaf, C.P. Prader-Willi Syndrome and Schaaf-Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene. Diseases 2016, 4, 2. [Google Scholar] [CrossRef]
- McCarthy, J.M.; McCann-Crosby, B.M.; Rech, M.E.; Yin, J.; Chen, C.A.; Ali, M.A.; Nguyen, H.N.; Miller, J.L.; Schaaf, C.P. Hormonal, metabolic and skeletal phenotype of Schaaf-Yang syndrome: A comparison to Prader-Willi syndrome. J. Med. Genet. 2018, 55, 307–315. [Google Scholar] [CrossRef]
- Hidalgo-Santos, A.D.; del Carmen DeMingo-Alemany, M.; Moreno-Macián, F.; Roselló, M.; Orellana, C.; Martínez, F.; Caro-Llopis, A.; León-Cariñena, S.; Tomás-Vila, M. A Novel Mutation of MAGEL2 in a Patient with Schaaf-Yang Syndrome and Hypopituitarism. Int. J. Endocrinol. Metab. 2018, 16, e67329. [Google Scholar] [CrossRef]
- Althammer, F.; Muscatelli, F.; Grinevich, V.; Schaaf, C.P. Oxytocin-based therapies for treatment of Prader-Willi and Schaaf-Yang syndromes: Evidence, disappointments, and future research strategies. Transl. Psychiatry 2022, 12, 318. [Google Scholar] [CrossRef]
- Crutcher, E.; Pal, R.; Naini, F.; Zhang, P.; Laugsch, M.; Kim, J.; Bajic, A.; Schaaf, C.P. mTOR and autophagy pathways are dysregulated in murine and human models of Schaaf-Yang syndrome. Sci. Rep. 2019, 9, 15935. [Google Scholar] [CrossRef] [PubMed]
- Tacer, K.F.; Potts, P.R. Cellular and disease functions of the Prader-Willi Syndrome gene MAGEL2. Biochem. J. 2017, 474, 2177–2190. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Basak, A. Proteins and proteases of Prader-Willi syndrome: A comprehensive review and perspectives. Biosci. Rep. 2022, 42, BSR20220610. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.R.; Fahlman, R.P.; Wevrick, R. The N-terminal domain of the Schaaf-Yang syndrome protein MAGEL2 likely has a role in RNA metabolism. J. Biol. Chem. 2021, 297, 100959. [Google Scholar] [CrossRef]
- McCarthy, J.; Lupo, P.J.; Kovar, E.; Rech, M.; Bostwick, B.; Scott, D.; Kraft, K.; Roscioli, T.; Charrow, J.; Schrier Vergano, S.A.; et al. Schaaf-Yang syndrome overview: Report of 78 individuals. Am. J. Med. Genet. Part A 2018, 176, 2564–2574. [Google Scholar] [CrossRef]
- Matuszewska, K.E.; Badura-Stronka, M.; Śmigiel, R.; Cabała, M.; Biernacka, A.; Kosinska, J.; Rydzanicz, M.; Winczewska-Wiktor, A.; Sasiadek, M.; Latos-Bieleńska, A.; et al. Phenotype of two Polish patients with Schaaf-Yang syndrome confirmed by identifying mutation in MAGEL2 gene. Clin. Dysmorphol. 2018, 27, 49–52. [Google Scholar] [CrossRef]
- Ieda, D.; Negishi, Y.; Miyamoto, T.; Johmura, Y.; Kumamoto, N.; Kato, K.; Miyoshi, I.; Nakanishi, M.; Ugawa, S.; Oishi, H.; et al. Two mouse models carrying truncating mutations in MAGEL2 show distinct phenotypes. PLoS ONE 2020, 15, e0237814. [Google Scholar] [CrossRef]
- Kozakova, L.; Vondrova, L.; Stejskal, K.; Charalabous, P.; Kolesar, P.; Lehmann, A.R.; Uldrijan, S.; Sanderson, C.M.; Zdrahal, Z.; Palecek, J.J. The melanoma-associated antigen 1 (MAGEA1) protein stimulates the E3 ubiquitin-ligase activity of TRIM31 within a TRIM31-MAGEA1-NSE4 complex. Cell Cycle 2015, 14, 920–930. [Google Scholar] [CrossRef]
- Wijesuriya, T.M.; De Ceuninck, L.; Masschaele, D.; Sanderson, M.R.; Carias, K.V.; Tavernier, J.; Wevrick, R. The Prader-Willi syndrome proteins MAGEL2 and necdin regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum. Mol. Genet. 2017, 26, 4215–4230. [Google Scholar] [CrossRef]
- Hao, Y.H.; Fountain, M.D., Jr.; Fon Tacer, K.; Xia, F.; Bi, W.; Kang, S.H.; Patel, A.; Rosenfeld, J.A.; Le Caignec, C.; Isidor, B.; et al. USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder. Mol. Cell 2015, 59, 956–969. [Google Scholar] [CrossRef]
- Carias, K.V.; Zoeteman, M.; Seewald, A.; Sanderson, M.R.; Bischof, J.M.; Wevrick, R. A MAGEL2-deubiquitinase complex modulates the ubiquitination of circadian rhythm protein CRY1. PLoS ONE 2020, 15, e0230874. [Google Scholar] [CrossRef]
- Tennese, A.A.; Wevrick, R. Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in MAGEL2-null mice. Endocrinology 2011, 152, 967–978. [Google Scholar] [CrossRef]
- Bischof, J.M.; Stewart, C.L.; Wevrick, R. Inactivation of the mouse MAGEL2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Genet. 2007, 16, 2713–2719. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.E.; Wevrick, R. Loss of MAGEL2, a candidate gene for features of Prader-Willi syndrome, impairs reproductive function in mice. PLoS ONE 2009, 4, e4291. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Bogenpohl, J.W.; Howell, M.P.; Wevrick, R.; Panda, S.; Hogenesch, J.B.; Muglia, L.J.; Van Gelder, R.N.; Herzog, E.D.; Stewart, C.L. The imprinted gene MAGEL2 regulates normal circadian output. Nat. Genet. 2007, 39, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Fountain, M.D.; Tao, H.; Chen, C.A.; Yin, J.; Schaaf, C.P. MAGEL2 knockout mice manifest altered social phenotypes and a deficit in preference for social novelty. Genes Brain Behav. 2017, 16, 592–600. [Google Scholar] [CrossRef]
- Devos, J.; Weselake, S.V.; Wevrick, R. MAGEL2, a Prader-Willi syndrome candidate gene, modulates the activities of circadian rhythm proteins in cultured cells. J. Circadian Rhythm. 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed]




| Family | Transcript ID | DNA_Variant | Amino Acid Changes | Classification of Variants # | Inheritance |
|---|---|---|---|---|---|
| 1 | NM_019066.4 | c.1996dupC | p.(Gln666Profs*47) | pathogenic | Pat |
| 2 | NM_019066.4 | c.2895G>A | p.(Trp965*) | pathogenic | Pat |
| 3 | NM_019066.5 | c.1996dupC | p.(Gln666Profs*47) | pathogenic | De novo |
| Sample No. | Embryo | PGT-M Result | PGT-A Result | Outcome | |
|---|---|---|---|---|---|
| Haplotype | Sanger Sequencing | ||||
| Family 1-II-2 | Embryo 1 | M0 | c.1996dupC | −2, −15, −12 | |
| Embryo 2 | M0 | c.1996dupC | N | ||
| Family 2-II-2 | Embryo 1 | M1/ | N | 46, XN | |
| Embryo 2 | M1/ | N | 46, XN | Transfer | |
| Embryo 3 | M0/ | c.2895G>A | 46, XN | ||
| Embryo 4 | M1/ | N | 46, XN | ||
| Embryo 5 | M1/ | N | 46, XN | ||
| Embryo 6 | M0/ | c.2895G>A | 46, XN | ||
| Embryo 7 | M1/ | N | 46, XN | ||
| Family 2-II-5 | Embryo 1 | M1/ | N | 47, XN, +22 | |
| Embryo 2 | M0/ | c.2895G>A | 46, XN | ||
| Embryo 3 | M1/ | N | 46, XN, −14q21.2qter | ||
| Embryo 4 | M1/ | N | 46, XN | Transfer | |
| Embryo 5 | M1/ | N | 46, XN | ||
| Sample No. | Gestation, Weeks | Fetus | Result | Outcome |
|---|---|---|---|---|
| Family 2-II-2 | 16 W | Family 2-III-3 | N | Born healthy |
| Family 2-II-5 | 17 + 4 W | Family 2-III-7 | N | Born healthy |
| Family 3-I-2 | 15 W | Family 3-II-2 | N | Born healthy |
| Patients and Source Publication | Totals d (n = 127) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1–91 | 92–95 | 96 | 97–102 | 103–104 | 105–108 | 109 | 110 | 111 | 112–115 | 116 | New | |||
| Sex (M/F) | N/A | 4/4 | 0/1 | 5/1 | 0/2 | 3/1 | 0/1 | 0/1 | 1/0 | 1/3 | 1/0 | 8/3 | 44/47 | |
| Prenatal symptoms | History of polyhydramnios | N/A | N/A | N/A | N/A | 0/2 | 1/4 | 0/1 | 0/1 | 0/1 | 1/3 | 1/1 | 0/10 | N/A |
| Decreased fetal movement | 8/23 | 2/8 | 0/1 | N/A | N/A | 0/4 | 1/1 | 0/1 | 0/1 | 0/4 | 0/1 | 0/7 | 11/51 (22%) | |
| Perinatal history | Temperature instability | 40/60 | N/A | N/A | 4/6 | N/A | N/A | 0/1 | 1/1 | 0/1 | 0/4 | N/A | 2/7 | 47/80 (59%) |
| Respiratory distress/defects | 49/70 | N/A | 1/1 | 0/6 | 2/2 | 3/4 | 1/1 | 1/1 | 0/1 | 2/4 | 1/1 | 11/11 | 71/102 (70%) | |
| Intubation | 38/64 | N/A | 1/1 | 1/6 | 2/2 | 2/4 | 1/1 | 1/1 | 0/1 | 2/4 | 1/1 | 11/11 | 60/96 (63%) | |
| Mechanical ventilator | 36/64 | 4/8 | 1/1 | 1/6 | 2/2 | 3/4 | 1/1 | 1/1 | 0/1 | 2/4 | 1/1 | 11/11 | 63/104 (61%) | |
| Tracheostomy | 13/58 | N/A | N/A | 0/6 | 0/2 | 0/4 | 0/1 | 1/1 | 0/1 | 2/4 | 0/1 | 0/10 | 16/88 (18%) | |
| Hypotonia | 72/78 | 6/7 | 1/1 | 6/6 | 2/2 | 3/4 | 1/1 | 1/1 | 0/1 | 1/1 | 1/1 | 9/11 | 103/114 (90%) | |
| Feeding issues | 71/80 | 7/7 | 1/1 | 4/6 | N/A | 1/4 | 1/1 | 1/1 | 0/1 | 1/4 | 1/1 | 10/10 | 98/116 (84%) | |
| Poor suck in infancy | 65/72 | 6/7 | N/A | 6/6 | 2/2 | 2/4 | 1/1 | 1/1 | 0/1 | N/A | N/A | 10/10 | 93/104 (89%) | |
| Dysphagia | 49/62 | N/A | N/A | 1/6 | 2/2 | 2/4 | 1/1 | 1/1 | 0/1 | N/A | N/A | 6/6 | 62/83 (75%) | |
| Use of nasogastric (NG) tube | 45/60 | 4/5 | 0/1 | 1/6 | 1/2 | 2/4 | 1/1 | 1/1 | 0/1 | N/A | N/A | 9/11 | 64/92 (70%) | |
| Use of G tube | 30/58 | N/A | 1/1 | N/A | 0/2 | 1/4 | 0/1 | 0/1 | 0/1 | N/A | N/A | 0/11 | 32/79 (41%) | |
| Dysmorphic features | Scoliosis | 29/54 | 5/8 | 0/1 | 0/6 | 1/2 | 0/4 | 0/1 | 0/1 | 0/1 | 3/4 | 0/1 | 0/11 | 38/94 (40%) |
| Kyphosis | 12/37 | N/A | 0/1 | 0/6 | 0/2 | 0/4 | 0/1 | 0/1 | 0/1 | 0/4 | 0/1 | 0/11 | 12/69 (17%) | |
| Contractures | 77/90 | 6/7 | 1/1 | 6/6 | 2/2 | 3/4 | 1/1 | N/A | 0/1 | 4/4 | 1/1 | 11/11 | 112/128 (88%) | |
| Small hands | 15/24 | N/A | N/A | 6/6 | 2/2 | 1/4 | 0/1 | 0/1 | 1/1 | 1/4 | N/A | 2/11 | 28/54 (52%) | |
| Small feet | 12/24 | N/A | N/A | N/A | 0/2 | 1/4 | 0/1 | 0/1 | 1/1 | 1/4 | N/A | 2/11 | 17/48 (35%) | |
| Gastrointestinal | Reflux/GERD | 36/66 | N/A | N/A | N/A | 1/2 | 1/4 | 0/1 | 0/1 | 0/1 | 2/4 | N/A | 0/5 | 40/84 (48%) |
| Chronic constipation | 40/60 | 6/8 | N/A | N/A | N/A | 0/4 | 0/1 | 0/1 | N/A | 3/4 | N/A | 0/5 | 49/83 (59%) | |
| Endocrine assessment | Excessive weight gain | 15/67 | 2/5 | N/A | 3/5 | N/A | 0/3 | 1/1 | 0/1 | 1/1 | N/A | N/A | 1/7 | 23/90 (26%) |
| Hyperphagia | 14/56 | N/A | N/A | N/A | N/A | N/A | 1/1 | 0/1 | 1/1 | N/A | N/A | 1/6 | 17/65 (26%) | |
| Hypogonadism | 29/74 | 3/7 | N/A | 4/6 | N/A | 1/2 | N/A | 1/1 | 1/1 | 1/4 | 1/1 | 3/7 | 44/103 (43%) | |
| Hypopituitarism | 3/9 | N/A | 1/1 | 3/3 | N/A | N/A | N/A | 1/1 | 1/1 | 3/4 | N/A | N/A | N/A | |
| Growth hormone deficiency | 3/8 | 2/8 | 1/1 | 3/3 | N/A | N/A | N/A | 1/1 | 1/1 | 4/4 | N/A | N/A | N/A | |
| Short stature | 13/27 | N/A | 1/1 | 5/5 | N/A | 3/4 | N/A | 1/1 | 1/1 | 4/4 | N/A | 1/1 | 29/44 (66%) | |
| Increased fatty tissue | 4/5 | N/A | 0/1 | N/A | 0/2 | 0/4 | 1/1 | 0/1 | 1/1 | 0/4 | N/A | 1/7 | N/A | |
| Cortical blindness | 1/2 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| Neurodevelopment | Developmental delay | 79/83 | 8/8 | 1/1 | 6/6 | N/A | 4/4 | 1/1 | 1/1 | 1/1 | 4/4 | N/A | 1/1 | 106/110 (96%) |
| Attention deficit disorder | N/A | 2/5 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| Obsessive compulsive disorder | N/A | 2/5 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| ASD diagnosis | 29/39 | 4/7 | N/A | 1/1 | N/A | 2/4 | N/A | N/A | N/A | 1/4 | N/A | N/A | 37/55 (67%) | |
| Seizures | 25/73 | N/A | 1/1 | 2/6 | 1/2 | 1/4 | 0/1 | 0/1 | 0/1 | 0/4 | 0/1 | 0/5 | 30/99 (30%) | |
| Self-injurious behavior | 9/16 | 4/8 | N/A | 4/6 a | N/A | N/A | N/A | N/A | 0/1 | 1/4 | N/A | 0/1 | 18/36 (50%) | |
| Intellectual disability | N/A | 5/5 | N/A | 5/6 | N/A | 4/4 | 1/1 | N/A | 1/1 | N/A | N/A | 1/1 | N/A | |
| Sleep problems | Abnormal sleep cycle b | N/A | 8/8 | N/A | 5/6 c | N/A | N/A | N/A | 0/1 | 0/1 | N/A | N/A | 0/1 | N/A |
| Sleep apnea | 43/58 | 5/7 | 1/1 | 1/5 | N/A | 2/4 | 1/1 | 1/1 | 0/1 | 2/4 | 1/1 | 9/11 | 66/94 (70%) | |
| Assistant examination | Echocardiography Findings | N/A | N/A | N/A | N/A | 2/2 | N/A | N/A | 0/1 | 0/1 | 1/4 | 1/1 | 2/2 | N/A |
| Brain MRI findings | 6/11 | N/A | 0/1 | 4/6 | 0/1 | 2/4 | N/A | 1/1 | N/A | 2/4 | N/A | 3/3 | 18/31 (58%) | |
| Others | Hypopigmentation | N/A | N/A | 0/1 | 4/6 | 0/2 | 0/4 | 0/1 | 0/1 | 0/1 | 0/4 | 0/1 | 0/11 | 4/32 (13%) |
| Hirsutism | 1/6 | N/A | N/A | N/A | N/A | 0/4 | 0/1 | N/A | 0/1 | N/A | N/A | 0/2 | N/A | |
| Eye abnormalities | 18/25 | N/A | 0/1 | 5/6 | 1/2 | 0/4 | 0/1 | 0/1 | 0/1 | 4/4 | 0/1 | 0/7 | 28/53 (53%) | |
| Patients with Any Variant Other Than c.1996dupC (n = 67) | Patients with the c.1996dupC Variant (n = 51) | p-Value | ||
|---|---|---|---|---|
| Prenatal symptoms | History of polyhydramnios | 2/11 (18%) | 1/12 (8%) | 0.590 |
| Decreased fetal movement | 3/16 (19%) | 0/12 (0%) | 0.112 | |
| Perinatal history | Temperature instability | 23/46 (50%) | 24/43 (56%) | 0.583 |
| Respiratory distress/defects | 29/52 (56%) | 35/43 (81%) | 0.008 * | |
| Intubation | 26/55 (47%) | 32/44 (73%) | 0.0111 * | |
| Mechanical ventilator | 22/47 (47%) | 32/44 (73%) | 0.012 * | |
| Tracheostomy | 6/45 (13%) | 9/52 (17%) | 0.589 | |
| Hypotonia | 57/63 (90%) | 45/47 (96%) | 0.426 | |
| Feeding issues | 55/65 (85%) | 45/50 (90%) | 0.395 | |
| Poor suck in infancy | 49/57 (86%) | 45/45 (100%) | 0.009 * | |
| Dysphagia | 28/44 (64%) | 36/40 (90%) | 0.005 * | |
| Use of nasogastric (NG) tube | 24/51 (47%) | 39/45 (87%) | 0.00004 * | |
| Use of G tube | 11/46 (24%) | 21/39 (54%) | 0.005 * | |
| Dysmorphic features | Scoliosis | 17/51 (33%) | 19/38 (50%) | 0.113 |
| Kyphosis | 5/38 (13%) | 7/35 (20%) | 0.431 | |
| Contractures | 54/66 (82%) | 51/52 (98%) | 0.038 * | |
| Small hands | 7/17 (41%) | 7/14 (50%) | 0.623 | |
| Small feet | 2/12 (17%) | 4/13 (31%) | 0.645 | |
| Gastrointestinal | Reflux/GERD | 15/40 (38%) | 25/43 (58%) | 0.351 |
| Chronic constipation | 23/44 (52%) | 27/42 (64%) | 0.259 | |
| Endocrine assessment | Excessive weight gain | 18/49 (37%) | 5/40 (13%) | 0.009 * |
| Hyperphagia | 13/37 (35%) | 4/34 (12%) | 0.021 * | |
| Hypogonadism | 25/53 (47%) | 18/44 (41%) | 0.537 | |
| Hypopituitarism | 7/7 (100%) | 3/4 (75%) | 0.364 | |
| Growth hormone deficiency | 22/43 (51%) | 17/30 (57%) | 0.643 | |
| Short stature | 30/48 (63%) | 24/37 (65%) | 0.822 | |
| Increased fatty tissue | 3/9 (33%) | 0/13 (0%) | 0.025 * | |
| Cortical blindness | 0/1 (0%) | N/A (/) | / | |
| Neurodevelopment | Developmental delay | 45/53 (85%) | 33/34 (97%) | 0.069 |
| Attention deficit disorder | 2/6 (33%) | N/A (/) | / | |
| Obsessive compulsive disorder | 2/6 (33%) | 1/1 (100%) | 0.429 | |
| ASD diagnosis Seizures | 24/34 (71%) | 14/21 (67%) | 0.760 | |
| Seizures | 17/53 (32%) | 12/43 (28%) | 0.658 | |
| Self-injurious behavior | 7/16 (44%) | 2/5 (40%) | 1.000 | |
| Intellectual disability | 15/15 (100%) | 3/3 (100%) | 1.000 | |
| Sleep problems | Abnormal sleep cycle | 11/15 (73%) | 2/2 (100%) | 1.000 |
| Sleep apnea | 36/52 (69%) | 27/40 (68%) | 0.859 | |
| Assistant examination | Echocardiography Findings | 2/4 (50%) | 4/7 (57%) | 1.000 |
| Brain MRI findings | 6/11 (55%) | 6/9 (67%) | 0.670 | |
| Others | Hypopigmentation | 4/19 (21%) | 0/14 (0%) | 0.119 |
| Hirsutism | 0/6 (0%) | 0/3 (0%) | 1.000 | |
| Eye abnormalities | 5/14 (36%) | 5/14 (36%) | 1.000 | |
| Total number of symptoms | 11.5 (0.54) | 13.6 (0.57) | 0.012 * | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, N.; Shi, W.; Cao, X.; Zhou, X.; Huang, H.; Chen, S.; Xu, C. Preimplantation Genetic Testing (PGT) and Prenatal Diagnosis of Schaaf-Yang Syndrome: A Report of Three Families and a Research on Genotype–Phenotype Correlations. J. Clin. Med. 2023, 12, 1688. https://doi.org/10.3390/jcm12041688
Xu N, Shi W, Cao X, Zhou X, Huang H, Chen S, Xu C. Preimplantation Genetic Testing (PGT) and Prenatal Diagnosis of Schaaf-Yang Syndrome: A Report of Three Families and a Research on Genotype–Phenotype Correlations. Journal of Clinical Medicine. 2023; 12(4):1688. https://doi.org/10.3390/jcm12041688
Chicago/Turabian StyleXu, Naixin, Weihui Shi, Xianling Cao, Xuanyou Zhou, Hefeng Huang, Songchang Chen, and Chenming Xu. 2023. "Preimplantation Genetic Testing (PGT) and Prenatal Diagnosis of Schaaf-Yang Syndrome: A Report of Three Families and a Research on Genotype–Phenotype Correlations" Journal of Clinical Medicine 12, no. 4: 1688. https://doi.org/10.3390/jcm12041688
APA StyleXu, N., Shi, W., Cao, X., Zhou, X., Huang, H., Chen, S., & Xu, C. (2023). Preimplantation Genetic Testing (PGT) and Prenatal Diagnosis of Schaaf-Yang Syndrome: A Report of Three Families and a Research on Genotype–Phenotype Correlations. Journal of Clinical Medicine, 12(4), 1688. https://doi.org/10.3390/jcm12041688