
Nephrological Complications in Hemoglobinopathies: SITE Good Practice

 , , ,
, , ,  , ,
, , 
{kind=link}
Abstract
:1. Introduction
- grade IA: strong recommendation based on strong evidence certainties and meta-analyses;
- grade IB: strong recommendation based on strong evidence certainties;
- grade IIA: strong recommendation based on moderate evidence certainties;
- grade IIB: strong recommendation based on moderate–weak evidence certainties;
- grade IIC: strong recommendation based on weak evidence certainties;
- grade IIIA: conditional recommendation based on strong evidence certainties;
- grade IIIB: conditional recommendation based on moderate evidence certainties;
- grade IIIC: conditional recommendation based on weak evidence certainties;
- grade IV: conditional recommendation based on expert indication.
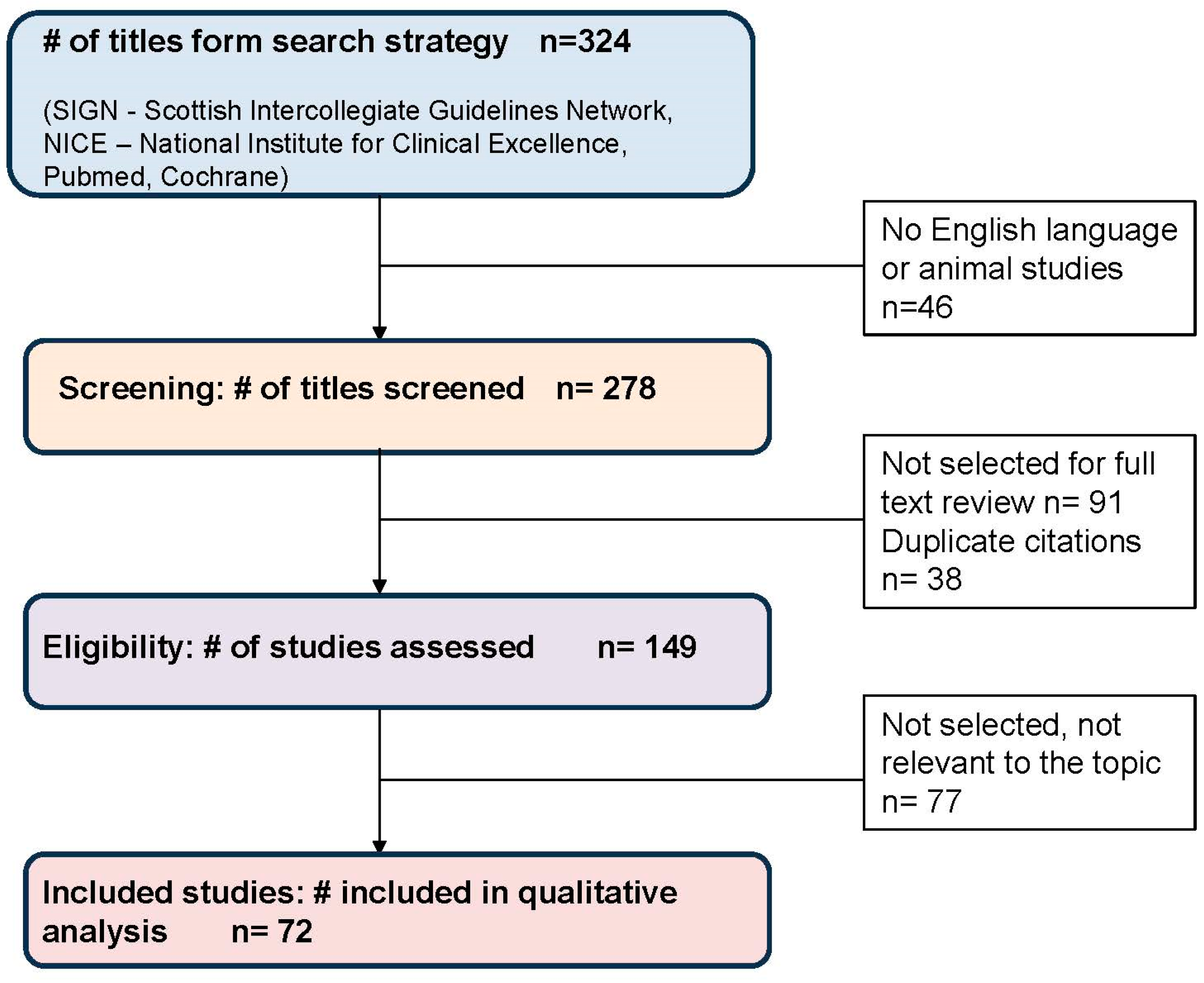
2. Methods
- -
- S.I.T.O. guidelines (https://www.societaitalianatrapiantidiorgano.com/linee-guida/) (accessed on 25 October 2022);
- -
- KDIGO guidelines (https://kdigo.org/guidelines/) (accessed on 12 Febrary 2023;
- -
- SITE good practice (https://manage.site-italia.org/scienza-e-formazione/buone-pratiche-site.html) (accessed on 2 July 2022)
Grading Scheme
- strong and meta-analyses—systematic reviews and meta-analyses;
- strong—clinical studies with a randomized control group (RTC);
- moderate—clinical studies with a control group;
- moderate weak—cohort studies, case control studies, and observational studies;
- weak—case reports, case studies, and expert opinion.
- A
- data derived from several systematic reviews and meta-analyses;
- B
- data derived from only one systematic review and meta-analysis or from various clinical studies with a randomized control group (RTC);
- C
- data derived from observational, retrospective clinical studies or expert opinion.
- grade IA: strong recommendation based on strong evidence certainties and meta-analyses;
- grade IB: strong recommendation based on strong evidence certainties;
- grade IIA: strong recommendation based on moderate evidence certainties;
- grade IIB: strong recommendation based on moderate–weak evidence certainties;
- grade IIC: strong recommendation based on weak evidence certainties;
- grade IIIA: conditional recommendation based on strong evidence certainties;
- grade IIIB: conditional recommendation based on moderate evidence certainties;
- grade IIIC: conditional recommendation based on weak evidence certainties;
- grade IV: conditional recommendation based on expert indication.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TDT | transfusion dependent thalassemia |
| NTDT | non-transfusion-dependent thalassemias |
| SCD | sickle cell disease |
| EEX | erythroexchange |
| UACR | urinary albumin/creatinine ratio |
| UPCR | urinary protein/creatinine ratio |
| ANA | anti-nuclear antibody |
| ENA-Ab | extractable nuclear antigen antibody |
| RF | rheumathoid factor |
| PCR | protein/creatinine ratio |
| ACR | albumin/creatinine ratio |
| ESKD | end-stage kidney disease |
| eGFR | estimated glomerular filtration rate |
| ACEi | angiotensin-converting enzyme inhibitor |
| MRA | mineral corticoid antagonist |
| SGLT2 | sodium–glucose cotransporter 2 inhibitor |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| EMA | European Medicines Agency |
| SITE | Società Italiana Talassemie ed Emoglobinopatie |
| SITO | Società Italiana Trapianti d’Organo |
| ARNI | angiotensin receptor–neprilysin inhibitors |
| ARBs | angiotensin receptor blockers |
| ANCAs | antineutrophil cytoplasmatic antibodies |
| C3-C4 | complement fraction 3 and fraction 4 |
References
- Piel, F.B.; Tatem, A.J.; Huang, Z.; Gupta, S.; Williams, T.N.; Weatherall, D.J. Global migration and the changing distribution of sickle haemoglobin: A quantitative study of temporal trends between 1960 and 2000. Lancet Glob. Health 2014, 2, e80–e89. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J.; Clegg, J.B. Inherited haemoglobin disorders: An increasing global health problem. Bull. World Health Organ. 2001, 79, 704–712. [Google Scholar] [PubMed]
- Modell, B.; Darlison, M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Lux, C.; Piel, F.B.; Gianesin, B.; Bonetti, F.; Casale, M.; Graziadei, G.; Lisi, R.; Pinto, V.; Putti, M.C.; et al. Access to emergency departments for acute events and identification of sickle cell disease in refugees. Blood 2019, 133, 2100–2103. [Google Scholar] [CrossRef]
- Forni, G.L.; Gianesin, B.; Musallam, K.M.; Longo, F.; Rosso, R.; Lisi, R.; Gamberini, M.R.; Pinto, V.M.; Graziadei, G.; Vitucci, A.; et al. Overall and complication-free survival in a large cohort of patients with beta-thalassemia major followed over 50 years. Am. J. Hematol. 2023, 98, 381–387. [Google Scholar] [CrossRef]
- Pinto, V.M.; Gianesin, B.; Piel, F.B.; Longo, F.; Rigano, P.; Quota, A.; Spadola, V.; Graziadei, G.; Mazzi, F.; Cappellini, M.D.; et al. Morbidity and mortality of sickle cell disease patients is unaffected by splenectomy: Evidence from 3 decades follow-up in a high-income setting. Haematologica 2022, 108, 1158–1162. [Google Scholar] [CrossRef]
- Elmariah, H.; Garrett, M.E.; De Castro, L.M.; Jonassaint, J.C.; Ataga, K.I.; Eckman, J.R.; Ashley-Koch, A.E.; Telen, M.J. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am. J. Hematol. 2014, 89, 530–535. [Google Scholar] [CrossRef]
- Lubeck, D.; Agodoa, I.; Bhakta, N.; Danese, M.; Pappu, K.; Howard, R.; Gleeson, M.; Halperin, M.; Lanzkron, S. Estimated Life Expectancy and Income of Patients With Sickle Cell Disease Compared With Those Without Sickle Cell Disease. JAMA Netw. Open 2019, 2, e1915374. [Google Scholar] [CrossRef]
- Origa, R.; Gianesin, B.; Longo, F.; Di Maggio, R.; Cassinerio, E.; Gamberini, M.R.; Pinto, V.M.; Quarta, A.; Casale, M.; La Nasa, G.; et al. Incidence of cancer and related deaths in hemoglobinopathies: A follow-up of 4631 patients between 1970 and 2021. Cancer 2023, 129, 107–117. [Google Scholar] [CrossRef]
- Sharpe, C.C.; Thein, S.L. How I treat renal complications in sickle cell disease. Blood 2014, 123, 3720–3726. [Google Scholar] [CrossRef]
- Olaniran, K.O.; Eneanya, N.D.; Nigwekar, S.U.; Vela-Parada, X.F.; Achebe, M.M.; Sharma, A.; Thadhani, R.I. Sickle Cell Nephropathy in the Pediatric Population. Blood Purif. 2019, 47, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Niss, O.; Lane, A.; Asnani, M.R.; Yee, M.E.; Raj, A.; Creary, S.; Fitzhugh, C.; Bodas, P.; Saraf, S.L.; Sarnaik, S.; et al. Progression of albuminuria in patients with sickle cell anemia: A multicenter, longitudinal study. Blood Adv. 2020, 4, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Saraf, S.L.; Derebail, V.K. The nephropathy of sickle cell trait and sickle cell disease. Nat. Rev. Nephrol. 2022, 18, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Saif, A.; Soliman, N.; Abdelhamid, A. Doppler assessment of renal hemodynamic alterations in homozygous sickle cell disease and sickle Beta-thalassemia. Ultrason. Imaging 2015, 37, 258–264. [Google Scholar] [CrossRef]
- Lafferty HMAnderson, S.; Brenner, B.M. Anemia: A potent modulator of renal hemodynamics in models of progressive renal disease. Am. J. Kidney Dis. 1991, 17, 2–7. [Google Scholar]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end stage renal disease. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef]
- Demosthenous, C.; Vlachaki, E.; Apostolou, C.; Eleftheriou, P.; Kotsiafti, A.; Vetsiou, E.; Mandala, E.; Perifanis, V.; Sarafidis, P. Beta-thalassemia: Renal complications and mechanisms: A narrative review. Hematology 2019, 24, 426–438. [Google Scholar] [CrossRef]
- Musallam, K.M.; Taher, A.T. Mechanisms of renal disease in beta-thalassemia. J. Am. Soc. Nephrol. 2012, 23, 1299–1302. [Google Scholar] [CrossRef]
- Meloni, A.; Barbuto, L.; Pistoia, L.; Positano, V.; Renne, S.; Peritore, G.; Fina, P.; Spasiano, A.; Allò, M.; Messina, G.; et al. Frequency, pattern, and associations of renal iron accumulation in sickle/β-thalassemia patients. Ann. Hematol. 2022, 101, 1941–1950. [Google Scholar] [CrossRef]
- Cianciulli, P.; Sorrentino, F.; Forte, L.; Palombi, M.; Papa, G.; Meloni, C.; Gallucci, M.T.; Casciani, C.U. Acute renal failure occuring during intravenous desferrioxamine therapy: Recovery after haemodialysis. Haematologica 1992, 77, 514–515. [Google Scholar]
- Rafat, C.; Fakhouri, F.; Ribeil, J.A.; Delarue, R.; Le Quintrec, M. Fanconi syndrome due to deferasirox. Am. J. Kidney Dis. 2009, 54, 931–934. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICI670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef]
- Ponticelli, C.; Musallam, K.M.; Cianciulli, P.; Cappellini, M.D. Renal complications in transfusion-dependent beta thalassaemia. Blood Rev. 2010, 24, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Díaz-García, J.D.; Gallegos-Villalobos, A.; Gonzalez-Espinoza, L.; Sanchez-Niño, M.D.; Villarrubia, J.; Ortiz, A. Deferasirox nephrotoxicity-the knowns and unknowns. Nat. Rev. Nephrol. 2014, 10, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012, 26 (Suppl. S1), S3–S6. [Google Scholar] [CrossRef] [PubMed]
- Ekwattanakit, S.; Siritanaratkul, N.; Viprakasit, V. A prospective analysis for prevalence of complications in Thai nontransfusion-dependent Hb E/β-thalassemia and α-thalassemia (Hb H disease). Am. J. Hematol. 2018, 93, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Gianesin, B.; Voi, V.; Motta, I.; Pinto, V.M.; Piolatto, A.; Spasiano, A.; Ruffo, G.B.; Gamberini, M.R.; Barella, S.; et al. Italian patients with hemoglobinopathies exhibit a 5-fold increase in age-standardized lethality due to SARS-CoV-2 infection. Am. J. Hematol. 2022, 97, E75–E78. [Google Scholar] [CrossRef]
- Matte, A.; Recchiuti, A.; Federti, E.; Koehl, B.; Mintz, T.; El Nemer, W.; Tharaux, P.-L.; Brousse, V.; Andolfo, I.; Lamolinara, A.; et al. Resolution of sickle cell disease-associated inflammation and tissue damage with 17R-resolvin D1. Blood 2019, 133, 252–265. [Google Scholar] [CrossRef]
- Schünemann, H.J.; Wiercioch, W.; Brozek, J.; Etxeandia-Ikobaltzeta, I.; Mustafa, R.A.; Manja, V.; Brignardello-Petersen, R.; Neumann, I.; Falavigna, M.; Alhazzani, W.; et al. GRADE Evidence to Decision (EtD) frameworks for adoption, adaptation, and de novo development of trustworthy recommendations: GRADE-ADOLOPMENT. J. Clin. Epidemiol. 2017, 81, 101–110. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruffo, G.B.; Russo, R.; Casini, T.; Lombardini, L.; Orecchia, V.; Voi, V.; Origa, R.; Forni, G.L.; Marchetti, M.; Gigante, A.; et al. Nephrological Complications in Hemoglobinopathies: SITE Good Practice. J. Clin. Med. 2023, 12, 7476. https://doi.org/10.3390/jcm12237476
Ruffo GB, Russo R, Casini T, Lombardini L, Orecchia V, Voi V, Origa R, Forni GL, Marchetti M, Gigante A, et al. Nephrological Complications in Hemoglobinopathies: SITE Good Practice. Journal of Clinical Medicine. 2023; 12(23):7476. https://doi.org/10.3390/jcm12237476
Chicago/Turabian StyleRuffo, Giovan Battista, Rodolfo Russo, Tommaso Casini, Letizia Lombardini, Valeria Orecchia, Vincenzo Voi, Raffaella Origa, Gian Luca Forni, Monia Marchetti, Antonia Gigante, and et al. 2023. "Nephrological Complications in Hemoglobinopathies: SITE Good Practice" Journal of Clinical Medicine 12, no. 23: 7476. https://doi.org/10.3390/jcm12237476
APA StyleRuffo, G. B., Russo, R., Casini, T., Lombardini, L., Orecchia, V., Voi, V., Origa, R., Forni, G. L., Marchetti, M., Gigante, A., Garibotto, G., Maggio, A., & De Franceschi, L. (2023). Nephrological Complications in Hemoglobinopathies: SITE Good Practice. Journal of Clinical Medicine, 12(23), 7476. https://doi.org/10.3390/jcm12237476