
Heart Failure in Patients with Chronic Kidney Disease

 ,
,  ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Definitions
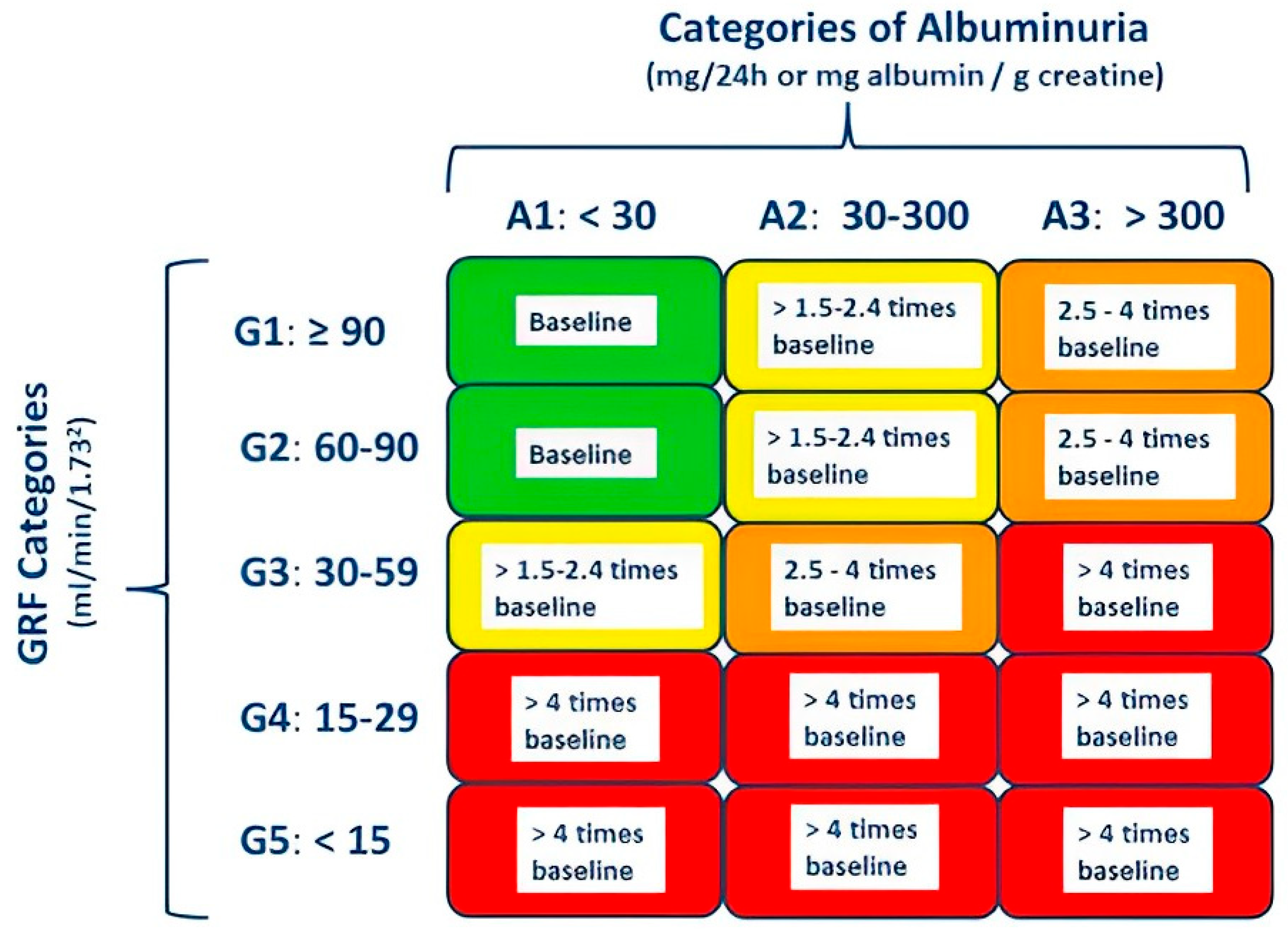
2.1. Chronic Kidney Disease
2.2. Acute Kidney Injury
3. Epidemiology
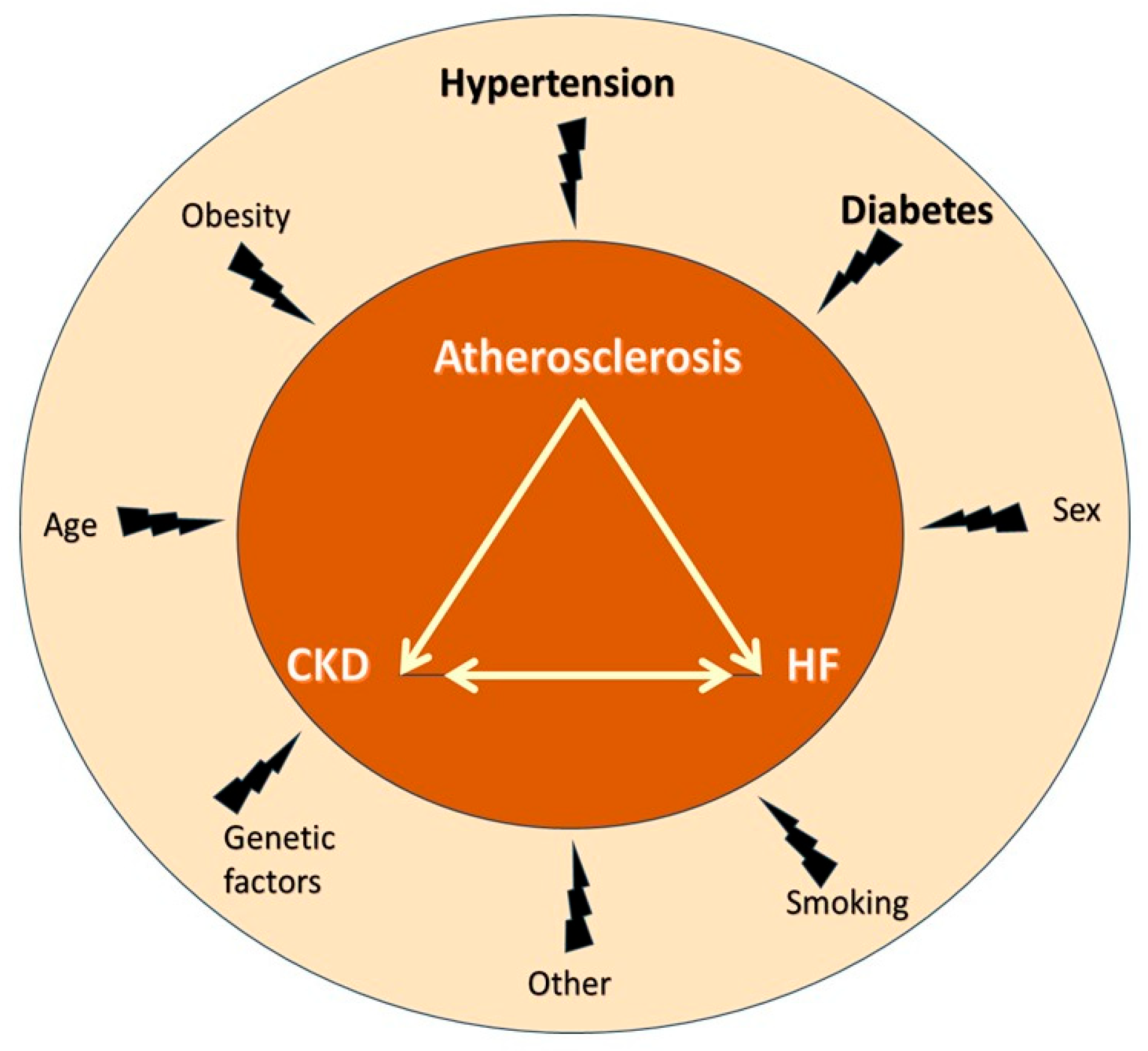
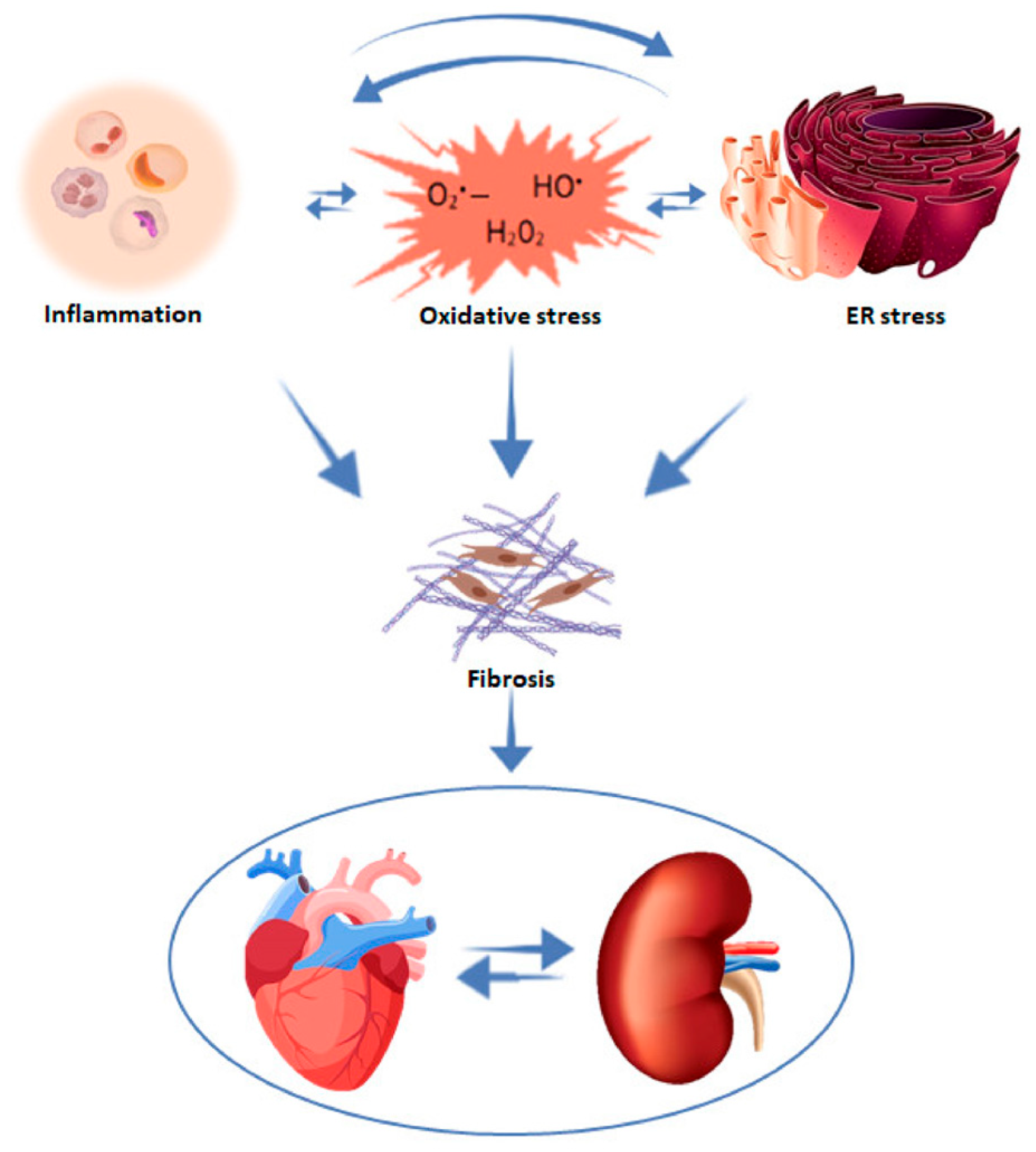
4. Mechanisms of HF Development in CKD
5. Major Limitation of Cardiovascular Trials: The Exclusion of Patients with Renal Disease
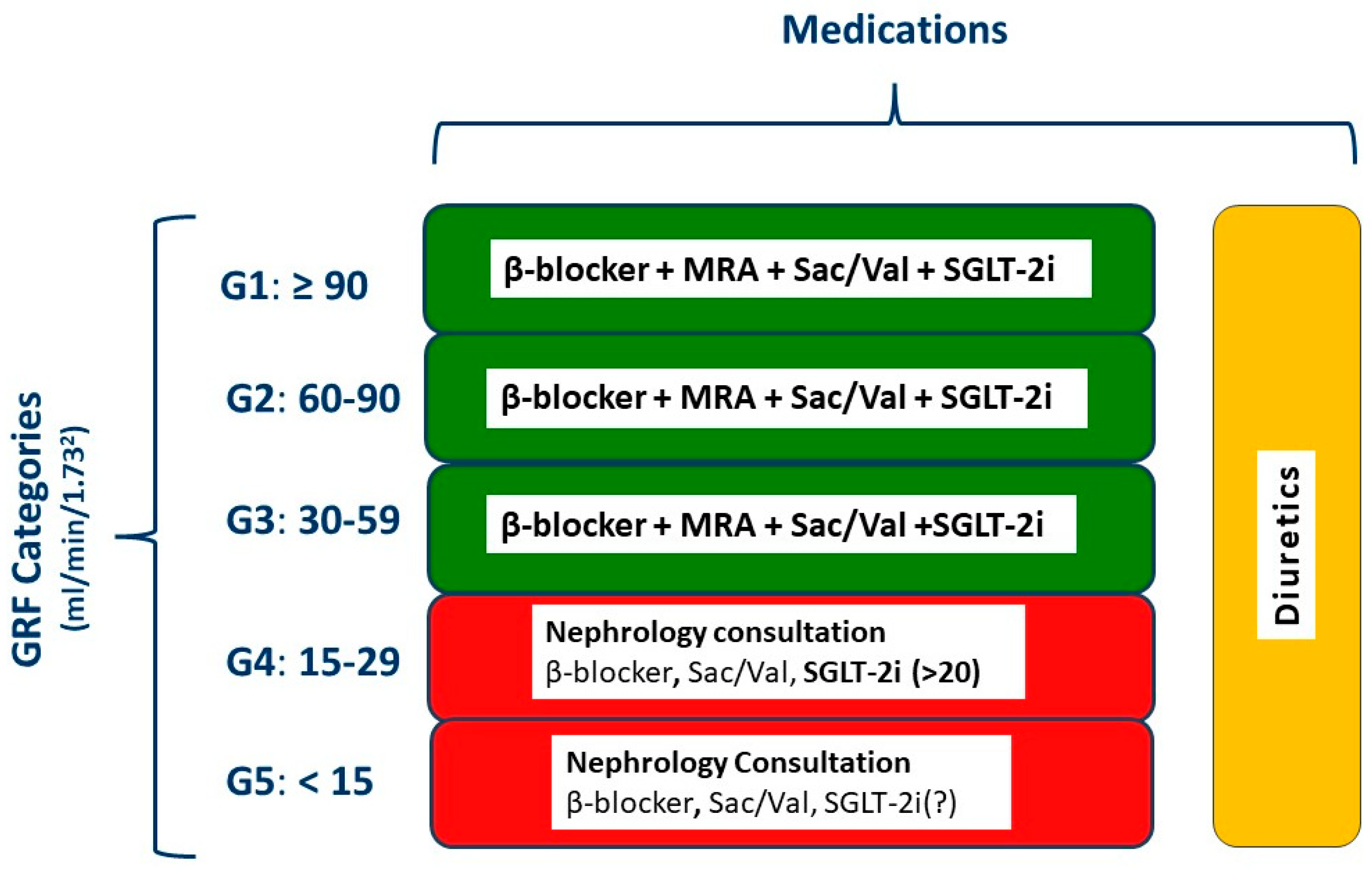
6. Medical Treatment of HF in CKD
6.1. β-Blockers
6.2. Renin–Angiotensin–Aldosterone System Inhibitors (RAASi)
6.2.1. Angiotensin Converting Enzyme Inhibitors/Angiotensin Receptor Blockers
6.2.2. Mineralocorticoid Receptor Antagonists (MRAs)
6.3. Sacubitril-Valsartan
6.4. Sodium-Glucose Cotransporter 2 Inhibitors (SGLT-2i)
6.5. Diuretics
7. Management of Specific Conditions
7.1. Worsening Renal Function
7.2. Hyperkalemia
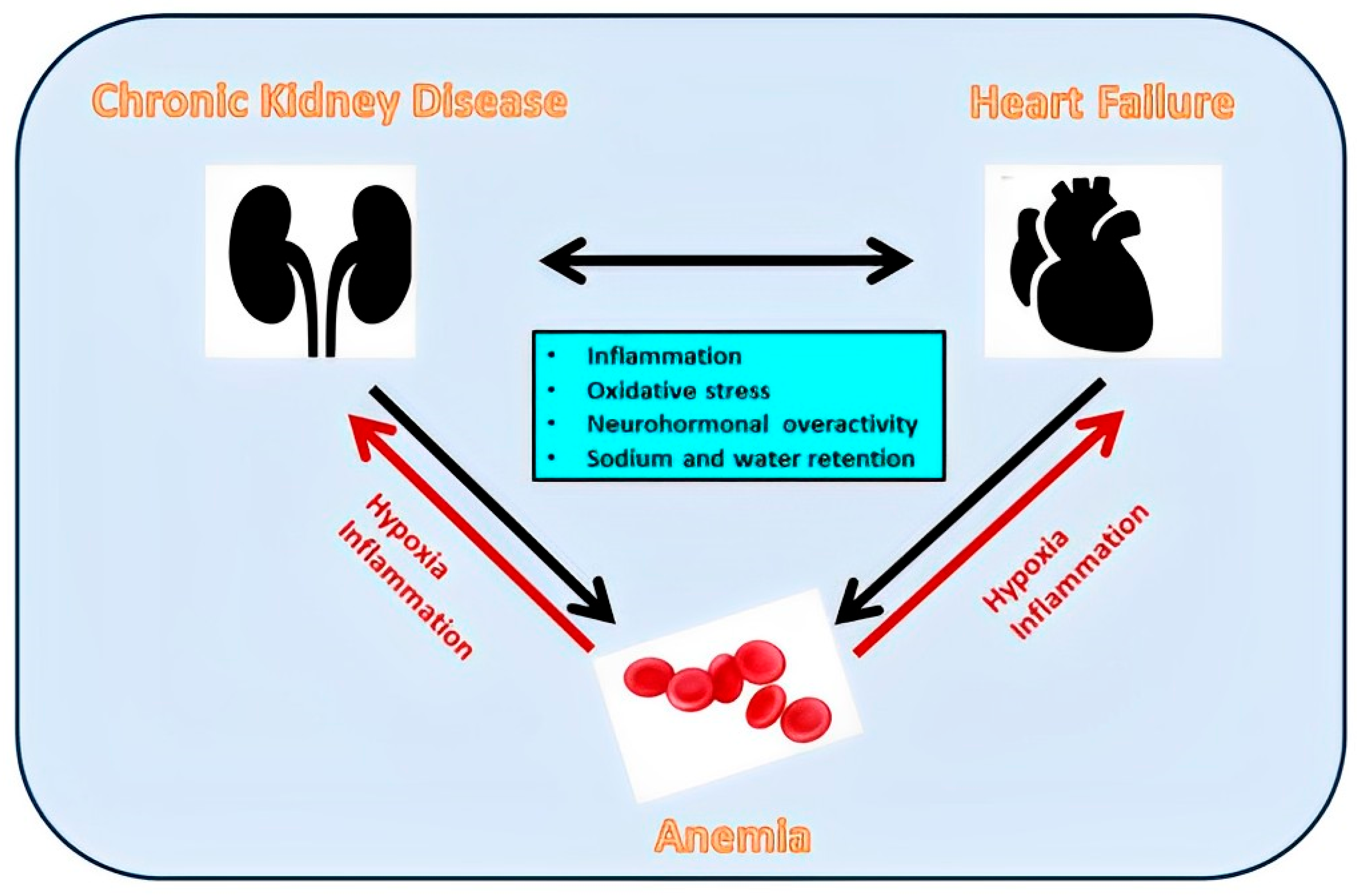
7.3. Anemia
8. Conclusions-Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Triposkiadis, F.; Giamouzis, G.; Parissis, J.; Starling, R.C.; Boudoulas, H.; Skoularigis, J.; Butler, J.; Filippatos, G. Reframing the association and significance of co-morbidities in heart failure. Eur. J. Heart Fail. 2016, 18, 744–758. [Google Scholar] [CrossRef]
- Szlagor, M.; Dybiec, J.; Mlynarska, E.; Rysz, J.; Franczyk, B. Chronic Kidney Disease as a Comorbidity in Heart Failure. Int. J. Mol. Sci. 2023, 24, 2988. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Xanthopoulos, A.; Parissis, J.; Butler, J.; Farmakis, D. Pathogenesis of chronic heart failure: Cardiovascular aging, risk factors, comorbidities, and disease modifiers. Heart Fail. Rev. 2022, 27, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Beldhuis, I.E.; Lam, C.S.P.; Testani, J.M.; Voors, A.A.; Van Spall, H.G.C.; Ter Maaten, J.M.; Damman, K. Evidence-Based Medical Therapy in Patients With Heart Failure With Reduced Ejection Fraction and Chronic Kidney Disease. Circulation 2022, 145, 693–712. [Google Scholar] [CrossRef] [PubMed]
- Ishida, J.H.; Johansen, K.L. Exclusion of Patients With Kidney Disease From Cardiovascular Trials. JAMA Intern. Med. 2016, 176, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Navarro-Gonzalez, J.F.; Nunez, J.; de la Espriella, R.; Cobo, M.; Santamaria, R.; de Sequera, P.; Diez, J. The unmet need of evidence-based therapy for patients with advanced chronic kidney disease and heart failure: Position paper from the Cardiorenal Working Groups of the Spanish Society of Nephrology and the Spanish Society of Cardiology. Clin. Kidney J. 2022, 15, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; de Jong, P.E.; Coresh, J.; El Nahas, M.; Astor, B.C.; Matsushita, K.; Gansevoort, R.T.; Kasiske, B.L.; Eckardt, K.U. The definition, classification, and prognosis of chronic kidney disease: A KDIGO Controversies Conference report. Kidney Int. 2011, 80, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Delgado, C.; Baweja, M.; Crews, D.C.; Eneanya, N.D.; Gadegbeku, C.A.; Inker, L.A.; Mendu, M.L.; Miller, W.G.; Moxey-Mims, M.M.; Roberts, G.V.; et al. A Unifying Approach for GFR Estimation: Recommendations of the NKF-ASN Task Force on Reassessing the Inclusion of Race in Diagnosing Kidney Disease. Am. J. Kidney Dis. 2022, 79, 268–288.e1. [Google Scholar] [CrossRef]
- Cockcroft, D.W.; Gault, M.H. Prediction of creatinine clearance from serum creatinine. Nephron 1976, 16, 31–41. [Google Scholar] [CrossRef]
- Doogue, M.P.; Polasek, T.M. Drug dosing in renal disease. Clin. Biochem. Rev. 2011, 32, 69–73. [Google Scholar]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Sileanu, F.E.; Bihorac, A.; Hoste, E.A.; Chawla, L.S. Recovery after Acute Kidney Injury. Am. J. Respir. Crit. Care Med. 2017, 195, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; O’Donoghue, D.J.; de Lusignan, S.; Van Vlymen, J.; Klebe, B.; Middleton, R.; Hague, N.; New, J.; Farmer, C.K. Chronic kidney disease management in the United Kingdom: NEOERICA project results. Kidney Int. 2007, 72, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M.; Alberta Kidney Disease, N. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, A.; Russell, S.D.; Loehr, L.R.; Crainiceanu, C.M.; Rosamond, W.D.; Chang, P.P.; Chambless, L.E.; Coresh, J. Reduced kidney function as a risk factor for incident heart failure: The atherosclerosis risk in communities (ARIC) study. J. Am. Soc. Nephrol. 2007, 18, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Waheed, S.; Matsushita, K.; Sang, Y.; Hoogeveen, R.; Ballantyne, C.; Coresh, J.; Astor, B.C. Combined association of albuminuria and cystatin C-based estimated GFR with mortality, coronary heart disease, and heart failure outcomes: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Kidney Dis. 2012, 60, 207–216. [Google Scholar] [CrossRef]
- House, A.A.; Wanner, C.; Sarnak, M.J.; Pina, I.L.; McIntyre, C.W.; Komenda, P.; Kasiske, B.L.; Deswal, A.; de Filippi, C.R.; Cleland, J.G.F.; et al. Heart failure in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019, 95, 1304–1317. [Google Scholar] [CrossRef]
- Damman, K.; Testani, J.M. The kidney in heart failure: An update. Eur. Heart J. 2015, 36, 1437–1444. [Google Scholar] [CrossRef]
- Boudoulas, K.D.; Triposkiadis, F.; Parissis, J.; Butler, J.; Boudoulas, H. The Cardio-Renal Interrelationship. Prog. Cardiovasc. Dis. 2017, 59, 636–648. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Schefold, J.C.; Filippatos, G.; Hasenfuss, G.; Anker, S.D.; von Haehling, S. Heart failure and kidney dysfunction: Epidemiology, mechanisms and management. Nat. Rev. Nephrol. 2016, 12, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Boorsma, E.M.; Ter Maaten, J.M.; Voors, A.A.; van Veldhuisen, D.J. Renal Compression in Heart Failure: The Renal Tamponade Hypothesis. JACC Heart Fail. 2022, 10, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Rossignol, P. Cardiorenal Syndrome Revisited. Circulation 2018, 138, 929–944. [Google Scholar] [CrossRef]
- Delgado-Valero, B.; Cachofeiro, V.; Martinez-Martinez, E. Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved. Cells 2021, 10, 1824. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef]
- Buliga-Finis, O.N.; Ouatu, A.; Tanase, D.M.; Gosav, E.M.; Seritean Isac, P.N.; Richter, P.; Rezus, C. Managing Anemia: Point of Convergence for Heart Failure and Chronic Kidney Disease? Life 2023, 13, 1311. [Google Scholar] [CrossRef]
- Romero-Gonzalez, G.; Ravassa, S.; Gonzalez, O.; Lorenzo, I.; Rojas, M.A.; Garcia-Trigo, I.; Garcia-Fernandez, N.; Lavilla, J.; Martin, P.L.; Lopez, B.; et al. Burden and challenges of heart failure in patients with chronic kidney disease. A call to action. Nefrología (Engl. Ed.) 2020, 40, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Pugh, D.; Gallacher, P.J.; Dhaun, N. Management of Hypertension in Chronic Kidney Disease. Drugs 2019, 79, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Bangalore, S.; Messerli, F.H.; Kostis, J.B.; Pepine, C.J. Cardiovascular protection using beta-blockers: A critical review of the evidence. J. Am. Coll. Cardiol. 2007, 50, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, D.; Jovovic, D.; Mihailovic-Stanojevic, N.; Miloradovic, Z.; Dimitrijevic, J.; Maksic, N.; Djukanovic, L. Influence of carvedilol on chronic renal failure progression in spontaneously hypertensive rats with adriamycin nephropathy. Clin. Nephrol. 2005, 63, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Salplachta, J.; Bartosikova, L.; Necas, J. Effects of carvedilol and BL-443 on kidney of rats with cyclosporine nephropathy. Gen. Physiol. Biophys. 2002, 21, 189–195. [Google Scholar] [PubMed]
- Cice, G.; Ferrara, L.; D’Andrea, A.; D’Isa, S.; Di Benedetto, A.; Cittadini, A.; Russo, P.E.; Golino, P.; Calabro, R. Carvedilol increases two-year survivalin dialysis patients with dilated cardiomyopathy: A prospective, placebo-controlled trial. J. Am. Coll. Cardiol. 2003, 41, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Herzog, C.A.; Collins, A.J.; United States Renal Data, S. Blood pressure and long-term mortality in United States hemodialysis patients: USRDS Waves 3 and 4 Study. Kidney Int. 2002, 62, 1784–1790. [Google Scholar] [CrossRef]
- Abbott, K.C.; Trespalacios, F.C.; Agodoa, L.Y.; Taylor, A.J.; Bakris, G.L. beta-Blocker use in long-term dialysis patients: Association with hospitalized heart failure and mortality. Arch. Intern. Med. 2004, 164, 2465–2471. [Google Scholar] [CrossRef]
- Bakris, G.L. Role for beta-blockers in the management of diabetic kidney disease. Am. J. Hypertens. 2003, 16, 7S–12S. [Google Scholar] [CrossRef][Green Version]
- Wright, R.S.; Reeder, G.S.; Herzog, C.A.; Albright, R.C.; Williams, B.A.; Dvorak, D.L.; Miller, W.L.; Murphy, J.G.; Kopecky, S.L.; Jaffe, A.S. Acute myocardial infarction and renal dysfunction: A high-risk combination. Ann. Intern. Med. 2002, 137, 563–570. [Google Scholar] [CrossRef]
- Bakris, G.L.; Hart, P.; Ritz, E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006, 70, 1905–1913. [Google Scholar] [CrossRef]
- Neves, D.V.; Lanchote, V.L.; Moyses Neto, M.; Cardeal da Costa, J.A.; Vieira, C.P.; Coelho, E.B. Influence of chronic kidney disease and haemodialysis treatment on pharmacokinetics of nebivolol enantiomers. Br. J. Clin. Pharmacol. 2016, 82, 83–91. [Google Scholar] [CrossRef]
- Apperloo, A.J.; de Zeeuw, D.; Sluiter, H.E.; de Jong, P.E. Differential effects of enalapril and atenolol on proteinuria and renal haemodynamics in non-diabetic renal disease. BMJ 1991, 303, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Hannedouche, T.; Landais, P.; Goldfarb, B.; el Esper, N.; Fournier, A.; Godin, M.; Durand, D.; Chanard, J.; Mignon, F.; Suo, J.M.; et al. Randomised controlled trial of enalapril and beta blockers in non-diabetic chronic renal failure. BMJ 1994, 309, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.T., Jr.; Bakris, G.; Greene, T.; Agodoa, L.Y.; Appel, L.J.; Charleston, J.; Cheek, D.; Douglas-Baltimore, J.G.; Gassman, J.; Glassock, R.; et al. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: Results from the AASK trial. JAMA 2002, 288, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Molnar, A.O.; Petrcich, W.; Weir, M.A.; Garg, A.X.; Walsh, M.; Sood, M.M. The association of beta-blocker use with mortality in elderly patients with congestive heart failure and advanced chronic kidney disease. Nephrol. Dial. Transplant. 2020, 35, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Fu, E.L.; Uijl, A.; Dekker, F.W.; Lund, L.H.; Savarese, G.; Carrero, J.J. Association between beta-Blocker Use and Mortality/Morbidity in Patients with Heart Failure with Reduced, Midrange, and Preserved Ejection Fraction and Advanced Chronic Kidney Disease. Circ. Heart Fail. 2020, 13, e007180. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S.; et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef]
- Remuzzi, G.; Chiurchiu, C.; Ruggenenti, P. Proteinuria predicting outcome in renal disease: Nondiabetic nephropathies (REIN). Kidney Int. Suppl. 2004, 66, S90–S96. [Google Scholar] [CrossRef]
- Qiao, Y.; Shin, J.I.; Sang, Y.; Inker, L.A.; Secora, A.; Luo, S.; Coresh, J.; Alexander, G.C.; Jackson, J.W.; Chang, A.R.; et al. Discontinuation of Angiotensin Converting Enzyme Inhibitors and Angiotensin Receptor Blockers in Chronic Kidney Disease. Mayo Clin. Proc. 2019, 94, 2220–2229. [Google Scholar] [CrossRef]
- Bhandari, S.; Mehta, S.; Khwaja, A.; Cleland, J.G.F.; Ives, N.; Brettell, E.; Chadburn, M.; Cockwell, P.; Investigators, S.A.T. Renin-Angiotensin System Inhibition in Advanced Chronic Kidney Disease. N. Engl. J. Med. 2022, 387, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Bernstein, K.; Celermajer, D.; Cohen, J.; Dahlof, B.; Deanfield, J.; Diez, J.; Drexler, H.; Ferrari, R.; van Gilst, W.; et al. The relevance of tissue angiotensin-converting enzyme: Manifestations in mechanistic and endpoint data. Am. J. Cardiol. 2001, 88, 1L–20L. [Google Scholar] [CrossRef]
- Levitt, D.G.; Schoemaker, R.C. Human physiologically based pharmacokinetic model for ACE inhibitors: Ramipril and ramiprilat. BMC Clin. Pharmacol. 2006, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, D.; Zhang, W.; Xing, Y.; Guo, Y.; Wang, F.; Jia, J.; Yan, T.; Liu, Y.; Lin, S. ACE Inhibitor Benefit to Kidney and Cardiovascular Outcomes for Patients with Non-Dialysis Chronic Kidney Disease Stages 3–5: A Network Meta-Analysis of Randomised Clinical Trials. Drugs 2020, 80, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Bowling, C.B.; Sanders, P.W.; Allman, R.M.; Rogers, W.J.; Patel, K.; Aban, I.B.; Rich, M.W.; Pitt, B.; White, M.; Bakris, G.C.; et al. Effects of enalapril in systolic heart failure patients with and without chronic kidney disease: Insights from the SOLVD Treatment trial. Int. J. Cardiol. 2013, 167, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.H.; Claggett, B.; Liu, J.; Lam, C.S.; Jhund, P.S.; Rosano, G.M.; Swedberg, K.; Yusuf, S.; Granger, C.B.; Pfeffer, M.A.; et al. Heart failure with mid-range ejection fraction in CHARM: Characteristics, outcomes and effect of candesartan across the entire ejection fraction spectrum. Eur. J. Heart Fail. 2018, 20, 1230–1239. [Google Scholar] [CrossRef]
- Ko, D.; Azizi, P.; Koh, M.; Chong, A.; Austin, P.; Stukel, T.; Jackevicius, C. Comparative effectiveness of ACE inhibitors and angiotensin receptor blockers in patients with prior myocardial infarction. Open Heart 2019, 6, e001010. [Google Scholar] [CrossRef]
- Alshahrani, S. Renin-angiotensin-aldosterone pathway modulators in chronic kidney disease: A comparative review. Front. Pharmacol. 2023, 14, 1101068. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Girerd, S.; Jaisser, F. Mineralocorticoid receptor antagonists and kidney diseases: Pathophysiological basis. Kidney Int. 2019, 96, 302–319. [Google Scholar] [CrossRef]
- Bauersachs, J.; Jaisser, F.; Toto, R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension 2015, 65, 257–263. [Google Scholar] [CrossRef]
- Agarwal, R.; Kolkhof, P.; Bakris, G.; Bauersachs, J.; Haller, H.; Wada, T.; Zannad, F. Steroidal and non-steroidal mineralocorticoid receptor antagonists in cardiorenal medicine. Eur. Heart J. 2021, 42, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M. Aldosterone breakthrough from a pharmacological perspective. Hypertens. Res. 2022, 45, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Horita, Y.; Taura, K.; Taguchi, T.; Furusu, A.; Kohno, S. Aldosterone breakthrough during therapy with angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers in proteinuric patients with immunoglobulin A nephropathy. Nephrology 2006, 11, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Saruta, T. Aldosterone breakthrough during angiotensin-converting enzyme inhibitor therapy. Am. J. Hypertens. 2003, 16, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Mori, Y.; Kageyama, S.; Arihara, K.; Sugiyama, T.; Ohmura, H.; Yakushigawa, T.; Sugiyama, H.; Shimada, Y.; Nojima, Y.; et al. Spironolactone reduces cardiovascular and cerebrovascular morbidity and mortality in hemodialysis patients. J. Am. Coll. Cardiol. 2014, 63, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Quach, K.; Lvtvyn, L.; Baigent, C.; Bueti, J.; Garg, A.X.; Hawley, C.; Haynes, R.; Manns, B.; Perkovic, V.; Rabbat, C.G.; et al. The Safety and Efficacy of Mineralocorticoid Receptor Antagonists in Patients Who Require Dialysis: A Systematic Review and Meta-analysis. Am. J. Kidney Dis. 2016, 68, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xie, N.; Liang, M. Aldosterone Antagonists Reduce the Risk of Cardiovascular Mortality in Dialysis Patients: A Meta-Analysis. Evid. Based Complement. Altern. Med. 2019, 2019, 1925243. [Google Scholar] [CrossRef] [PubMed]
- Charytan, D.M.; Himmelfarb, J.; Ikizler, T.A.; Raj, D.S.; Hsu, J.Y.; Landis, J.R.; Anderson, A.H.; Hung, A.M.; Mehrotra, R.; Sharma, S.; et al. Safety and cardiovascular efficacy of spironolactone in dialysis-dependent ESRD (SPin-D): A randomized, placebo-controlled, multiple dosage trial. Kidney Int. 2019, 95, 973–982. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef]
- Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B.; Group, E.-H.S. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Claggett, B.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; Gordeev, I.; et al. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial. Circulation 2015, 131, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Vardeny, O.; Wu, D.H.; Desai, A.; Rossignol, P.; Zannad, F.; Pitt, B.; Solomon, S.D.; Investigators, R. Influence of baseline and worsening renal function on efficacy of spironolactone in patients With severe heart failure: Insights from RALES (Randomized Aldactone Evaluation Study). J. Am. Coll. Cardiol. 2012, 60, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Marcath, L.A. Finerenone. Clin. Diabetes 2021, 39, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Fioretto, P.; Kovesdy, C.P.; Malyszko, J.; Pecoits-Filho, R.; Schnell, O.; Rossignol, P. Potassium management with finerenone: Practical aspects. Endocrinol. Diabetes Metab. 2022, 5, e360. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.; Agarwal, R.; Pitt, B.; Wanner, C.; Filippatos, G.; Boletis, J.; Tuttle, K.R.; Ruilope, L.M.; Rossing, P.; Toto, R.; et al. Outcomes with Finerenone in Participants with Stage 4 CKD and Type 2 Diabetes: A FIDELITY Subgroup Analysis. Clin. J. Am. Soc. Nephrol. 2023, 18, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Edelmann, F. The non-steroidal mineralocorticoid receptor antagonist finerenone and heart failure with preserved ejection fraction. Cardiovasc. Diabetol. 2023, 22, 162. [Google Scholar] [CrossRef]
- Pei, H.; Wang, W.; Zhao, D.; Wang, L.; Su, G.H.; Zhao, Z. The use of a novel non-steroidal mineralocorticoid receptor antagonist finerenone for the treatment of chronic heart failure: A systematic review and meta-analysis. Medicine 2018, 97, e0254. [Google Scholar] [CrossRef]
- Singh, J.S.S.; Burrell, L.M.; Cherif, M.; Squire, I.B.; Clark, A.L.; Lang, C.C. Sacubitril/valsartan: Beyond natriuretic peptides. Heart 2017, 103, 1569–1577. [Google Scholar] [CrossRef]
- Haynes, R.; Judge, P.K.; Staplin, N.; Herrington, W.G.; Storey, B.C.; Bethel, A.; Bowman, L.; Brunskill, N.; Cockwell, P.; Hill, M.; et al. Effects of Sacubitril/Valsartan Versus Irbesartan in Patients With Chronic Kidney Disease. Circulation 2018, 138, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Vaduganathan, M.; Claggett, B.L.; Packer, M.; Zile, M.; Swedberg, K.; Rouleau, J.; Pfeffer, M.A.; Desai, A.; Lund, L.H.; et al. Sacubitril/Valsartan Across the Spectrum of Ejection Fraction in Heart Failure. Circulation 2020, 141, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Zhang, J.; Zhang, X.; Qin, G.; Wang, K.; Deng, Z.; Fang, Y.; Chen, G. Effects of sacubitril/valsartan in patients with heart failure and chronic kidney disease: A meta-analysis. Eur. J. Pharmacol. 2020, 884, 173444. [Google Scholar] [CrossRef] [PubMed]
- Spannella, F.; Giulietti, F.; Filipponi, A.; Sarzani, R. Effect of sacubitril/valsartan on renal function: A systematic review and meta-analysis of randomized controlled trials. ESC Heart Fail. 2020, 7, 3487–3496. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.Y.; Yang, S.F.; Ou, S.M.; Wu, C.H.; Huang, P.H.; Hung, C.L.; Lin, C.C.; Li, S.Y. Sacubitril/Valsartan in Patients With Heart Failure and Concomitant End-Stage Kidney Disease. J. Am. Heart Assoc. 2022, 11, e026407. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Singh, T.; Newby, D.E.; Singh, J. Sodium-glucose co-transporter 2 inhibitor therapy: Mechanisms of action in heart failure. Heart 2021, 107, 1032–1038. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- The EMPA-KIDNEY Collaborative Group; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Bohm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Voors, A.A.; Angermann, C.E.; Teerlink, J.R.; Collins, S.P.; Kosiborod, M.; Biegus, J.; Ferreira, J.P.; Nassif, M.E.; Psotka, M.A.; Tromp, J.; et al. The SGLT2 inhibitor empagliflozin in patients hospitalized for acute heart failure: A multinational randomized trial. Nat. Med. 2022, 28, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Van der Aart-van der Beek, A.B.; de Boer, R.A.; Heerspink, H.J.L. Kidney and heart failure outcomes associated with SGLT2 inhibitor use. Nat. Rev. Nephrol. 2022, 18, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Keener, A.B. SGLT2 inhibitors breathe life into kidney-disease care. Nature 2023, 615, S2–S4. [Google Scholar] [CrossRef]
- Song, C.C.; Brown, A.; Winstead, R.; Yakubu, I.; Demehin, M.; Kumar, D.; Gupta, G. Early initiation of sodium-glucose linked transporter inhibitors (SGLT-2i) and associated metabolic and electrolyte outcomes in diabetic kidney transplant recipients. Endocrinol Diabetes Metab 2020, 4, e00185. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Bell, R.; Mandalia, R. Diuretics and the kidney. BJA Educ. 2022, 22, 216–223. [Google Scholar] [CrossRef]
- Ellison, D.H.; Felker, G.M. Diuretic Treatment in Heart Failure. N. Engl. J. Med. 2017, 377, 1964–1975. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Dauw, J.; Martens, P.; Verbrugge, F.H.; Nijst, P.; Meekers, E.; Tartaglia, K.; Chenot, F.; Moubayed, S.; Dierckx, R.; et al. Acetazolamide in Acute Decompensated Heart Failure with Volume Overload. N. Engl. J. Med. 2022, 387, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Sibbel, S.; Walker, A.G.; Colson, C.; Tentori, F.; Brunelli, S.M.; Flythe, J. Association of Continuation of Loop Diuretics at Hemodialysis Initiation with Clinical Outcomes. Clin. J. Am. Soc. Nephrol. 2019, 14, 95–102. [Google Scholar] [CrossRef]
- Ahmed, S.; Guffey, D.; Minard, C.; Workeneh, B. Efficacy of loop diuretics in the management of undocumented patients with end-stage renal disease. Am. J. Emerg. Med. 2016, 34, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Navaneethan, S.D.; Virani, S.S.; Gregg, L.P. Revisiting diuretic choice in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2022, 31, 406–413. [Google Scholar] [CrossRef]
- Damman, K.; Tang, W.H.; Testani, J.M.; McMurray, J.J. Terminology and definition of changes renal function in heart failure. Eur. Heart J. 2014, 35, 3413–3416. [Google Scholar] [CrossRef]
- Clark, H.; Krum, H.; Hopper, I. Worsening renal function during renin-angiotensin-aldosterone system inhibitor initiation and long-term outcomes in patients with left ventricular systolic dysfunction. Eur. J. Heart Fail. 2014, 16, 41–48. [Google Scholar] [CrossRef]
- Kotecha, D.; Gill, S.K.; Flather, M.D.; Holmes, J.; Packer, M.; Rosano, G.; Bohm, M.; McMurray, J.J.V.; Wikstrand, J.; Anker, S.D.; et al. Impact of Renal Impairment on Beta-Blocker Efficacy in Patients With Heart Failure. J. Am. Coll. Cardiol. 2019, 74, 2893–2904. [Google Scholar] [CrossRef]
- Mewton, N.; Girerd, N.; Boffa, J.J.; Courivaud, C.; Isnard, R.; Juillard, L.; Lamblin, N.; Legrand, M.; Logeart, D.; Mariat, C.; et al. Practical management of worsening renal function in outpatients with heart failure and reduced ejection fraction: Statement from a panel of multidisciplinary experts and the Heart Failure Working Group of the French Society of Cardiology. Arch. Cardiovasc. Dis. 2020, 113, 660–670. [Google Scholar] [CrossRef]
- Juurlink, D.N.; Mamdani, M.M.; Lee, D.S.; Kopp, A.; Austin, P.C.; Laupacis, A.; Redelmeier, D.A. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N. Engl. J. Med. 2004, 351, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.J.; Headley, S.A.; Germain, M.J. Impact of Dietary Potassium Restrictions in CKD on Clinical Outcomes: Benefits of a Plant-Based Diet. Kidney Med. 2020, 2, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, S.; Matarazzo, I.; Lembo, E.; Peccarino, L.; Annoiato, C.; Scognamiglio, M.R.; Foderini, A.; Ruotolo, C.; Franculli, A.; Capozzi, F.; et al. Chronic Hyperkaliemia in Chronic Kidney Disease: An Old Concern with New Answers. Int. J. Mol. Sci. 2022, 23, 6378. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, K.; Sanjanwala, R.; Zieroth, S. Hyperkalemia in heart failure. Curr. Opin. Cardiol. 2020, 35, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Mushiyakh, Y.; Dangaria, H.; Qavi, S.; Ali, N.; Pannone, J.; Tompkins, D. Treatment and pathogenesis of acute hyperkalemia. J. Community Hosp. Intern. Med. Perspect. 2011, 1, 7372. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Kalra, P.R.; Petrie, M.C.; Mark, P.B.; Tomlinson, L.A.; Tomson, C.R. Change in renal function associated with drug treatment in heart failure: National guidance. Heart 2019, 105, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.K.; Kapitsinou, P.; Pergola, P.E.; Kovesdy, C.P.; Jalal, D.I. Iron Deficiency in Chronic Kidney Disease: Updates on Pathophysiology, Diagnosis, and Treatment. J. Am. Soc. Nephrol. 2020, 31, 456–468. [Google Scholar] [CrossRef]
- Fishbane, S.; Spinowitz, B. Update on Anemia in ESRD and Earlier Stages of CKD: Core Curriculum 2018. Am J Kidney Dis 2018, 71, 423–435. [Google Scholar] [CrossRef]
- Wish, J.B. The KDIGO anemia guideline: Can reason triumph over regulation? Nephrol. News Issues 2012, 26, 20, 22–23. [Google Scholar]
- Chertow, G.M.; Liu, J.; Monda, K.L.; Gilbertson, D.T.; Brookhart, M.A.; Beaubrun, A.C.; Winkelmayer, W.C.; Pollock, A.; Herzog, C.A.; Ashfaq, A.; et al. Epoetin Alfa and Outcomes in Dialysis amid Regulatory and Payment Reform. J. Am. Soc. Nephrol. 2016, 27, 3129–3138. [Google Scholar] [CrossRef]
- Emrich, I.E.; Bohm, M.; Heine, G.H. Anemia and iron deficiency—Treatment options in chronic kidney disease and in chronic heart failure. Dtsch. Med. Wochenschr. 2020, 145, 1775–1780. [Google Scholar] [CrossRef]
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation 2018, 138, 80–98. [Google Scholar] [CrossRef]
- Macdougall, I.C.; White, C.; Anker, S.D.; Bhandari, S.; Farrington, K.; Kalra, P.A.; McMurray, J.J.V.; Murray, H.; Tomson, C.R.V.; Wheeler, D.C.; et al. Intravenous Iron in Patients Undergoing Maintenance Hemodialysis. N. Engl. J. Med. 2019, 380, 447–458. [Google Scholar] [CrossRef]
- Von Haehling, S.; Ebner, N.; Evertz, R.; Ponikowski, P.; Anker, S.D. Iron Deficiency in Heart Failure: An Overview. JACC Heart Fail. 2019, 7, 36–46. [Google Scholar] [CrossRef]
- Packer, M. How can sodium-glucose cotransporter 2 inhibitors stimulate erythrocytosis in patients who are iron-deficient? Implications for understanding iron homeostasis in heart failure. Eur. J. Heart Fail. 2022, 24, 2287–2296. [Google Scholar] [CrossRef]
- Packer, M. Mechanistic and Clinical Comparison of the Erythropoietic Effects of SGLT2 Inhibitors and Prolyl Hydroxylase Inhibitors in Patients With Chronic Kidney Disease and Renal Anemia. Am. J. Nephrol. 2023. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Mechanism of Action | Dose | Side Effects |
|---|---|---|---|
| Mild/Moderate Hyperkalemia (Serum K+ > 5 mEq to <6 mEq/L without ECG changes) | |||
| Sodium polystyrene sulfate (SPS): Oldest of potassium binding resins. Use limited by side effects. | SPS binds to K+ in the intestine in exchange for Na+. | SPS oral: 15 g 1–4 times daily. SPS rectal: 30–50 g every 6 h. The 30 g dose lowers K+ by ≈1 mEq/L. Action appears at 2–6 h. | Nausea Vomiting Constipation Diarrhea |
| Patiromer: K+ binder | It is nonabsorbable, binds more K+ than SPS and exchanges K+ for Ca++ and therefore is suitable for patients with heart failure. | The effect appears 4–7 h from first dose. Initial dose: 8.4 g orally once daily. Serum K+ should be monitored and dose adjusted in 8.4 mg increments at 1-week intervals depending on serum K+ level and target range. Maintenance dose: 8.4 to 25.2 mg/day. Maximum dose 25.2 g/day. -All medications should be spaced apart by 3 h from patiromer. | Hypomagnesemia Constipation Flatulence Diarrhea |
| Sodium zirconium cyclosilicate (SZC): K+ binder | Insoluble compound working throughout the gastrointestinal tract by binding K+ and exchanging it for Na+ and H+. | Initial dose: 10 g orally 3 times a day for up to 48 h, then 10 g orally once daily. Maintenance dose: 5 g every other day to 15 g once a day | Hypertension Peripheral edema Urinary tract infections |
| Severe Hyperkalemia (Serum K+ > 5 mEq/L to < 6.0 mEq with ECG changes or K+ ≥ 6.0 mEq/L (even without ECG changes)) | |||
| Calcium: Rapid response. Intravenous (IV) Ca++ salts should be administered immediately in hyperkalemic patients presenting with electrocardiographic (ECG) changes suggesting hyperkalemia. Ca++ is also indicated when K+ > 6.5 mEq/L regardless of the presence or absence of ECG changes. | Cardiomyocyte protection. Membrane stabilization with Ca++ is essential due to the cardiotoxic effects of hyperkalemia. Ca++ does not reduce the K+ level and must be combined with potassium-lowering interventions. | Calcium chloride: 0.5–1 g IV over 2–5 min. The effect appears within 1–2 min and lasts 30–60 min. Calcium gluconate: 1–3 g IV over 2–5 min. The effects appear within 5 min, and the dose can be repeated at this interval in cases with sustained, life-threatening ECG changes. | Hypotension Bradycardia |
| Insulin: Intermediate response | Intracellular shift of K+. Insulin acts on the glucose transporter type 4 promoting intracellular movement of potassium through the Na+/K+ ATPase pump. | Ten-unit bolus of regular insulin IV together with 1 ampule of 25 g dextrose to prevent the hypoglycemic effects. IV insulin lowers the serum potassium level by ≈1 mEq/L. The effect appears within 10–20 min and lasts about 4–6 h. | Hypoglycemia |
| Salbutamol: Intermediate response | Intracellular shift of K+. Salbutamol is a β2 agonist that also activates the Na+/K+ ATPase transporter on muscle and liver cells. | Amount of 10–20 mg of nebulized salbutamol will lower the K+ by 0.5 to 1.0 mEq/L. The effect appears within 15–30 min and lasts at least 2 h. | Trembling Palpitations |
| Sodium bicarbonate: Intermediate response. Only in patients with metabolic acidosis or in the setting of cardiac arrest. | Intracellular shift of K+ by serum alkalinization, and direct bicarbonate transport into muscle cells along with K+. | IV push of 50 mEq. The effect appears 15–30 min and lasts 2–6 h | Hypernatremia Volume overload |
| Furosemide: Delayed response and inconsistent effect | Elimination of K+ from the body. | Furosemide 40–80 mg IV (large doses may be needed in renal failure). The effect appears within 5–30 min and lasts 2–6 h. | Hypotension Worsening renal function |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xanthopoulos, A.; Papamichail, A.; Briasoulis, A.; Loritis, K.; Bourazana, A.; Magouliotis, D.E.; Sarafidis, P.; Stefanidis, I.; Skoularigis, J.; Triposkiadis, F. Heart Failure in Patients with Chronic Kidney Disease. J. Clin. Med. 2023, 12, 6105. https://doi.org/10.3390/jcm12186105
Xanthopoulos A, Papamichail A, Briasoulis A, Loritis K, Bourazana A, Magouliotis DE, Sarafidis P, Stefanidis I, Skoularigis J, Triposkiadis F. Heart Failure in Patients with Chronic Kidney Disease. Journal of Clinical Medicine. 2023; 12(18):6105. https://doi.org/10.3390/jcm12186105
Chicago/Turabian StyleXanthopoulos, Andrew, Adamantia Papamichail, Alexandros Briasoulis, Konstantinos Loritis, Angeliki Bourazana, Dimitrios E. Magouliotis, Pantelis Sarafidis, Ioannis Stefanidis, John Skoularigis, and Filippos Triposkiadis. 2023. "Heart Failure in Patients with Chronic Kidney Disease" Journal of Clinical Medicine 12, no. 18: 6105. https://doi.org/10.3390/jcm12186105
APA StyleXanthopoulos, A., Papamichail, A., Briasoulis, A., Loritis, K., Bourazana, A., Magouliotis, D. E., Sarafidis, P., Stefanidis, I., Skoularigis, J., & Triposkiadis, F. (2023). Heart Failure in Patients with Chronic Kidney Disease. Journal of Clinical Medicine, 12(18), 6105. https://doi.org/10.3390/jcm12186105