
Targeting Collagen Pathways as an HFpEF Therapeutic Strategy

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
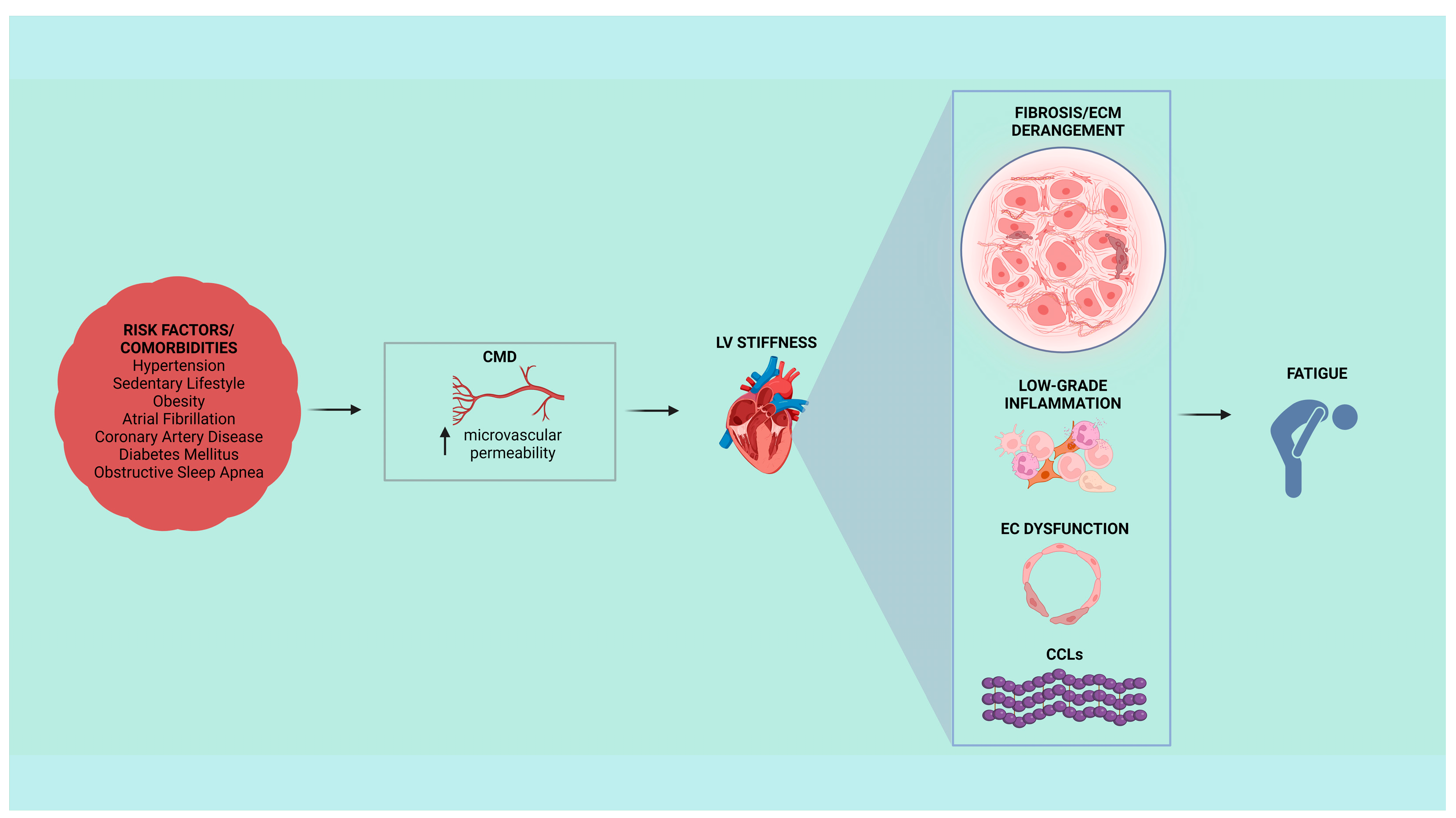
2. Endothelial Dysfunction and the HFpEF Microvascular Paradigm
3. Fibrosis and Extracellular Matrix Derangement in the Onset of HFpEF Stiffness
4. Qualitative and Quantitative Changes of Cardiac Collagen Fibers
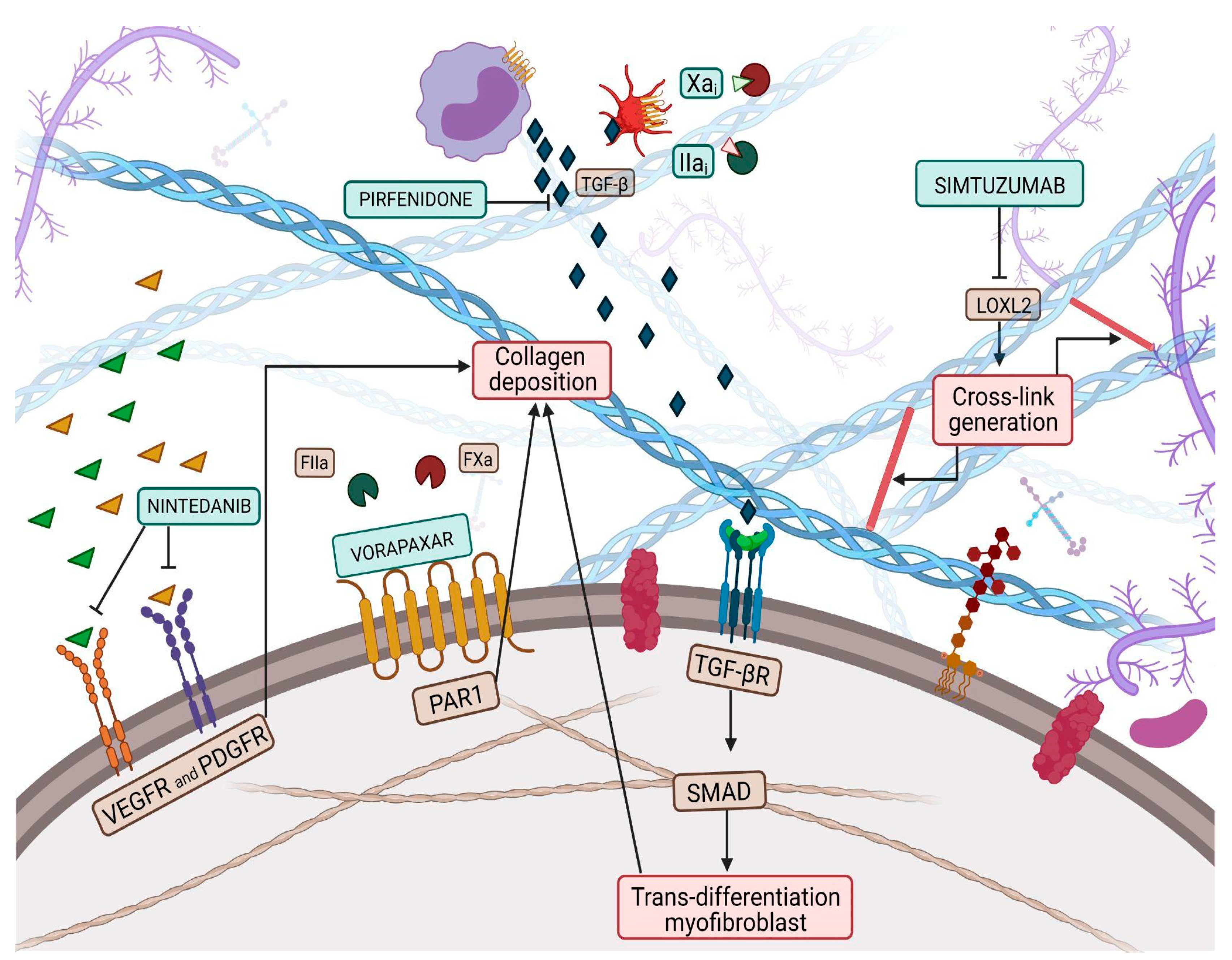
5. Myocardial Collagen Cross-Linking and Therapeutic Involvement in the Pharmacological Treatment of HFpEF
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.D.; Wei, J.; Bairey Merz, C.N. Coronary microvascular dysfunction and heart failure with preserved ejection fraction as female-pattern cardiovascular disease: The chicken or the egg? Eur. Heart J. 2018, 39, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.A.; Schelbert, E.B.; Williams, S.G.; Cunnington, C.; Ahmed, F.; McDonagh, T.A.; Miller, C.A. Biological Phenotypes of Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2017, 70, 2186–2200. [Google Scholar] [CrossRef] [PubMed]
- Del Buono, M.G.; Iannaccone, G.; Scacciavillani, R.; Carbone, S.; Camilli, M.; Niccoli, G.; Borlaug, B.A.; Lavie, C.J.; Arena, R.; Crea, F.; et al. Heart failure with preserved ejection fraction diagnosis and treatment: An updated review of the evidence. Prog. Cardiovasc. Dis. 2020, 63, 570–584. [Google Scholar] [CrossRef]
- Sunderji, I.; Singh, V.; Fraser, A.G. When does the E/e’ index not work? The pitfalls of oversimplifying diastolic function. Echocardiography 2020, 37, 1897–1907. [Google Scholar] [CrossRef]
- Pieske, B.; Tschöpe, C.; de Boer, R.A.; Fraser, A.G.; Anker, S.D.; Donal, E.; Edelmann, F.; Fu, M.; Guazzi, M.; Lam, C.S.P.; et al. How to diagnose heart failure with preserved ejection fraction: The HFA-PEFF diagnostic algorithm: A consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. Heart J. 2019, 40, 3297–3317. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef]
- Rocco, E.; Grimaldi, M.C.; Maino, A.; Cappannoli, L.; Pedicino, D.; Liuzzo, G.; Biasucci, L.M. Advances and Challenges in Biomarkers Use for Coronary Microvascular Dysfunction: From Bench to Clinical Practice. J. Clin. Med. 2022, 11, 2055. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Rodolico, D.; Hill, J.A. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 423–434. [Google Scholar] [CrossRef]
- Canonico, F.; Pedicino, D.; Severino, A.; Vinci, R.; Flego, D.; Pisano, E.; d’Aiello, A.; Ciampi, P.; Ponzo, M.; Bonanni, A.; et al. GLUT-1/PKM2 loop dysregulation in patients with non-ST-segment elevation myocardial infarction promotes metainflammation. Cardiovasc. Res. 2022, 2022, cvac184. [Google Scholar] [CrossRef]
- Rosenblatt-Velin, N.; Montessuit, C.; Papageorgiou, I.; Terrand, J.; Lerch, R. Postinfarction heart failure in rats is associated with upregulation of GLUT-1 and downregulation of genes of fatty acid metabolism. Cardiovasc. Res. 2001, 52, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Kenny, H.C.; Abel, E.D. Heart Failure in Type 2 Diabetes Mellitus. Circ. Res. 2019, 124, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- D’Amario, D.; Migliaro, S.; Borovac, J.A.; Restivo, A.; Vergallo, R.; Galli, M.; Leone, A.M.; Montone, R.A.; Niccoli, G.; Aspromonte, N.; et al. Microvascular Dysfunction in Heart Failure with Preserved Ejection Fraction. Front. Physiol. 2019, 10, 1347. [Google Scholar] [CrossRef]
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenes. Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef]
- Mohindra, R.; Agrawal, D.K.; Thankam, F.G. Altered Vascular Extracellular Matrix in the Pathogenesis of Atherosclerosis. J. Cardiovasc. Transl. Res. 2021, 14, 647–660. [Google Scholar] [CrossRef]
- Pedicino, D.; Giglio, A.F.; Ruggio, A.; Massaro, G.; D’Aiello, A.; Trotta, F.; Lucci, C.; Graziani, F.; Biasucci, L.M.; Crea, F.; et al. Inflammasome, T Lymphocytes and Innate-Adaptive Immunity Crosstalk: Role in Cardiovascular Disease and Therapeutic Perspectives. Thromb. Haemost. 2018, 118, 1352–1369. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Goshorn, D.K.; Squires, C.E.; Escobar, G.P.; Hendrick, J.W.; Mingoia, J.T.; Sweterlitsch, S.E.; Spinale, F.G. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc. Res. 2005, 66, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Bonnema, D.D.; Webb, C.S.; Pennington, W.R.; Stroud, R.E.; Leonardi, A.E.; Clark, L.L.; McClure, C.D.; Finklea, L.; Spinale, F.G.; Zile, M.R. Effects of age on plasma matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs). J. Card. Fail. 2007, 13, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Wilson, S.L.; Guilbert, M.; Sulé-Suso, J.; Torbet, J.; Jeannesson, P.; Sockalingum, G.D.; Yang, Y. A microscopic and macroscopic study of aging collagen on its molecular structure, mechanical properties, and cellular response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 14–25. [Google Scholar] [CrossRef]
- Dworatzek, E.; Mahmoodzadeh, S.; Schriever, C.; Kusumoto, K.; Kramer, L.; Santos, G.; Fliegner, D.; Leung, Y.K.; Ho, S.M.; Zimmermann, W.H.; et al. Sex-specific regulation of collagen I and III expression by 17β-Estradiol in cardiac fibroblasts: Role of estrogen receptors. Cardiovasc. Res. 2019, 115, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Schulze, C.; Wetzel, F.; Kueper, T.; Malsen, A.; Muhr, G.; Jaspers, S.; Blatt, T.; Wittern, K.P.; Wenck, H.; Käs, J.A. Stiffening of human skin fibroblasts with age. Biophys. J. 2010, 99, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Mikulíková, K.; Eckhardt, A.; Pataridis, S.; Miksík, I. Study of posttranslational non-enzymatic modifications of collagen using capillary electrophoresis/mass spectrometry and high performance liquid chromatography/mass spectrometry. J. Chromatogr. A 2007, 1155, 125–133. [Google Scholar] [CrossRef]
- Guilbert, M.; Roig, B.; Terryn, C.; Garnotel, R.; Jeannesson, P.; Sockalingum, G.D.; Manfait, M.; Perraut, F.; Dinten, J.M.; Koenig, A.; et al. Highlighting the impact of aging on type I collagen: Label-free investigation using confocal reflectance microscopy and diffuse reflectance spectroscopy in 3D matrix model. Oncotarget 2016, 7, 8546–8555. [Google Scholar] [CrossRef]
- Lovell, C.R.; Smolenski, K.A.; Duance, V.C.; Light, N.D.; Young, S.; Dyson, M. Type I and III collagen content and fibre distribution in normal human skin during ageing. Br. J. Dermatol. 1987, 117, 419–428. [Google Scholar] [CrossRef]
- Echegaray, K.; Andreu, I.; Lazkano, A.; Villanueva, I.; Sáenz, A.; Elizalde, M.R.; Echeverría, T.; López, B.; Garro, A.; González, A.; et al. Role of Myocardial Collagen in Severe Aortic Stenosis With Preserved Ejection Fraction and Symptoms of Heart Failure. Rev. Esp. Cardiol. 2017, 70, 832–840. [Google Scholar] [CrossRef]
- Collier, P.; Watson, C.J.; van Es, M.H.; Phelan, D.; McGorrian, C.; Tolan, M.; Ledwidge, M.T.; McDonald, K.M.; Baugh, J.A. Getting to the heart of cardiac remodeling; how collagen subtypes may contribute to phenotype. J. Mol. Cell. Cardiol. 2012, 52, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Graziani, F.; Varone, F.; Crea, F.; Richeldi, L. Treating heart failure with preserved ejection fraction: Learning from pulmonary fibrosis. Eur. J. Heart Fail. 2018, 20, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- López, B.; González, A.; Hermida, N.; Valencia, F.; de Teresa, E.; Díez, J. Role of lysyl oxidase in myocardial fibrosis: From basic science to clinical aspects. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1–H9. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure with Preserved Ejection Fraction Paradigm Revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef]
- Marcos-Garcés, V.; Molina Aguilar, P.; Bea Serrano, C.; García Bustos, V.; Benavent Seguí, J.; Ferrández Izquierdo, A.; Ruiz-Saurí, A. Age-related dermal collagen changes during development, maturation and ageing—A morphometric and comparative study. J. Anat. 2014, 225, 98–108. [Google Scholar] [CrossRef]
- Molnar, J.; Fong, K.S.; He, Q.P.; Hayashi, K.; Kim, Y.; Fong, S.F.; Fogelgren, B.; Szauter, K.M.; Mink, M.; Csiszar, K. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim. Biophys. Acta 2003, 1647, 220–224. [Google Scholar] [CrossRef]
- Rodríguez, C.; Martínez-González, J. The Role of Lysyl Oxidase Enzymes in Cardiac Function and Remodeling. Cells 2019, 8, 1483. [Google Scholar] [CrossRef]
- Kasner, M.; Westermann, D.; Lopez, B.; Gaub, R.; Escher, F.; Kühl, U.; Schultheiss, H.P.; Tschöpe, C. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J. Am. Coll. Cardiol. 2011, 57, 977–985. [Google Scholar] [CrossRef]
- Yang, J.; Savvatis, K.; Kang, J.S.; Fan, P.; Zhong, H.; Schwartz, K.; Barry, V.; Mikels-Vigdal, A.; Karpinski, S.; Kornyeyev, D.; et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 2016, 7, 13710. [Google Scholar] [CrossRef]
- Raghu, G.; Brown, K.K.; Collard, H.R.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Martinez, F.J.; Noble, P.W.; Song, J.W.; et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: A randomised, double-blind, controlled, phase 2 trial. Lancet. Respir. Med. 2017, 5, 22–32. [Google Scholar] [CrossRef]
- Friebel, J.; Weithauser, A.; Witkowski, M.; Rauch, B.H.; Savvatis, K.; Dörner, A.; Tabaraie, T.; Kasner, M.; Moos, V.; Bösel, D.; et al. Protease-activated receptor 2 deficiency mediates cardiac fibrosis and diastolic dysfunction. Eur. Heart J. 2019, 40, 3318–3332. [Google Scholar] [CrossRef] [PubMed]
- The TORAFIC Investigators Group. Effects of prolonged-release torasemide versus furosemide on myocardial fibrosis in hypertensive patients with chronic heart failure: A randomized, blinded-end point, active-controlled study. Clin. Ther. 2011, 33, 1204–1213.e3. [Google Scholar] [CrossRef] [PubMed]
- Gautieri, A.; Passini, F.S.; Silván, U.; Guizar-Sicairos, M.; Carimati, G.; Volpi, P.; Moretti, M.; Schoenhuber, H.; Redaelli, A.; Berli, M.; et al. Advanced glycation end-products: Mechanics of aged collagen from molecule to tissue. Matrix Biol. J. Int. Soc. Matrix Biol. 2017, 59, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M. Nonenzymatic glycosylation, the Maillard reaction and the aging process. J. Gerontol. 1990, 45, B105–B111. [Google Scholar] [CrossRef]
- Van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.J.; Schalkwijk, C.G.; Bronzwaer, J.G.; Diamant, M.; et al. Diastolic stiffness of the failing diabetic heart: Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 2008, 117, 43–51. [Google Scholar] [CrossRef]
- Hartog, J.W.; Voors, A.A.; Bakker, S.J.; Smit, A.J.; van Veldhuisen, D.J. Advanced glycation end-products (AGEs) and heart failure: Pathophysiology and clinical implications. Eur. J. Heart Fail. 2007, 9, 1146–1155. [Google Scholar] [CrossRef]
- Okamoto, T.; Yamagishi, S.; Inagaki, Y.; Amano, S.; Koga, K.; Abe, R.; Takeuchi, M.; Ohno, S.; Yoshimura, A.; Makita, Z. Angiogenesis induced by advanced glycation end products and its prevention by cerivastatin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1928–1930. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E.; Thallas, V.; Burns, W.C.; Thomas, M.C.; Brammar, G.C.; Lee, F.; Grant, S.L.; Burrell, L.M.; Jerums, G.; et al. Reduction of the accumulation of advanced glycation end products by ACE inhibition in experimental diabetic nephropathy. Diabetes 2002, 51, 3274–3282. [Google Scholar] [CrossRef]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020, 396, 819–829. [Google Scholar] [CrossRef]
- d’Aiello, A.; Bonanni, A.; Vinci, R.; Pedicino, D.; Severino, A.; De Vita, A.; Filomia, S.; Brecciaroli, M.; Liuzzo, G. Meta-Inflammation and New Anti-Diabetic Drugs: A New Chance to Knock Down Residual Cardiovascular Risk. Int. J. Mol. Sci. 2023, 24, 8643. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef]
- Solomon, S.D.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; Shah, S.J.; Lindholm, D.; et al. Dapagliflozin in heart failure with preserved and mildly reduced ejection fraction: Rationale and design of the DELIVER trial. Eur. J. Heart Fail. 2021, 23, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Ghionzoli, N.; Gentile, F.; Del Franco, A.M.; Castiglione, V.; Aimo, A.; Giannoni, A.; Burchielli, S.; Cameli, M.; Emdin, M.; Vergaro, G. Current and emerging drug targets in heart failure treatment. Heart Fail Rev. 2022, 27, 1119–1136. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Bairey Merz, C.N.; Beltrame, J.F.; Kaski, J.C.; Ogawa, H.; Ong, P.; Sechtem, U.; Shimokawa, H.; Camici, P.G. Coronary Vasomotion Disorders International Study Group (COVADIS). The parallel tales of microvascular angina and heart failure with preserved ejection fraction: A paradigm shift. Eur. Heart J. 2017, 38, 473–477. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonanni, A.; Vinci, R.; d’Aiello, A.; Grimaldi, M.C.; Di Sario, M.; Tarquini, D.; Proto, L.; Severino, A.; Pedicino, D.; Liuzzo, G. Targeting Collagen Pathways as an HFpEF Therapeutic Strategy. J. Clin. Med. 2023, 12, 5862. https://doi.org/10.3390/jcm12185862
Bonanni A, Vinci R, d’Aiello A, Grimaldi MC, Di Sario M, Tarquini D, Proto L, Severino A, Pedicino D, Liuzzo G. Targeting Collagen Pathways as an HFpEF Therapeutic Strategy. Journal of Clinical Medicine. 2023; 12(18):5862. https://doi.org/10.3390/jcm12185862
Chicago/Turabian StyleBonanni, Alice, Ramona Vinci, Alessia d’Aiello, Maria Chiara Grimaldi, Marianna Di Sario, Dalila Tarquini, Luca Proto, Anna Severino, Daniela Pedicino, and Giovanna Liuzzo. 2023. "Targeting Collagen Pathways as an HFpEF Therapeutic Strategy" Journal of Clinical Medicine 12, no. 18: 5862. https://doi.org/10.3390/jcm12185862
APA StyleBonanni, A., Vinci, R., d’Aiello, A., Grimaldi, M. C., Di Sario, M., Tarquini, D., Proto, L., Severino, A., Pedicino, D., & Liuzzo, G. (2023). Targeting Collagen Pathways as an HFpEF Therapeutic Strategy. Journal of Clinical Medicine, 12(18), 5862. https://doi.org/10.3390/jcm12185862