Hypertensive Heart Disease: A Narrative Review Series—Part 2: Macrostructural and Functional Abnormalities



Abstract
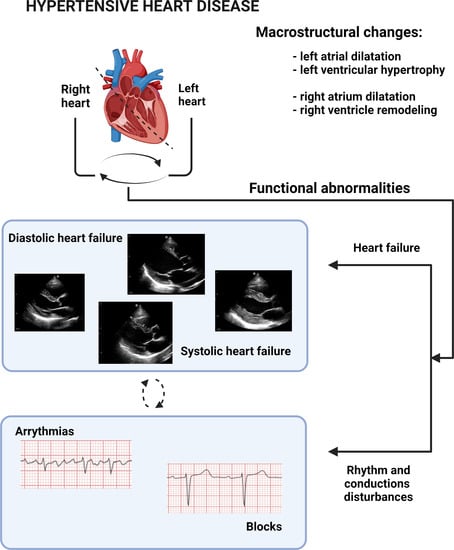
1. Introduction
2. Methods
3. Review
3.1. Left Atrium
3.1.1. Physiological Significance of the Left Atrium
3.1.2. Hypertension-Mediated Left Atrial Remodelling
3.1.3. Clinical Significance of Left Atrium Dysfunction
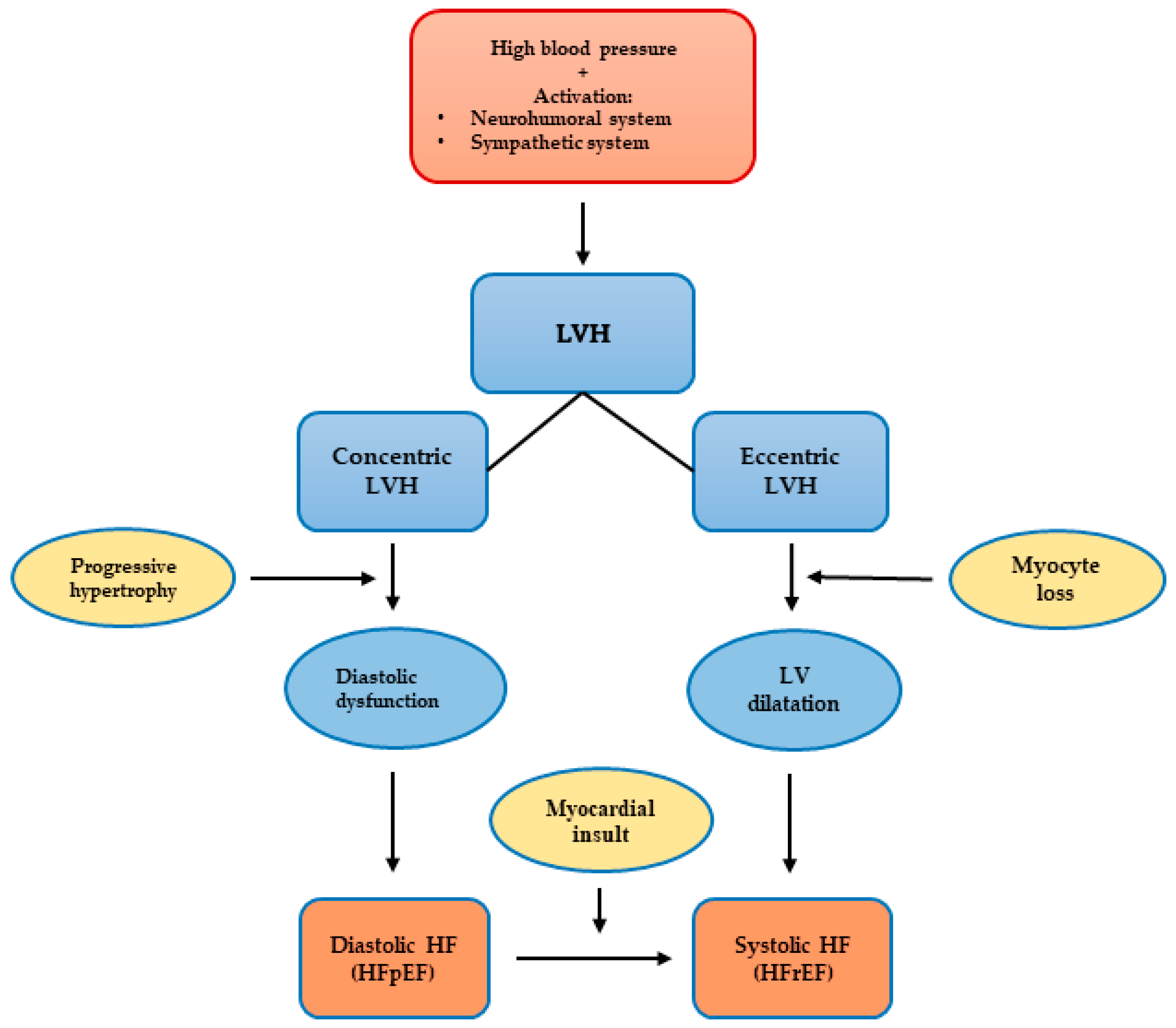
3.2. Left Ventricular Hypertrophy
3.2.1. Left Ventricular Hypertrophy Classification and Patterns
3.2.2. Clinical Significance of Hypertensive Left Ventricular Hypertrophy
3.2.3. Left Ventricular Hypertrophy as a Risk Factor for Cardiovascular Events
3.2.4. Left Ventricular Hypertrophy and Arrhythmias
3.2.5. Left Ventricular Hypertrophy and Heart Failure
3.2.6. Left Ventricular Hypertrophy as Therapeutic Target
3.3. Heart Failure
3.3.1. Heart Failure Definition and Classification
3.3.2. Heart Failure and Hypertensive Heart Disease
3.3.3. First-Line Antihypertensive Drugs and Heart Failure in the Setting of Hypertensive Heart Disease
3.4. Right Heart (Right Atrium and Ventricle)
3.4.1. Involvement of Right Heart in Hypertensive Heart Disease
3.4.2. Right Heart Adaptions as a Predictor of Cardiovascular Outcomes
3.4.3. First-Line Antihypertensive Drugs and Right Heart in the Setting of Hypertensive Heart Disease
3.5. Arrhythmias and Conductions Disturbances
3.5.1. Pathophysiological Substrate for Arrhythmogenic and Conduction Disturbances in Hypertensive Heart Disease
3.5.2. Hypertensive Heart Disease and Atrial Fibrillation
3.5.3. Other Arrhythmias and Conductions Disturbances and Hypertensive Heart Disease
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Kontaridis, M.I.; Geladari, E.V.; Geladari, C.V. Structural Alterations in the Hypertensive Heart Disease Result in Intercalated Disc Remodeling and Arrhythmias. In Hypertension and Cardiovascular Disease; Andreadis, E., Ed.; Springer: Cham, Switzerland, 2016; pp. 97–120. [Google Scholar]
- Vischer, A.S.; Burkard, T. How Should We Measure and Deal with Office Blood Pressure in 2021? Diagnostics 2021, 11, 235. [Google Scholar] [CrossRef]
- Saheera, S.; Krishnamurthy, P. Cardiovascular Changes Associated with Hypertensive Heart Disease and Aging. Cell Transplant. 2020, 29, 963689720920830. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, 1269–1324. [Google Scholar] [CrossRef]
- Nemtsova, V.; Vischer, A.S.; Burkard, T. Hypertensive Heart Disease: A Narrative Review Series-Part 1: Pathophysiology and Microstructural Changes. J. Clin. Med. 2023, 12, 2606. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 16, 233–271. [Google Scholar] [CrossRef]
- Blume, G.G.; McLeod, C.J.; Barnes, M.E.; Seward, J.B.; Pellikka, P.A.; Bastiansen, P.M.; Tsang, T.S. Left atrial function: Physiology, assessment, and clinical implications. Eur. J. Echocardiogr. 2011, 12, 421–430. [Google Scholar] [CrossRef]
- Kockskamper, J.; Pluteanu, F. Left Atrial Myocardium in Arterial Hypertension. Cells 2022, 11, 3157. [Google Scholar] [CrossRef]
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Europace 2016, 18, 1455–1490. [Google Scholar] [CrossRef]
- Shanks, J.; Manou-Stathopoulou, S.; Lu, C.J.; Li, D.; Paterson, D.J.; Herring, N. Cardiac sympathetic dysfunction in the prehypertensive spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H980–H986. [Google Scholar] [CrossRef] [PubMed]
- Cameli, M.; Mandoli, G.E.; Lisi, E.; Ibrahim, A.; Incampo, E.; Buccoliero, G.; Rizzo, C.; Devito, F.; Ciccone, M.M.; Mondillo, S. Left atrial, ventricular and atrio-ventricular strain in patients with subclinical heart dysfunction. Int. J. Cardiovasc. Imaging 2019, 35, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Kockskamper, J.; von Lewinski, D.; Khafaga, M.; Elgner, A.; Grimm, M.; Eschenhagen, T.; Gottlieb, P.A.; Sachs, F.; Pieske, B. The slow force response to stretch in atrial and ventricular myocardium from human heart: Functional relevance and subcellular mechanisms. Prog. Biophys. Mol. Biol. 2008, 97, 250–267. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Breyer, T.; Dragu, A.; Wakili, R.; Burkard, T.; Schmidt-Schweda, S.; Fuchtbauer, E.M.; Dohrmann, U.; Beyersdorf, F.; Radicke, D.; et al. Gene expression of brain natriuretic peptide in isolated atrial and ventricular human myocardium: Influence of angiotensin II and diastolic fiber length. Circulation 2000, 102, 3074–3079. [Google Scholar] [CrossRef]
- Vaziri, S.M.; Larson, M.G.; Lauer, M.S.; Benjamin, E.J.; Levy, D. Influence of blood pressure on left atrial size. The Framingham Heart Study. Hypertension 1995, 25, 1155–1160. [Google Scholar] [CrossRef]
- Gerdts, E.; Oikarinen, L.; Palmieri, V.; Otterstad, J.E.; Wachtell, K.; Boman, K.; Dahlof, B.; Devereux, R.B. Correlates of left atrial size in hypertensive patients with left ventricular hypertrophy: The Losartan Intervention For Endpoint Reduction in Hypertension (LIFE) Study. Hypertension 2002, 39, 739–743. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C.; Radojkovic, J.; Rihor, B.; Kocijanic, V.; Celic, V. Masked Hypertension and Left Atrial Dysfunction: A Hidden Association. J. Clin. Hypertens. 2017, 19, 305–311. [Google Scholar] [CrossRef]
- Cameli, M.; Pastore, M.C.; Henein, M.Y.; Mondillo, S. The left atrium and the right ventricle: Two supporting chambers to the failing left ventricle. Heart Fail. Rev. 2019, 24, 661–669. [Google Scholar] [CrossRef]
- D’Andrea, A.; De Corato, G.; Scarafile, R.; Romano, S.; Reigler, L.; Mita, C.; Allocca, F.; Limongelli, G.; Gigantino, G.; Liccardo, B.; et al. Left atrial myocardial function in either physiological or pathological left ventricular hypertrophy: A two-dimensional speckle strain study. Br. J. Sports Med. 2008, 42, 696–702. [Google Scholar] [CrossRef]
- Bode, D.; Lindner, D.; Schwarzl, M.; Westermann, D.; Deissler, P.; Primessnig, U.; Hegemann, N.; Blatter, L.A.; van Linthout, S.; Tschope, C.; et al. The role of fibroblast—Cardiomyocyte interaction for atrial dysfunction in HFpEF and hypertensive heart disease. J. Mol. Cell Cardiol. 2019, 131, 53–65. [Google Scholar] [CrossRef]
- Lau, D.H.; Mackenzie, L.; Rajendram, A.; Psaltis, P.J.; Kelly, D.R.; Spyropoulos, P.; Zhang, Y.; Olakkengil, S.A.; Russell, C.H.; Brooks, A.G.; et al. Characterization of cardiac remodeling in a large animal “one-kidney, one-clip” hypertensive model. Blood Press. 2010, 19, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Cuspidi, C.; Sala, C.; Tadic, M.; Gherbesi, E.; Grassi, G.; Mancia, G. Pre-hypertension and subclinical cardiac damage: A meta-analysis of echocardiographic studies. Int. J. Cardiol. 2018, 270, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, E.; Wachtell, K.; Omvik, P.; Otterstad, J.E.; Oikarinen, L.; Boman, K.; Dahlof, B.; Devereux, R.B. Left atrial size and risk of major cardiovascular events during antihypertensive treatment: Losartan intervention for endpoint reduction in hypertension trial. Hypertension 2007, 49, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, M.; Steel, K.; Jerosch-Herold, M.; Khin, M.; Tsang, S.; Hauser, T.; Kwong, R.Y. Strong cardiovascular prognostic implication of quantitative left atrial contractile function assessed by cardiac magnetic resonance imaging in patients with chronic hypertension. J. Cardiovasc. Magn. Reson. 2011, 13, 42. [Google Scholar] [CrossRef]
- Di Tullio, M.R.; Qian, M.; Thompson, J.L.P.; Labovitz, A.J.; Mann, D.L.; Sacco, R.L.; Pullicino, P.M.; Freudenberger, R.S.; Teerlink, J.R.; Graham, S.; et al. Left atrial volume and cardiovascular outcomes in systolic heart failure: Effect of antithrombotic treatment. ESC Heart Fail. 2018, 5, 800–808. [Google Scholar] [CrossRef]
- Pellicori, P.; Zhang, J.; Lukaschuk, E.; Joseph, A.C.; Bourantas, C.V.; Loh, H.; Bragadeesh, T.; Clark, A.L.; Cleland, J.G. Left atrial function measured by cardiac magnetic resonance imaging in patients with heart failure: Clinical associations and prognostic value. Eur. Heart J. 2015, 36, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.M.; Lam, C.S. Function over form? Assessing the left atrium in heart failure. Eur. Heart J. 2015, 36, 711–714. [Google Scholar] [CrossRef]
- Buggey, J.; Hoit, B.D. Left atrial strain. Curr. Opin. Cardiol. 2018, 33, 479–485. [Google Scholar] [CrossRef]
- Okin, P.M.; Kamel, H.; Kjeldsen, S.E.; Devereux, R.B. Electrocardiographic left atrial abnormalities and risk of incident stroke in hypertensive patients with electrocardiographic left ventricular hypertrophy. J. Hypertens. 2016, 34, 1831–1837. [Google Scholar] [CrossRef]
- Parikh, A.; Patel, D.; McTiernan, C.F.; Xiang, W.; Haney, J.; Yang, L.; Lin, B.; Kaplan, A.D.; Bett, G.C.; Rasmusson, R.L.; et al. Relaxin suppresses atrial fibrillation by reversing fibrosis and myocyte hypertrophy and increasing conduction velocity and sodium current in spontaneously hypertensive rat hearts. Circ. Res. 2013, 113, 313–321. [Google Scholar] [CrossRef]
- Lieb, W.; Gona, P.; Larson, M.G.; Aragam, J.; Zile, M.R.; Cheng, S.; Benjamin, E.J.; Vasan, R.S. The natural history of left ventricular geometry in the community: Clinical correlates and prognostic significance of change in LV geometric pattern. JACC Cardiovasc. Imaging 2014, 7, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.F.; Frey, S.; Kaufmann, B.A.; Winkel, D.J.; Boll, D.T.; Zellweger, M.J.; Haaf, P. Hypertensive Heart Disease—The Imaging Perspective. J. Clin. Med. 2023, 12, 3122. [Google Scholar] [CrossRef] [PubMed]
- Hoey, E.T.; Pakala, V.; Teoh, J.K.; Simpson, H. The role of imaging in hypertensive heart disease. Int. J. Angiol. 2014, 23, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Devereux, R.B.; Pickering, T.G.; Alderman, M.H.; Chien, S.; Borer, J.S.; Laragh, J.H. Left-Ventricular Hypertrophy in Hypertension—Prevalence and Relationship to Pathophysiologic Variables. Hypertension 1987, 9, 53–60. [Google Scholar] [CrossRef]
- Kahan, T.; Bergfeldt, L. Left ventricular hypertrophy in hypertension: Its arrhythmogenic potential. Heart 2005, 91, 250–256. [Google Scholar] [CrossRef]
- Cuspidi, C.; Sala, C.; Negri, F.; Mancia, G.; Morganti, A.; Italian Society of Hypertension. Prevalence of left-ventricular hypertrophy in hypertension: An updated review of echocardiographic studies. J. Hum. Hypertens. 2012, 26, 343–349. [Google Scholar] [CrossRef]
- Ganau, A.; Devereux, R.B.; Roman, M.J.; de Simone, G.; Pickering, T.G.; Saba, P.S.; Vargiu, P.; Simongini, I.; Laragh, J.H. Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J. Am. Coll. Cardiol. 1992, 19, 1550–1558. [Google Scholar] [CrossRef]
- Barbieri, A.; Albini, A.; Maisano, A.; De Mitri, G.; Camaioni, G.; Bonini, N.; Mantovani, F.; Boriani, G. Clinical Value of Complex Echocardiographic Left Ventricular Hypertrophy Classification Based on Concentricity, Mass, and Volume Quantification. Front. Cardiovasc. Med. 2021, 8, 667984. [Google Scholar] [CrossRef]
- Lang, R.M.; Bierig, M.; Devereux, R.B.; Flachskampf, F.A.; Foster, E.; Pellikka, P.A.; Picard, M.H.; Roman, M.J.; Seward, J.; Shanewise, J.S.; et al. Recommendations for chamber quantification: A report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J. Am. Soc. Echocardiogr. 2005, 18, 1440–1463. [Google Scholar] [CrossRef]
- Simone, G.d.; Devereux, R.B.; Roman, M.J.; Alderman, M.H.; Laragh, J.H. Relation of obesity and gender to left ventricular hypertrophy in normotensive and hypertensive adults. Hypertension 1994, 23, 600–606. [Google Scholar] [CrossRef]
- Cuspidi, C.; Meani, S.; Negri, F.; Giudici, V.; Valerio, C.; Sala, C.; Zanchetti, A.; Mancia, G. Indexation of left ventricular mass to body surface area and height to allometric power of 2.7: Is the difference limited to obese hypertensives? J. Hum. Hypertens. 2009, 23, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Drazner, M.H. The progression of hypertensive heart disease. Circulation 2011, 123, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Gaasch, W.H.; Zile, M.R. Left ventricular structural remodeling in health and disease: With special emphasis on volume, mass, and geometry. J. Am. Coll. Cardiol. 2011, 58, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Drazner, M.H. Refining the classification of left ventricular hypertrophy to provide new insights into the progression from hypertension to heart failure. Curr. Opin. Cardiol. 2016, 31, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Patel, D.A.; Milani, R.V.; Ventura, H.O.; Shah, S.; Gilliland, Y. Impact of echocardiographic left ventricular geometry on clinical prognosis. Prog. Cardiovasc. Dis. 2014, 57, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, S.; Katsi, V.; Vartela, V.; Noutsias, M.; Markousis-Mavrogenis, G.; Kolovou, G.; Manolis, A. The emerging role of Cardiovascular Magnetic Resonance in the evaluation of hypertensive heart disease. BMC Cardiovasc. Disord. 2017, 17, 132. [Google Scholar] [CrossRef]
- Keller, K.; Hartung, K.; Del Castillo Carillo, L.; Treiber, J.; Stock, F.; Schroder, C.; Hugenschmidt, F.; Friedmann-Bette, B. Exercise Hypertension in Athletes. J. Clin. Med. 2022, 11, 4870. [Google Scholar] [CrossRef]
- Slivnick, J.; Lampert, B.C. Hypertension and Heart Failure. Heart Fail. Clin. 2019, 15, 531–541. [Google Scholar] [CrossRef]
- D’Ascenzi, F.; Fiorentini, C.; Anselmi, F.; Mondillo, S. Left ventricular hypertrophy in athletes: How to differentiate between hypertensive heart disease and athlete’s heart. Eur. J. Prev. Cardiol. 2020, 28, 1125–1133. [Google Scholar] [CrossRef]
- Yalcin, F.; Kucukler, N.; Cingolani, O.; Mbiyangandu, B.; Sorensen, L.; Pinherio, A.; Abraham, M.R.; Abraham, T.P. Evolution of ventricular hypertrophy and myocardial mechanics in physiological and pathological hypertrophy. J. Appl. Physiol. 2019, 126, 354–362. [Google Scholar] [CrossRef]
- Yalcin, F.; Yalcin, H.; Abraham, M.R.; Abraham, T.P. Ultimate phases of hypertensive heart disease and stressed heart morphology by conventional and novel cardiac imaging. Am. J. Cardiovasc. Dis. 2021, 11, 628–634. [Google Scholar] [PubMed]
- Yalcin, F.; Topaloglu, C.; Kucukler, N.; Ofgeli, M.; Abraham, T.P. Could early septal involvement in the remodeling process be related to the advance hypertensive heart disease? Int. J. Cardiol. Heart Vasc. 2015, 7, 141–145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paoletti, E.; De Nicola, L.; Gabbai, F.B.; Chiodini, P.; Ravera, M.; Pieracci, L.; Marre, S.; Cassottana, P.; Luca, S.; Vettoretti, S.; et al. Associations of Left Ventricular Hypertrophy and Geometry with Adverse Outcomes in Patients with CKD and Hypertension. Clin. J. Am. Soc. Nephrol. 2016, 11, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Shah, A.M. Alterations in cardiac structure and function in hypertension. Curr. Hypertens. Rep. 2014, 16, 428. [Google Scholar] [CrossRef] [PubMed]
- Nunez, D.J.; Clifford, C.P.; al-Mahdawi, S.; Dutka, D. Hypertensive cardiac hypertrophy--is genetic variance the missing link? Br. J. Clin. Pharmacol. 1996, 42, 107–117. [Google Scholar] [CrossRef]
- Do, A.N.; Zhao, W.; Baldridge, A.S.; Raffield, L.M.; Wiggins, K.L.; Shah, S.J.; Aslibekyan, S.; Tiwari, H.K.; Limdi, N.; Zhi, D.; et al. Genome-wide meta-analysis of SNP and antihypertensive medication interactions on left ventricular traits in African Americans. Mol. Genet. Genom. Med. 2019, 7, e00788. [Google Scholar] [CrossRef]
- Drazner, M.H.; Dries, D.L.; Peshock, R.M.; Cooper, R.S.; Klassen, C.; Kazi, F.; Willett, D.; Victor, R.G. Left ventricular hypertrophy is more prevalent in blacks than whites in the general population: The Dallas Heart Study. Hypertension 2005, 46, 124–129. [Google Scholar] [CrossRef]
- Lewis, A.A.; Ayers, C.R.; Selvin, E.; Neeland, I.; Ballantyne, C.M.; Nambi, V.; Pandey, A.; Powell-Wiley, T.M.; Drazner, M.H.; Carnethon, M.R.; et al. Racial Differences in Malignant Left Ventricular Hypertrophy and Incidence of Heart Failure: A Multicohort Study. Circulation 2020, 141, 957–967. [Google Scholar] [CrossRef]
- Garg, P.; Assadi, H.; Jones, R.; Chan, W.B.; Metherall, P.; Thomas, R.; van der Geest, R.; Swift, A.J.; Al-Mohammad, A. Left ventricular fibrosis and hypertrophy are associated with mortality in heart failure with preserved ejection fraction. Sci. Rep. 2021, 11, 617. [Google Scholar] [CrossRef]
- Frohlich, E.D. Ischemia and fibrosis: The risk mechanisms of hypertensive heart disease. Braz. J. Med. Biol. Res. 2000, 33, 693–700. [Google Scholar] [CrossRef]
- Verdecchia, P.; Carini, G.; Circo, A.; Dovellini, E.; Giovannini, E.; Lombardo, M.; Solinas, P.; Gorini, M.; Maggioni, A.P.; the MAVI Study Group. Left ventricular mass and cardiovascular morbidity in essential hypertension: The MAVI study. J. Am. Coll. Cardiol. 2001, 38, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Koracevic, G.; Perisic, Z.; Stanojkovic, M.; Stojanovic, M.; Zdravkovic, M.; Tomasevic, M.; Djordjevic, D.; Mladenovic, K.; Koracevic, M.; Trkulja, J. A Discrepancy: Calcium Channel Blockers Are Effective for the Treatment of Hypertensive Left Ventricular Hypertrophy but Not as Effective for Prevention of Heart Failure. Med. Princ. Pract. 2022, 31, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.S.; Spears, D.A.; Care, M. Evaluation of cardiac hypertrophy in the setting of sudden cardiac death. Forensic Sci. Res. 2019, 4, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Porthan, K.; Kentta, T.; Niiranen, T.J.; Nieminen, M.S.; Oikarinen, L.; Viitasalo, M.; Hernesniemi, J.; Jula, A.M.; Salomaa, V.; Huikuri, H.V.; et al. ECG left ventricular hypertrophy as a risk predictor of sudden cardiac death. Int. J. Cardiol. 2019, 276, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Levy, D. Why is left ventricular hypertrophy so predictive of morbidity and mortality? Am. J. Med. Sci. 1999, 317, 168–175. [Google Scholar] [CrossRef]
- Movahed, M.R.; Ramaraj, R.; Manrique, C.; Hashemzadeh, M. Left ventricular hypertrophy is independently associated with all-cause mortality. Am. J. Cardiovasc. Dis. 2022, 12, 38–41. [Google Scholar] [PubMed]
- Gluba, A.; Bielecka-Dabrowa, A.; Mikhailidis, D.P.; Wong, N.D.; Franklin, S.S.; Rysz, J.; Banach, M. An update on biomarkers of heart failure in hypertensive patients. J. Hypertens. 2012, 30, 1681–1689. [Google Scholar] [CrossRef]
- Heckbert, S.R.; Post, W.; Pearson, G.D.; Arnett, D.K.; Gomes, A.S.; Jerosch-Herold, M.; Hundley, W.G.; Lima, J.A.; Bluemke, D.A. Traditional cardiovascular risk factors in relation to left ventricular mass, volume, and systolic function by cardiac magnetic resonance imaging: The Multiethnic Study of Atherosclerosis. J. Am. Coll. Cardiol. 2006, 48, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Apitz, A.; Socrates, T.; Burkard, T.; Mayr, M.; Vischer, A.S. Prevalence and Characterisation of Severe Left Ventricular Hypertrophy Diagnosed by Echocardiography in Hypertensive Patients. J. Clin. Med. 2022, 12, 228. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bavishi, C.; Sardar, P.; Agarwal, V.; Krishnamoorthy, P.; Grodzicki, T.; Messerli, F.H. Meta-analysis of left ventricular hypertrophy and sustained arrhythmias. Am. J. Cardiol. 2014, 114, 1049–1052. [Google Scholar] [CrossRef]
- Verdecchia, P.; Reboldi, G.; Gattobigio, R.; Bentivoglio, M.; Borgioni, C.; Angeli, F.; Carluccio, E.; Sardone, M.G.; Porcellati, C. Atrial Fibrillation in Hypertension. Hypertension 2003, 41, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Wachtell, K.; Lehto, M.; Gerdts, E.; Olsen, M.H.; Hornestam, B.; Dahlof, B.; Ibsen, H.; Julius, S.; Kjeldsen, S.E.; Lindholm, L.H.; et al. Angiotensin II receptor blockade reduces new-onset atrial fibrillation and subsequent stroke compared to atenolol: The Losartan Intervention For End Point Reduction in Hypertension (LIFE) study. J. Am. Coll. Cardiol. 2005, 45, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.N.; Altara, R.; Amin, G.; Habeichi, N.J.; Thomas, D.G.; Jun, S.; Kaplan, A.; Booz, G.W.; Zouein, F.A. Genomic, Proteomic, and Metabolic Comparisons of Small Animal Models of Heart Failure With Preserved Ejection Fraction: A Tale of Mice, Rats, and Cats. J. Am. Heart Assoc. 2022, 11, e026071. [Google Scholar] [CrossRef] [PubMed]
- Okin, P.M.; Devereux, R.B.; Jern, S.; Kjeldsen, S.E.; Julius, S.; Nieminen, M.S.; Snapinn, S.; Harris, K.E.; Aurup, P.; Edelman, J.M.; et al. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 2004, 292, 2343–2349. [Google Scholar] [CrossRef]
- Mathew, J.; Sleight, P.; Lonn, E.; Johnstone, D.; Pogue, J.; Yi, Q.; Bosch, J.; Sussex, B.; Probstfield, J.; Yusuf, S.; et al. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation 2001, 104, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Pierdomenico, S.D.; Cuccurullo, F. Risk reduction after regression of echocardiographic left ventricular hypertrophy in hypertension: A meta-analysis. Am. J. Hypertens. 2010, 23, 876–881. [Google Scholar] [CrossRef]
- Jekell, A.; Nilsson, P.M.; Kahan, T. Treatment of Hypertensive Left Ventricular Hypertrophy. Curr. Pharm. Des. 2018, 24, 4391–4396. [Google Scholar] [CrossRef]
- Fagard, R.H.; Celis, H.; Thijs, L.; Wouters, S. Regression of left ventricular mass by antihypertensive treatment: A meta-analysis of randomized comparative studies. Hypertension 2009, 54, 1084–1091. [Google Scholar] [CrossRef]
- Klingbeil, A.U.; Schneider, M.; Martus, P.; Messerli, F.H.; Schmieder, R.E. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am. J. Med. 2003, 115, 41–46. [Google Scholar] [CrossRef]
- Pitt, B.; Reichek, N.; Willenbrock, R.; Zannad, F.; Phillips, R.A.; Roniker, B.; Kleiman, J.; Krause, S.; Burns, D.; Williams, G.H. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: The 4E-left ventricular hypertrophy study. Circulation 2003, 108, 1831–1838. [Google Scholar] [CrossRef]
- Schmieder, R.E.; Wagner, F.; Mayr, M.; Delles, C.; Ott, C.; Keicher, C.; Hrabak-Paar, M.; Heye, T.; Aichner, S.; Khder, Y.; et al. The effect of sacubitril/valsartan compared to olmesartan on cardiovascular remodelling in subjects with essential hypertension: The results of a randomized, double-blind, active-controlled study. Eur. Heart J. 2017, 38, 3308–3317. [Google Scholar] [CrossRef] [PubMed]
- Omboni, S.; Malacco, E.; Napoli, C.; Modesti, P.A.; Manolis, A.; Parati, G.; Agabiti-Rosei, E.; Borghi, C. Efficacy of Zofenopril vs. Irbesartan in Combination with a Thiazide Diuretic in Hypertensive Patients with Multiple Risk Factors not Controlled by a Previous Monotherapy: A Review of the Double-Blind, Randomized “Z” Studies. Adv. Ther. 2017, 34, 784–798. [Google Scholar] [CrossRef] [PubMed]
- Neutel, J.M. Prescribing patterns in hypertension: The emerging role of fixed-dose combinations for attaining BP goals in hypertensive patients. Curr. Med. Res. Opin. 2008, 24, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Dahlof, B.; Gosse, P.; Gueret, P.; Dubourg, O.; de Simone, G.; Schmieder, R.; Karpov, Y.; Garcia-Puig, J.; Matos, L.; De Leeuw, P.W.; et al. Perindopril/indapamide combination more effective than enalapril in reducing blood pressure and left ventricular mass: The PICXEL study. J. Hypertens. 2005, 23, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- de Luca, N.; Mallion, J.M.; O’Rourke, M.F.; O’Brien, E.; Rahn, K.H.; Trimarco, B.; Romero, R.; De Leeuw, P.W.; Hitzenberger, G.; Battegay, E.; et al. Regression of left ventricular mass in hypertensive patients treated with perindopril/indapamide as a first-line combination: The REASON echocardiography study. Am. J. Hypertens. 2004, 17, 660–667. [Google Scholar] [CrossRef]
- Paczkowska-Walendowska, M.; Sip, S.; Staszewski, R.; Cielecka-Piontek, J. Single-Pill Combination to Improve Hypertension Treatment: Pharmaceutical Industry Development. Int. J. Environ. Res. Public Health 2022, 19, 4156. [Google Scholar] [CrossRef]
- Perrone, V.; Veronesi, C.; Gambera, M.; Nati, G.; Perone, F.; Tagliabue, P.F.; Degli Esposti, L.; Volpe, M. Treatment with Free Triple Combination Therapy of Atorvastatin, Perindopril, Amlodipine in Hypertensive Patients: A Real-World Population Study in Italy. High Blood Press. Cardiovasc. Prev. 2019, 26, 399–404. [Google Scholar] [CrossRef]
- Degli Esposti, L.; Perrone, V.; Veronesi, C.; Gambera, M.; Nati, G.; Perone, F.; Tagliabue, P.F.; Buda, S.; Borghi, C. Modifications in drug adherence after switch to fixed-dose combination of perindopril/amlodipine in clinical practice. Results of a large-scale Italian experience. The amlodipine-perindopril in real settings (AMPERES) study. Curr. Med. Res. Opin. 2018, 34, 1571–1577. [Google Scholar] [CrossRef]
- Gaciong, Z. Preference and Adherence to a Fixed-Dose Combination of Bisoprolol-Aspirin and Blood Pressure Control: Results of an Open-Label, Multicentre Study. J. Clin. Med. 2022, 12, 17. [Google Scholar] [CrossRef]
- Roman, M.J.; Pickering, T.G.; Schwartz, J.E.; Pini, R.; Devereux, R.B. Association of carotid atherosclerosis and left ventricular hypertrophy. J. Am. Coll. Cardiol. 1995, 25, 83–90. [Google Scholar] [CrossRef]
- Emmons-Bell, S.; Johnson, C.; Roth, G. Prevalence, incidence and survival of heart failure: A systematic review. Heart 2022, 108, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Coats, A.J.S.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Chioncel, O.; Lainscak, M.; Seferovic, P.M.; Anker, S.D.; Crespo-Leiro, M.G.; Harjola, V.P.; Parissis, J.; Laroche, C.; Piepoli, M.F.; Fonseca, C.; et al. Epidemiology and one-year outcomes in patients with chronic heart failure and preserved, mid-range and reduced ejection fraction: An analysis of the ESC Heart Failure Long-Term Registry. Eur. J. Heart Fail. 2017, 19, 1574–1585. [Google Scholar] [CrossRef]
- Kalogeropoulos, A.P.; Goulbourne, C.; Butler, J. Diagnosis and Prevention of Hypertensive Heart Failure. Heart Fail. Clin. 2019, 15, 435–445. [Google Scholar] [CrossRef]
- Tackling, G.; Borhade, M.B. Hypertensive Heart Disease; StatPearls: Tampa, FL, USA, 2022. [Google Scholar]
- Levy, D.; Larson, M.G.; Vasan, R.S.; Kannel, W.B.; Ho, K.K.L. The progression from hypertension to congestive heart failure. JAMA 1996, 275, 1557–1562. [Google Scholar] [CrossRef]
- Maeda, D.; Dotare, T.; Matsue, Y.; Teramoto, K.; Sunayama, T.; Tromp, J.; Minamino, T. Blood pressure in heart failure management and prevention. Hypertens. Res. 2023, 46, 817–833. [Google Scholar] [CrossRef]
- Gonzalez, A.; Ravassa, S.; Lopez, B.; Moreno, M.U.; Beaumont, J.; San Jose, G.; Querejeta, R.; Bayes-Genis, A.; Diez, J. Myocardial Remodeling in Hypertension. Hypertension 2018, 72, 549–558. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure With Preserved Ejection Fraction Paradigm Revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, J.H.; Zhen, Z.; Zuo, Y.; Lin, Q.; Liu, M.; Zhao, C.; Wu, M.; Cao, G.; Wang, R.; et al. Assessment of left ventricular function and peripheral vascular arterial stiffness in patients with dipper and non-dipper hypertension. J. Investig. Med. 2018, 66, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Shalimova, A.; Fadieienko, G.; Kolesnikova, O.; Isayeva, A.; Zlatkina, V.; Nemtsova, V.; Prosolenko, K.; Psarova, V.; Kyrychenko, N.; Kochuieva, M. The Role of Genetic Polymorphism in the Formation of Arterial Hypertension, Type 2 Diabetes and their Comorbidity. Curr. Pharm. Des. 2019, 25, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Messerli, F.H.; Rimoldi, S.F.; Bangalore, S. The Transition From Hypertension to Heart Failure: Contemporary Update. JACC Heart Fail. 2017, 5, 543–551. [Google Scholar] [CrossRef]
- Tam, M.C.; Lee, R.; Cascino, T.M.; Konerman, M.C.; Hummel, S.L. Current Perspectives on Systemic Hypertension in Heart Failure with Preserved Ejection Fraction. Curr. Hypertens. Rep. 2017, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Hill, J.A.; Singh, A.; Valero-Munoz, M.; Sam, F. Heart Failure With Preserved Ejection Fraction: Heterogeneous Syndrome, Diverse Preclinical Models. Circ. Res. 2022, 130, 1906–1925. [Google Scholar] [CrossRef]
- Seko, Y.; Kato, T.; Shiba, M.; Morita, Y.; Yamaji, Y.; Haruna, Y.; Nakane, E.; Haruna, T.; Inoko, M. Staging Cardiac Damage in Patients With Hypertension. Hypertension 2019, 74, 1357–1365. [Google Scholar] [CrossRef]
- Pavlopoulos, H.; Nihoyannopoulos, P. The constellation of hypertensive heart disease. Hell. J. Cardiol. 2008, 49, 92–99. [Google Scholar]
- Hill, J.A.; Rothermel, B.; Yoo, K.D.; Cabuay, B.; Demetroulis, E.; Weiss, R.M.; Kutschke, W.; Bassel-Duby, R.; Williams, R.S. Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy. Preservation of systolic function. J. Biol. Chem. 2002, 277, 10251–10255. [Google Scholar] [CrossRef]
- Hill, J.A.; Karimi, M.; Kutschke, W.; Davisson, R.L.; Zimmerman, K.; Wang, Z.; Kerber, R.E.; Weiss, R.M. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation 2000, 101, 2863–2869. [Google Scholar] [CrossRef]
- Velagaleti, R.S.; Gona, P.; Pencina, M.J.; Aragam, J.; Wang, T.J.; Levy, D.; D’Agostino, R.B.; Lee, D.S.; Kannel, W.B.; Benjamin, E.J.; et al. Left ventricular hypertrophy patterns and incidence of heart failure with preserved versus reduced ejection fraction. Am. J. Cardiol. 2014, 113, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Redfield, M.M. Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation 2011, 123, 2006–2013; discussion 2014. [Google Scholar] [CrossRef] [PubMed]
- Rame, J.E.; Ramilo, M.; Spencer, N.; Blewett, C.; Mehta, S.K.; Dries, D.L.; Drazner, M.H. Development of a depressed left ventricular ejection fraction in patients with left ventricular hypertrophy and a normal ejection fraction. Am. J. Cardiol. 2004, 93, 234–237. [Google Scholar] [CrossRef]
- Drazner, M.H.; Rame, J.E.; Marino, E.K.; Gottdiener, J.S.; Kitzman, D.W.; Gardin, J.M.; Manolio, T.A.; Dries, D.L.; Siscovick, D.S. Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: The Cardiovascular Health Study. J. Am. Coll. Cardiol. 2004, 43, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Black, H.R.; Elliott, W.J.; Grandits, G.; Grambsch, P.; Lucente, T.; White, W.B.; Neaton, J.D.; Grimm, R.H., Jr.; Hansson, L.; Lacourciere, Y.; et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) trial. JAMA 2003, 289, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Chaugai, S.; Sherpa, L.Y.; Sepehry, A.A.; Kerman, S.R.J.; Arima, H. Effects of Long- and Intermediate-Acting Dihydropyridine Calcium Channel Blockers in Hypertension: A Systematic Review and Meta-Analysis of 18 Prospective, Randomized, Actively Controlled Trials. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 433–445. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C. Right Ventricle in Arterial Hypertension: Did We Forget Something? J. Clin. Med. 2022, 11, 6257. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, C.E.; Ye, P.; Sheng, L.; Luo, L. Right ventricle may be involved in regional diastolic dysfunction earliest in primary hypertension patients. J. Cell. Biochem. 2019, 120, 18088–18093. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C.; Kocijancic, V.; Celic, V.; Vukomanovic, V. Does Left Ventricular Geometric Patterns Impact Right Atrial Phasic Function? Findings from the Hypertensive Population. Echocardiography 2016, 33, 1186–1194. [Google Scholar] [CrossRef]
- Liu, S.; Liao, Y.; Zhu, Z.; Wang, S.; Li, Y.; Liang, D.; Xie, Y.; Zhang, Z. Association between cumulative blood pressure in early adulthood and right ventricular structure and function in middle age: The CARDIA study. Clin. Cardiol. 2022, 45, 83–90. [Google Scholar] [CrossRef]
- Pechlivanidis, G.; Mantziari, L.; Giannakoulas, G.; Dimitroula, H.; Styliadis, H.; Karvounis, H.; Styliadis, I.H.; Parharidis, G. Effects of renin-angiotensin system inhibition on right ventricular function in patients with mild essential hypertension. J. Renin-Angiotensin-Aldosterone Syst. 2011, 12, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Borgdorff, M.A.; Dickinson, M.G.; Berger, R.M.; Bartelds, B. Right ventricular failure due to chronic pressure load: What have we learned in animal models since the NIH working group statement? Heart Fail. Rev. 2015, 20, 475–491. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tadic, M.; Cuspidi, C.; Vukomanovic, V.; Celic, V.; Pavlovic, T.; Kocijancic, V. The influence of masked hypertension on the right ventricle: Is everything really masked? J. Am. Soc. Hypertens. 2016, 10, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Xie, E.; Yu, R.; Ambale-Venkatesh, B.; Bakhshi, H.; Heckbert, S.R.; Soliman, E.Z.; Bluemke, D.A.; Kawut, S.M.; Wu, C.O.; Nazarian, S.; et al. Association of right atrial structure with incident atrial fibrillation: A longitudinal cohort cardiovascular magnetic resonance study from the Multi-Ethnic Study of Atherosclerosis (MESA). J. Cardiovasc. Magn. Reson. 2020, 22, 36. [Google Scholar] [CrossRef] [PubMed]
- Santamore, W.P.; Dell’Italia, L.J. Ventricular interdependence: Significant left ventricular contributions to right ventricular systolic function. Prog. Cardiovasc. Dis. 1998, 40, 289–308. [Google Scholar] [CrossRef]
- Pedrinelli, R.; Canale, M.L.; Giannini, C.; Talini, E.; Penno, G.; Dell’Omo, G.; Di Bello, V. Right ventricular dysfunction in early systemic hypertension: A tissue Doppler imaging study in patients with high-normal and mildly increased arterial blood pressure. J. Hypertens. 2010, 28, 615–621. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C.; Celic, V.; Petrovic, O.; Pencic, B.; Mancia, G.; Grassi, G.; Ivanovic, B. The prognostic importance of right ventricular remodeling and the circadian blood pressure pattern on the long-term cardiovascular outcome. J. Hypertens. 2020, 38, 1525–1530. [Google Scholar] [CrossRef]
- Mantziari, L.; Kamperidis, V.; Ventoulis, I.; Damvopoulou, E.; Giannakoulas, G.; Efthimiadis, G.; Paraskevaidis, S.; Vassilikos, V.; Ziakas, A.; Karvounis, H.; et al. Increased right atrial volume index predicts low Duke activity status index in patients with chronic heart failure. Hell. J. Cardiol. 2013, 54, 32–38. [Google Scholar]
- Chow, V.; Ng, A.C.; Chung, T.; Thomas, L.; Kritharides, L. Right atrial to left atrial area ratio on early echocardiography predicts long-term survival after acute pulmonary embolism. Cardiovasc. Ultrasound 2013, 11, 17. [Google Scholar] [CrossRef]
- Kawut, S.M.; Barr, R.G.; Lima, J.A.; Praestgaard, A.; Johnson, W.C.; Chahal, H.; Ogunyankin, K.O.; Bristow, M.R.; Kizer, J.R.; Tandri, H.; et al. Right ventricular structure is associated with the risk of heart failure and cardiovascular death: The Multi-Ethnic Study of Atherosclerosis (MESA)—Right ventricle study. Circulation 2012, 126, 1681–1688. [Google Scholar] [CrossRef]
- Zhang, J.; Du, L.; Qin, X.; Guo, X. Effect of Sacubitril/Valsartan on the Right Ventricular Function and Pulmonary Hypertension in Patients With Heart Failure With Reduced Ejection Fraction: A Systematic Review and Meta-Analysis of Observational Studies. J. Am. Heart Assoc. 2022, 11, e024449. [Google Scholar] [CrossRef] [PubMed]
- Norozi, K.; Bahlmann, J.; Raab, B.; Alpers, V.; Arnhold, J.O.; Kuehne, T.; Klimes, K.; Zoege, M.; Geyer, S.; Wessel, A.; et al. A prospective, randomized, double-blind, placebo controlled trial of beta-blockade in patients who have undergone surgical correction of tetralogy of Fallot. Cardiol. Young 2007, 17, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.R.; McConnell, M.E.; Book, W.M. Effect of beta blockers (carvedilol or metoprolol XL) in patients with transposition of great arteries and dysfunction of the systemic right ventricle. Am. J. Cardiol. 2007, 99, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Ross, G. Effect of Hypertension on the P Wave of the Electrocardiogram. Br. Heart J. 1963, 25, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Dunn, F.G.; Pfeffer, M.A.; Frohlich, E.D. ECG alterations with progressive left ventricular hypertrophy in spontaneous hypertension. Clin. Exp. Hypertens. 1978, 1, 67–86. [Google Scholar] [CrossRef]
- Kallistratos, M.S.; Poulimenos, L.E.; Manolis, A.J. Atrial fibrillation and arterial hypertension. Pharmacol. Res. 2018, 128, 322–326. [Google Scholar] [CrossRef]
- Ogunsua, A.A.; Shaikh, A.Y.; Ahmed, M.; McManus, D.D. Atrial Fibrillation and Hypertension: Mechanistic, Epidemiologic, and Treatment Parallels. Methodist DeBakey Cardiovasc. J. 2015, 11, 228–234. [Google Scholar] [CrossRef]
- Prisant, L.M. Hypertensive heart disease. J. Clin. Hypertens. 2005, 7, 231–238. [Google Scholar] [CrossRef]
- Pluteanu, F.; Nikonova, Y.; Holzapfel, A.; Herzog, B.; Scherer, A.; Preisenberger, J.; Plackic, J.; Scheer, K.; Ivanova, T.; Bukowska, A.; et al. Progressive impairment of atrial myocyte function during left ventricular hypertrophy and heart failure. J. Mol. Cell Cardiol. 2018, 114, 253–263. [Google Scholar] [CrossRef]
- Zehender, M.; Faber, T.; Koscheck, U.; Meinertz, T.; Just, H. Ventricular tachyarrhythmias, myocardial ischemia, and sudden cardiac death in patients with hypertensive heart disease. Clin. Cardiol. 1995, 18, 377–383. [Google Scholar] [CrossRef]
- Marazzato, J.; Blasi, F.; Golino, M.; Verdecchia, P.; Angeli, F.; De Ponti, R. Hypertension and Arrhythmias: A Clinical Overview of the Pathophysiology-Driven Management of Cardiac Arrhythmias in Hypertensive Patients. J. Cardiovasc. Dev. Dis. 2022, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, K.; Ichimura, K.; Reddy, S.; Haddad, F.; Spiekerkoetter, E. Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front. Cardiovasc. Med. 2022, 9, 886553. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Schelbert, E.B.; Diez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Vischer, A.S.; Kuster, G.M.; Twerenbold, R.; Pfister, O.; Zhou, Q.; Villiger, A.; Poglitsch, M.; Krahenbuhl, S.; Mayr, M.; Osswald, S.; et al. Influence of Antihypertensive Treatment on RAAS Peptides in Newly Diagnosed Hypertensive Patients. Cells 2021, 10, 534. [Google Scholar] [CrossRef]
- Verdecchia, P.; Angeli, F.; Reboldi, G. Hypertension and Atrial Fibrillation: Doubts and Certainties From Basic and Clinical Studies. Circ. Res. 2018, 122, 352–368. [Google Scholar] [CrossRef]
- Krishnan, A.; Chilton, E.; Raman, J.; Saxena, P.; McFarlane, C.; Trollope, A.F.; Kinobe, R.; Chilton, L. Are Interactions between Epicardial Adipose Tissue, Cardiac Fibroblasts and Cardiac Myocytes Instrumental in Atrial Fibrosis and Atrial Fibrillation? Cells 2021, 10, 2501. [Google Scholar] [CrossRef]
- Bassand, J.P.; Accetta, G.; Al Mahmeed, W.; Corbalan, R.; Eikelboom, J.; Fitzmaurice, D.A.; Fox, K.A.A.; Gao, H.; Goldhaber, S.Z.; Goto, S.; et al. Risk factors for death, stroke, and bleeding in 28,628 patients from the GARFIELD-AF registry: Rationale for comprehensive management of atrial fibrillation. PLoS ONE 2018, 13, e0191592. [Google Scholar] [CrossRef]
- Okin, P.M.; Wachtell, K.; Kjeldsen, S.E.; Devereux, R.B. Relationship of incident atrial fibrillation to the electrocardiographic strain pattern in hypertensive patients with electrocardiographic left ventricular hypertrophy. In Proceedings of the American Heart Association’s 2016 Scientific Sessions and Resuscitation Science Symposium, New Orleans, LA, USA, 12–16 November 2016; Volume 134, p. A11359. [Google Scholar]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef]
- Grundvold, I.; Skretteberg, P.T.; Liestol, K.; Erikssen, G.; Kjeldsen, S.E.; Arnesen, H.; Erikssen, J.; Bodegard, J. Upper normal blood pressures predict incident atrial fibrillation in healthy middle-aged men: A 35-year follow-up study. Hypertension 2012, 59, 198–204. [Google Scholar] [CrossRef]
- Webb, A.J.; Rothwell, P.M. Blood pressure variability and risk of new-onset atrial fibrillation: A systematic review of randomized trials of antihypertensive drugs. Stroke 2010, 41, 2091–2093. [Google Scholar] [CrossRef]
- Palareti, G.; Cosmi, B. Bleeding with anticoagulation therapy—Who is at risk, and how best to identify such patients. Thromb. Haemost. 2009, 102, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Rienstra, M.; Van Veldhuisen, D.J.; Crijns, H.J.; Van Gelder, I.C.; Investigators, R. Enhanced cardiovascular morbidity and mortality during rhythm control treatment in persistent atrial fibrillation in hypertensives: Data of the RACE study. Eur. Heart J. 2007, 28, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.H.; Coca, A.; Kahan, T.; Boriani, G.; Manolis, A.S.; Olsen, M.H.; Oto, A.; Potpara, T.S.; Steffel, J.; Marin, F.; et al. Hypertension and cardiac arrhythmias: A consensus document from the European Heart Rhythm Association (EHRA) and ESC Council on Hypertension, endorsed by the Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS) and Sociedad Latinoamericana de Estimulacion Cardiaca y Electrofisiologia (SOLEACE). Europace 2017, 19, 891–911. [Google Scholar] [CrossRef]
- Li, Z.; Dahlof, B.; Okin, P.M.; Kjeldsen, S.E.; Wachtell, K.; Ibsen, H.; Nieminen, M.S.; Jern, S.; Devereux, R.B. Left bundle branch block and cardiovascular morbidity and mortality in hypertensive patients with left ventricular hypertrophy: The Losartan Intervention For Endpoint Reduction in Hypertension study. J. Hypertens. 2008, 26, 1244–1249. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Relative Wall Thickness | >0.42 | Concentric remodelling | Concentric hypertrophy |
| ≤0.42 | Normal geometry | Eccentric hypertrophy | |
| m ≤ 115 f ≤ 95 | m > 115 f > 95 | ||
| Left Ventricular Mass Index (g/m2) | |||
| LVH Subgroups (by CMR) | LVH Subgroups (by Echo) | LV Mass | LV EDV | RWT |
|---|---|---|---|---|
| Indeterminate LVH | Eccentric non-dilated | + | - | - |
| Dilated LVH | Eccentric dilated | + | + | - |
| Thick LVH | Concentric non-dilated | + | - | + |
| Both thick and dilated LVH | Concentric dilated | + | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemtsova, V.; Burkard, T.; Vischer, A.S. Hypertensive Heart Disease: A Narrative Review Series—Part 2: Macrostructural and Functional Abnormalities. J. Clin. Med. 2023, 12, 5723. https://doi.org/10.3390/jcm12175723
Nemtsova V, Burkard T, Vischer AS. Hypertensive Heart Disease: A Narrative Review Series—Part 2: Macrostructural and Functional Abnormalities. Journal of Clinical Medicine. 2023; 12(17):5723. https://doi.org/10.3390/jcm12175723
Chicago/Turabian StyleNemtsova, Valeriya, Thilo Burkard, and Annina S. Vischer. 2023. "Hypertensive Heart Disease: A Narrative Review Series—Part 2: Macrostructural and Functional Abnormalities" Journal of Clinical Medicine 12, no. 17: 5723. https://doi.org/10.3390/jcm12175723
APA StyleNemtsova, V., Burkard, T., & Vischer, A. S. (2023). Hypertensive Heart Disease: A Narrative Review Series—Part 2: Macrostructural and Functional Abnormalities. Journal of Clinical Medicine, 12(17), 5723. https://doi.org/10.3390/jcm12175723