
Bronchopulmonary Dysplasia: Pathogenesis and Pathophysiology

Abstract
{kind=link}
1. Introduction
2. Pathology
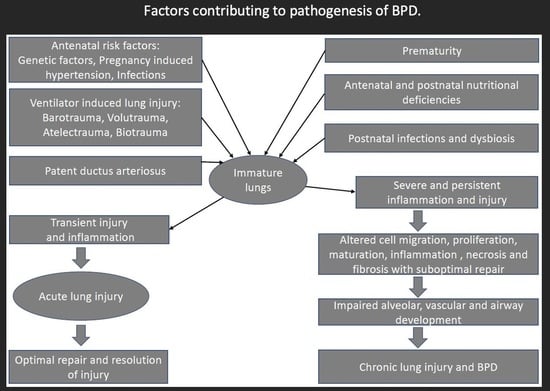
3. Pathogenesis of Bronchopulmonary Dysplasia
3.1. Prenatal Risk Factors
3.1.1. Maternal Smoking
3.1.2. Chorioamnionitis
3.1.3. Pregnancy-Induced Hypertension (PIH)
3.1.4. Intrauterine Growth Restriction (IUGR)
3.1.5. Genetics
3.2. Postnatal Risk Factors
3.2.1. Mechanical Ventilation and Ventilator-Induced Lung Injury (VILI)
- Barotrauma
- Volutrauma
- Which is more important in causing lung injury: barotrauma or volutrauma?
- Atelectrauma
- Oxygen toxicity
- Biotrauma
- Mechanical power, Stress, and Strain
- Lung deflation injury
- Pre-existing lung disease
3.2.2. Patent Ductus Arteriosus (PDA)
3.2.3. Sepsis
3.2.4. Dysbiosis
3.2.5. Nutrition
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abman, S.H.; Bancalari, E.; Jobe, A. The Evolution of Bronchopulmonary Dysplasia after 50 Years. Am. J. Respir. Crit. Care Med. 2017, 195, 421–424. [Google Scholar] [CrossRef]
- Northway, W.H.; Rosan, R.C.; Porter, D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N. Engl. J. Med. 1967, 276, 357–368. [Google Scholar] [CrossRef]
- Jobe, A.; Ikegami, M. Surfactant for the Treatment of Respiratory Distress Syndrome. Am. Rev. Respir. Dis. 1987, 136, 1256–1275. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M. An Official American Thoracic Society Workshop Report: Features and Measurements of Experimental Acute Lung Injury in Animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef]
- Thekkeveedu, R.K.; El-Saie, A.; Prakash, V.; Katakam, L.; Shivanna, B. Ventilation-Induced Lung Injury (VILI) in Neonates: Evidence-Based Concepts and Lung-Protective Strategies. J. Clin. Med. 2022, 11, 557. [Google Scholar] [CrossRef]
- Tomashefski, J.F., Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin. Chest Med. 2000, 21, 435–466. [Google Scholar] [CrossRef]
- Jobe, A.H.; Bancalari, E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 163, 1723–1729. [Google Scholar] [CrossRef]
- Bhandari, A.; Bhandari, V. Pathogenesis, pathology and pathophysiology of pulmonary sequelae of bronchopulmonary dysplasia in premature infants. Front. Biosci. 2003, 8, e370-80. [Google Scholar] [CrossRef]
- Hoo, A.-F.; Henschen, M.; Dezateux, C.; Costeloe, K.; Stocks, J. Respiratory function among preterm infants whose mothers smoked during pregnancy. Am. J. Respir. Crit. Care Med. 1998, 158, 700–705. [Google Scholar] [CrossRef]
- McEvoy, C.T.; Spindel, E.R. Pulmonary Effects of Maternal Smoking on the Fetus and Child: Effects on Lung Development, Respiratory Morbidities, and Life Long Lung Health. Paediatr. Respir. Rev. 2017, 21, 27–33. [Google Scholar] [CrossRef]
- Antonucci, R.; Contu, P.; Porcella, A.; Atzeni, C.; Chiappe, S. Intrauterine smoke exposure: A new risk factor for bronchopulmonary dysplasia? J. Perinat. Med. 2004, 32, 272–277. [Google Scholar] [CrossRef]
- Morrow, L.A.; Wagner, B.D.; Ingram, D.A.; Poindexter, B.B.; Schibler, K.; Cotten, C.M.; Dagle, J.; Sontag, M.K.; Mourani, P.M.; Abman, S.H. Antenatal Determinants of Bronchopulmonary Dysplasia and Late Respiratory Disease in Preterm Infants. Am. J. Respir. Crit. Care Med. 2017, 196, 364–374. [Google Scholar] [CrossRef]
- González-Luis, G.E.; van Westering-Kroon, E.; Villamor-Martinez, E.; Huizing, M.J.; Kilani, M.A.; Kramer, B.W.; Villamor, E. Tobacco Smoking during Pregnancy Is Associated with Increased Risk of Moderate/Severe Bronchopulmonary Dysplasia: A Systematic Review and Meta-Analysis. Front. Pediatr. 2020, 8, 160. [Google Scholar] [CrossRef]
- Thekkeveedu, R.K.; Guaman, M.C.; Shivanna, B. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir. Med. 2017, 132, 170–177. [Google Scholar] [CrossRef]
- Choi, C.W. Chorioamnionitis: Is a major player in the development of bronchopulmonary dysplasia? Korean J. Pediatr. 2017, 60, 203–207. [Google Scholar] [CrossRef][Green Version]
- Villamor-Martinez, E.; Álvarez-Fuente, M.; Ghazi, A.M.T.; Degraeuwe, P.; Zimmermann, L.J.I.; Kramer, B.W.; Villamor, E. Association of Chorioamnionitis with Bronchopulmonary Dysplasia among Preterm Infants: A Systematic Review, Meta-analysis, and Metaregression. JAMA Netw. Open 2019, 2, e1914611. [Google Scholar] [CrossRef] [PubMed]
- Dessardo, N.S.; Mustać, E.; Dessardo, S.; Banac, S.; Peter, B.; Finderle, A.; Marić, M.; Haller, H. Chorioamnionitis and chronic lung disease of prematurity: A path analysis of causality. Am. J. Perinatol. 2012, 29, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ballard, A.R.; Mallett, L.H.; Pruszynski, J.; Cantey, J.B. chorioamnionitis and subsequent bronchopulmonary dysplasia in very-low-birth weight infants: A 25-year cohort. J. Perinatol. 2016, 36, 1045–1048. [Google Scholar] [CrossRef]
- Collins, J.J.P.; Kallapur, S.G.; Knox, C.L.; Nitsos, I.; Polglase, G.R.; Pillow, J.J.; Kuypers, E.; Newnham, J.P.; Jobe, A.H.; Kramer, B.W.; et al. Inflammation in fetal sheep from intra-amniotic injection of Ureaplasma parvum. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L852–L860. [Google Scholar] [CrossRef]
- Viscardi, R.; Manimtim, W.; He, J.R.; Hasday, J.D.; Sun, C.-C.J.; Joyce, B.; Pierce, R.A. Disordered pulmonary myofibroblast distribution and elastin expression in preterm infants with Ureaplasma urealyticum pneumonitis. Pediatr. Dev. Pathol. 2006, 9, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Schelonka, R.L.; Katz, B.; Waites, K.B.; Benjamin, D.K. Critical appraisal of the role of Ureaplasma in the development of bronchopulmonary dysplasia with metaanalytic techniques. Pediatr. Infect. Dis. J. 2005, 24, 1033–1039. [Google Scholar] [CrossRef]
- Burton, G.J.; Charnock-Jones, D.S.; Jauniaux, E. Regulation of vascular growth and function in the human placenta. Reproduction 2009, 138, 895–902. [Google Scholar] [CrossRef]
- Shin, S.H.; Kim, S.H.; Kim, Y.-J.; Cho, H.; Kim, E.-K.; Kim, H.-S. The Association of Pregnancy-induced Hypertension with Bronchopulmonary Dysplasia—A Retrospective Study Based on the Korean Neonatal Network database. Sci. Rep. 2020, 10, 5600. [Google Scholar] [CrossRef]
- Razak, A.; Florendo-Chin, A.; Banfield, L.; Wahab, M.G.A.; McDonald, S.; Shah, P.S.; Mukerji, A. Pregnancy-induced hypertension and neonatal outcomes: A systematic review and meta-analysis. J. Perinatol. 2018, 38, 46–53. [Google Scholar] [CrossRef]
- Pierro, M.; Villamor-Martinez, E.; van Westering-Kroon, E.; Alvarez-Fuente, M.; Abman, S.H.; Villamor, E. Association of the dysfunctional placentation endotype of prematurity with bronchopulmonary dysplasia: A systematic review, meta-analysis and meta-regression. Thorax 2022, 77, 268–275. [Google Scholar] [CrossRef]
- Sehgal, A.; Gwini, S.M.; Menahem, S.; Allison, B.J.; Miller, S.L.; Polglase, G.R. Preterm growth restriction and bronchopulmonary dysplasia: The vascular hypothesis and related physiology. J. Physiol. 2019, 597, 1209–1220. [Google Scholar] [CrossRef]
- Groene, S.G.; Spekman, J.A.; Pas, A.B.T.; Heijmans, B.T.; Haak, M.C.; van Klink, J.M.; Roest, A.A.; Lopriore, E. Respiratory distress syndrome and bronchopulmonary dysplasia after fetal growth restriction: Lessons from a natural experiment in identical twins. Eclinicalmedicine 2021, 32, 100725. [Google Scholar] [CrossRef]
- Le Cras, T.D.; Markham, N.E.; Tuder, R.M.; Voelkel, N.F.; Abman, S.H. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L555–L562. [Google Scholar] [CrossRef]
- Rozance, P.J.; Seedorf, G.J.; Brown, A.; Roe, G.; O’Meara, M.C.; Gien, J.; Tang, J.-R.; Abman, S.H.; Lai, P.Y.; Jing, X.; et al. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L860–L871. [Google Scholar] [CrossRef]
- Bhandari, V.; Bizzarro, M.J.; Shetty, A.; Zhong, X.; Page, G.P.; Zhang, H.; Ment, L.R.; Gruen, J.R.; for the Neonatal Genetics Study Group. Familial and Genetic Susceptibility to Major Neonatal Morbidities in Preterm Twins. Pediatrics 2006, 117, 1901–1906. [Google Scholar] [CrossRef]
- Lavoie, P.M.; Pham, C.; Jang, K.L. Heritability of Bronchopulmonary Dysplasia, Defined According to the Consensus Statement of the National Institutes of Health. Pediatrics 2008, 122, 479–485. [Google Scholar] [CrossRef]
- Yu, K.-H.; Li, J.; Snyder, M.; Shaw, G.M.; O’brodovich, H.M. The genetic predisposition to bronchopulmonary dysplasia. Curr. Opin. Pediatr. 2016, 28, 318–323. [Google Scholar] [CrossRef]
- Mailaparambil, B.; Krueger, M.; Heizmann, U.; Schlegel, K.; Heinze, J.; Heinzmann, A. Genetic and epidemiological risk factors in the development of bronchopulmonary dysplasia. Dis. Mark. 2010, 29, 1–9. [Google Scholar] [CrossRef]
- Floros, J.; Londono, D.; Gordon, D.; Silveyra, P.; Diangelo, S.L.; Viscardi, R.M.; Worthen, G.S.; Shenberger, J.; Wang, G.; Lin, Z.; et al. IL-18R1 and IL-18RAP SNPs may be associated with bronchopulmonary dysplasia in African-American infants. Pediatr. Res. 2012, 71, 107–114. [Google Scholar] [CrossRef]
- Hadchouel, A.; Decobert, F.; Franco-Montoya, M.L.; Halphen, I.; Jarreau, P.H.; Boucherat, O.; Martin, E.; Benachi, A.; Amselem, S.; Bourbon, J.; et al. Matrix metalloproteinase gene polymorphisms and bronchopulmonary dysplasia: Identification of MMP16 as a new player in lung development. PLoS ONE 2008, 3, e3188. [Google Scholar] [CrossRef]
- Ambalavanan, N.; Cotten, C.M.; Page, G.P.; Carlo, W.A.; Murray, J.C.; Bhattacharya, S.; Mariani, T.J.; Cuna, A.C.; Faye-Petersen, O.M.; Kelly, D.; et al. Integrated Genomic Analyses in Bronchopulmonary Dysplasia. J. Pediatr. 2015, 166, 531–537.e13. [Google Scholar] [CrossRef]
- Dreyfuss, D.; Basset, G.; Soler, P.; Saumon, G. Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am. Rev. Respir. Dis. 1985, 132, 880–884. [Google Scholar]
- Webb, H.H.; Tierney, D.F. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am. Rev. Respir. Dis. 1974, 110, 556–565. [Google Scholar]
- Kolobow, T.; Moretti, M.P.; Fumagalli, R.; Mascheroni, D.; Prato, P.; Chen, V.; Joris, M. Severe impairment in lung function induced by high peak airway pressure during mechanical ventilation. An experimental study. Am. Rev. Respir. Dis. 1987, 135, 312–315. [Google Scholar]
- Petersen, G.W.; Baier, H. Incidence of pulmonary barotrauma in a medical ICU. Crit. Care Med. 1983, 11, 67–69. [Google Scholar] [CrossRef]
- Slutsky, A.S. Lung Injury Caused by Mechanical Ventilation. Chest 1999, 116 (Suppl. 1), 9s–15s. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Jobe, A.H.; Ikegami, M. Tidal volume effects on surfactant treatment responses with the initiation of ventilation in preterm lambs. J. Appl. Physiol. 1997, 83, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Björklund, L.J.; Ingimarsson, J.; Curstedt, T.; John, J.; Robertson, B.; Werner, O.; Vilstrup, C.T.; Bj, L.J. Manual Ventilation with a Few Large Breaths at Birth Compromises the Therapeutic Effect of Subsequent Surfactant Replacement in Immature Lambs. Pediatr. Res. 1997, 42, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.A.; Gutierrez, J.A.; Jones, K.D.; Allen, L.; Dobbs, L.; Matthay, M.A. Low Tidal Volume Reduces Epithelial and Endothelial Injury in Acid-injured Rat Lungs. Am. J. Respir. Crit. Care Med. 2002, 165, 242–249. [Google Scholar] [CrossRef]
- Acute Respiratory Distress Syndrome Network; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar]
- Lista, G.; Colnaghi, M.; Castoldi, F.; Condò, V.; Reali, R.; Compagnoni, G.; Mosca, F. Impact of targeted-volume ventilation on lung inflammatory response in preterm infants with respiratory distress syndrome (RDS). Pediatr. Pulmonol. 2004, 37, 510–514. [Google Scholar] [CrossRef]
- Dreyfuss, D.; Soler, P.; Basset, G.; Saumon, G. High Inflation Pressure Pulmonary Edema: Respective Effects of High Airway Pressure, High Tidal Volume, and Positive End-expiratory Pressure. Am. Rev. Respir. Dis. 1988, 137, 1159–1164. [Google Scholar] [CrossRef]
- Hernandez, L.A.; Peevy, K.J.; Moise, A.A.; Parker, J.C. Chest wall restriction limits high airway pressure-induced lung injury in young rabbits. J. Appl. Physiol. 1989, 66, 2364–2368. [Google Scholar] [CrossRef]
- Carlton, D.P.; Cummings, J.J.; Scheerer, R.G.; Poulain, F.R.; Bland, R.D. Lung overexpansion increases pulmonary microvascular protein permeability in young lambs. J. Appl. Physiol. 1990, 69, 577–583. [Google Scholar] [CrossRef]
- Klingenberg, C.; Wheeler, K.I.; McCallion, N.; Morley, C.J.; Davis, P.G. Volume-targeted versus pressure-limited ventilation in neonates. Cochrane Database Syst. Rev. 2017, 10, Cd003666. [Google Scholar] [CrossRef]
- Beitler, J.R.; Malhotra, A.; Thompson, B.T. Ventilator-induced Lung Injury. Clin. Chest Med. 2016, 37, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Taskar, V.; John, J.; Evander, E.; Robertson, B.; Jonson, B. Surfactant dysfunction makes lungs vulnerable to repetitive collapse and reexpansion. Am. J. Respir. Crit. Care Med. 1997, 155, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.; Takishima, T.; Leith, D. Stress distribution in lungs: A model of pulmonary elasticity. J. Appl. Physiol. 1970, 28, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.H.; Gerstmann, D.R.; Jobe, A.H.; Moffitt, S.T.; Slutsky, A.S.; Yoder, B.A. Lung injury in neonates: Causes, strategies for prevention, and long-term consequences. J. Pediatr. 2001, 139, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.B.; Mcculloch, P.R.; Sugiura, M.; Vaclavik, S.; Possmayer, F.; Moller, F. Optimizing Alveolar Expansion Prolongs the Effectiveness of Exogenous Surfactant Therapy in the Adult Rabbit. Am. Rev. Respir. Dis. 1993, 148, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Sandhar, B.K.; Niblett, D.J.; Argiras, E.P.; Dunnill, M.S.; Sykes, M.K. Effects of positive end-expiratory pressure on hyaline membrane formation in a rabbit model of the neonatal respiratory distress syndrome. Intensive Care Med. 1988, 14, 538–546. [Google Scholar] [CrossRef]
- Muscedere, J.G.; Mullen, J.B.; Gan, K.; Slutsky, A.S. Tidal ventilation at low airway pressures can augment lung injury. Am. J. Respir. Crit. Care Med. 1994, 149, 1327–1334. [Google Scholar] [CrossRef]
- Bhandari, V. Hyperoxia-derived lung damage in preterm infants. Semin. Fetal Neonatal Med. 2010, 15, 223–229. [Google Scholar] [CrossRef]
- Frank, L. Antioxidants, nutrition, and bronchopulmonary dysplasia. Clin. Perinatol. 1992, 19, 541–562. [Google Scholar] [CrossRef]
- Berkelhamer, S.K.; Kim, G.A.; Radder, J.E.; Wedgwood, S.; Czech, L.; Steinhorn, R.H.; Schumacker, P.T. Developmental differences in hyperoxia-induced oxidative stress and cellular responses in the murine lung. Free Radic. Biol. Med. 2013, 61, 51–60. [Google Scholar] [CrossRef]
- Irwin, D.; Helm, K.; Campbell, N.; Imamura, M.; Fagan, K.; Harral, J.; Carr, M.; Young, K.A.; Klemm, D.; Gebb, S.; et al. Neonatal lung side population cells demonstrate endothelial potential and are altered in response to hyperoxia-induced lung simplification. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L941–L951. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.C.; Kuppusamy, P.; Parinandi, N.L.; Malireddy, S.; Kotha, S.R.; Secor, J.D.; Gurney, T.O.; Abbott, J.L.; Maulik, G.; Maddipati, K.R.; et al. Oxygen, the Lead Actor in the Pathophysiologic Drama: Enactment of the Trinity of Normoxia, Hypoxia, and Hyperoxia in Disease and Therapy. Antioxid. Redox Signal. 2007, 9, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, V.S.; Chalak, L.F.; Sparks, J.E.; Allen, J.R.; Savani, R.C.; Wyckoff, M.H. Resuscitation of Preterm Neonates with Limited Versus High Oxygen Strategy. Pediatrics 2013, 132, e1488–e1496. [Google Scholar] [CrossRef] [PubMed]
- Saugstad, O.D.; Aune, D. Optimal Oxygenation of Extremely Low Birth Weight Infants: A Meta-Analysis and Systematic Review of the Oxygen Saturation Target Studies. Neonatology 2014, 105, 55–63. [Google Scholar] [CrossRef]
- Vogel, E.R.; Britt, R.D., Jr.; Trinidad, M.C.; Faksh, A.; Martin, R.J.; Macfarlane, P.M.; Pabelick, C.M.; Prakash, Y. Perinatal oxygen in the developing lung. Can. J. Physiol. Pharmacol. 2015, 93, 119–127. [Google Scholar] [CrossRef]
- Ranieri, V.M.; Suter, P.M.; Tortorella, C.; De Tullio, R.; Dayer, J.M.; Brienza, A.; Bruno, F.; Slutsky, A.S. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: A randomized controlled trial. JAMA 1999, 282, 54–61. [Google Scholar] [CrossRef]
- Tremblay, L.; Valenza, F.; Ribeiro, S.P.; Li, J.; Slutsky, A.S. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J. Clin. Investig. 1997, 99, 944–952. [Google Scholar] [CrossRef]
- Chiumello, D.; Pristine, G.; Slutsky, A.S. Mechanical Ventilation Affects Local and Systemic Cytokines in an Animal Model of Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 1999, 160, 109–116. [Google Scholar] [CrossRef]
- Curley, G.F.; Laffey, J.G.; Zhang, H.; Slutsky, A.S. Biotrauma and Ventilator-Induced Lung Injury: Clinical Implications. Chest 2016, 150, 1109–1117. [Google Scholar] [CrossRef]
- Vasques, F.; Duscio, E.; Cipulli, F.; Romitti, F.; Quintel, M.; Gattinoni, L. Determinants and Prevention of Ventilator-Induced Lung Injury. Crit. Care Clin. 2018, 34, 343–356. [Google Scholar] [CrossRef]
- Cressoni, M.; Gotti, M.; Chiurazzi, C.; Massari, D.; Algieri, I.; Amini, M.; Cammaroto, A.; Brioni, M.; Montaruli, C.; Nikolla, K.; et al. Mechanical Power and Development of Ventilator-induced Lung Injury. Anesthesiology 2016, 124, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Chiumello, D.; Carlesso, E.; Cadringher, P.; Caironi, P.; Valenza, F.; Polli, F.; Tallarini, F.; Cozzi, P.; Cressoni, M.; Colombo, A.; et al. Lung Stress and Strain during Mechanical Ventilation for Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2008, 178, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.J.; Rocco, P.R.M.; Gattinoni, L. Static and Dynamic Contributors to Ventilator-induced Lung Injury in Clinical Practice. Pressure, Energy, and Power. Am. J. Respir. Crit. Care Med. 2020, 201, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Cressoni, M.; Cadringher, P.; Chiurazzi, C.; Amini, M.; Gallazzi, E.; Marino, A.; Brioni, M.; Carlesso, E.; Chiumello, D.; Quintel, M.; et al. Lung Inhomogeneity in Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2014, 189, 149–158. [Google Scholar] [CrossRef]
- Protti, A.; Andreis, D.T.; Monti, M.; Santini, A.; Sparacino, C.C.; Langer, T.; Votta, E.; Gatti, S.; Lombardi, L.; Leopardi, O.; et al. Lung stress and strain during mechanical ventilation: Any difference between statics and dynamics? Crit. Care Med. 2013, 41, 1046–1055. [Google Scholar] [CrossRef]
- Katira, B.H.; Engelberts, D.; Otulakowski, G.; Giesinger, R.E.; Yoshida, T.; Post, M.; Kuebler, W.M.; Connelly, K.A.; Kavanagh, B.P. Abrupt Deflation after Sustained Inflation Causes Lung Injury. Am. J. Respir. Crit. Care Med. 2018, 198, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Varsila, E.; Hallman, M.; Venge, P.; Andersson, S. Closure of Patent Ductus arteriosus Decreases Pulmonary Myeloperoxidase in Premature Infants with Respiratory Distress Syndrome. Neonatology 1995, 67, 167–171. [Google Scholar] [CrossRef]
- Slaughter, J.L.; Reagan, P.B.; Newman, T.B.; Klebanoff, M.A. Comparative Effectiveness of Nonsteroidal Anti-inflammatory Drug Treatment vs No Treatment for Patent Ductus Arteriosus in Preterm Infants. JAMA Pediatr. 2017, 171, e164354. [Google Scholar] [CrossRef]
- McCurnin, D.; Seidner, S.; Chang, L.-Y.; Waleh, N.; Ikegami, M.; Petershack, J.; Yoder, B.; Giavedoni, L.; Albertine, K.H.; Dahl, M.J.; et al. Ibuprofen-Induced Patent Ductus Arteriosus Closure: Physiologic, Histologic, and Biochemical Effects on the Premature Lung. Pediatrics 2008, 121, 945–956. [Google Scholar] [CrossRef]
- Brooks, J.M.; Travadi, J.N.; Patole, S.K.; Doherty, D.A.; Simmer, K. Is surgical ligation of patent ductus arteriosus necessary? The Western Australian experience of conservative management. Arch. Dis. Child. Fetal Neonatal Ed. 2005, 90, F235–F239. [Google Scholar] [CrossRef]
- Madan, J.C.; Kendrick, D.; Hagadorn, J.I.; Frantz, I.D.; the National Institute of Child Health and Human Development Neonatal Research Network. Patent Ductus Arteriosus Therapy: Impact on Neonatal and 18-Month Outcome. Pediatrics 2009, 123, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Sellmer, A.; Bjerre, J.V.; Schmidt, M.R.; McNamara, P.J.; Hjortdal, V.E.; Høst, B.; Bech, B.H.; Henriksen, T.B. Morbidity and mortality in preterm neonates with patent ductus arteriosus on day 3. Arch. Dis. Child. Fetal Neonatal Ed. 2013, 98, F505–F510. [Google Scholar] [CrossRef] [PubMed]
- Clyman, R.I.; Kaempf, J.; Liebowitz, M.; Erdeve, O.; Bulbul, A.; Håkansson, S.; Lindqvist, J.; Farooqi, A.; Katheria, A.; Sauberan, J.; et al. Prolonged Tracheal Intubation and the Association Between Patent Ductus Arteriosus and Bronchopulmonary Dysplasia: A Secondary Analysis of the PDA-TOLERATE trial. J. Pediatr. 2021, 229, 283–288.e2. [Google Scholar] [CrossRef] [PubMed]
- Mirza, H.; Garcia, J.; McKinley, G.; Hubbard, L.; Sensing, W.; Schneider, J.; Oh, W.; Wadhawan, R. Duration of significant patent ductus arteriosus and bronchopulmonary dysplasia in extremely preterm infants. J. Perinatol. 2019, 39, 1648–1655. [Google Scholar] [CrossRef]
- Hundscheid, T.; Onland, W.; Kooi, E.M.; Vijlbrief, D.C.; de Vries, W.B.; Dijkman, K.P.; van Kaam, A.H.; Villamor, E.; Kroon, A.A.; Visser, R.; et al. Expectant Management or Early Ibuprofen for Patent Ductus Arteriosus. N. Engl. J. Med. 2023, 388, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Han, D.; Wei, Y.; Lin, B.; Zeng, D.; Zhang, Y.; Wei, B.; Huang, Z.; Chen, X.; Yang, C. Decreased plasma levels of PDGF-BB, VEGF-A, and HIF-2alpha in preterm infants after ibuprofen treatment. Front. Pediatr. 2022, 10, 919879. [Google Scholar] [CrossRef] [PubMed]
- Ambalavanan, N.; Carlo, W.A.; D’Angio, C.T.; McDonald, S.A.; Das, A.; Schendel, D.; Thorsen, P.; Higgins, R.D.; for the Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Cytokines Associated with Bronchopulmonary Dysplasia or Death in Extremely Low Birth Weight Infants. Pediatrics 2009, 123, 1132–1141. [Google Scholar] [CrossRef]
- Franco, M.-L.; Waszak, P.; Banalec, G.; Levame, M.; Lafuma, C.; Harf, A.; Delacourt, C. LPS-induced lung injury in neonatal rats: Changes in gelatinase activities and consequences on lung growth. Am. J. Physiol. Cell. Mol. Physiol. 2002, 282, L491–L500. [Google Scholar] [CrossRef]
- Choi, C.W.; Lee, J.; Oh, J.Y.; Lee, S.H.; Lee, H.J.; Kim, B.I. Protective effect of chorioamnionitis on the development of bronchopulmonary dysplasia triggered by postnatal systemic inflammation in neonatal rats. Pediatr. Res. 2016, 79, 287–294. [Google Scholar] [CrossRef]
- Shrestha, A.K.; Bettini, M.L.; Menon, R.T.; Gopal, V.Y.N.; Huang, S.; Edwards, D.P.; Pammi, M.; Barrios, R.; Shivanna, B. Consequences of early postnatal lipopolysaccharide exposure on developing lungs in mice. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L229–L244. [Google Scholar] [CrossRef]
- Oh, W.; Poindexter, B.B.; Perritt, R.; Lemons, J.A.; Bauer, C.R.; Ehrenkranz, R.A.; Stoll, B.J.; Poole, K.; Wright, L.L. Association between Fluid Intake and Weight Loss during the First Ten Days of Life and Risk of Bronchopulmonary Dysplasia in Extremely Low Birth Weight Infants. J. Pediatr. 2005, 147, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Lahra, M.M.; Beeby, P.J.; Jeffery, H.E. Intrauterine Inflammation, Neonatal Sepsis, and Chronic Lung Disease: A 13-Year Hospital Cohort Study. Pediatrics 2009, 123, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Zemanick, E.T.; Wagner, B.D.; Robertson, C.E.; Stevens, M.J.; Szefler, S.J.; Accurso, F.J.; Sagel, S.D.; Harris, J.K. Assessment of Airway Microbiota and Inflammation in Cystic Fibrosis Using Multiple Sampling Methods. Ann. Am. Thorac. Soc. 2015, 12, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Pammi, M.; Lal, C.V.; Wagner, B.D.; Mourani, P.M.; Lohmann, P.; Luna, R.A.; Sisson, A.; Shivanna, B.; Hollister, E.B.; Abman, S.H.; et al. Airway Microbiome and Development of Bronchopulmonary Dysplasia in Preterm Infants: A Systematic Review. J. Pediatr. 2019, 204, 126–133.e2. [Google Scholar] [CrossRef]
- Taft, D.; Ambalavanan, N.; Schibler, K.R.; Yu, Z.; Newburg, D.S.; Deshmukh, H.; Ward, D.V.; Morrow, A.L. Center Variation in Intestinal Microbiota Prior to Late-Onset Sepsis in Preterm Infants. PLoS ONE 2015, 10, e0130604. [Google Scholar] [CrossRef]
- Wagner, B.D.; Sontag, M.K.; Harris, J.K.; Miller, J.I.; Morrow, L.; Robertson, C.E.; Stephens, M.; Poindexter, B.B.; Abman, S.H.; Mourani, P.M. Airway Microbial Community Turnover Differs by BPD Severity in Ventilated Preterm Infants. PLoS ONE 2017, 12, e0170120. [Google Scholar] [CrossRef]
- Lohmann, P.; Luna, R.A.; Hollister, E.B.; Devaraj, S.; Mistretta, T.-A.; Welty, S.E.; Versalovic, J. The airway microbiome of intubated premature infants: Characteristics and changes that predict the development of bronchopulmonary dysplasia. Pediatr. Res. 2014, 76, 294–301. [Google Scholar] [CrossRef]
- Sato, M.; Go, H.; Ogasawara, K.; Kanai, Y.; Maeda, H.; Chishiki, M.; Shimizu, H.; Mashiyama, F.; Goto, A.; Momoi, N.; et al. The Microbiome of the Lower Respiratory Tract in Premature Infants with and without Severe Bronchopulmonary Dysplasia. Am. J. Perinatol. 2017, 34, 80–87. [Google Scholar] [CrossRef]
- Surana, N.K.; Kasper, D.L. Deciphering the tete-a-tete between the microbiota and the immune system. J. Clin. Investig. 2014, 124, 4197–4203. [Google Scholar] [CrossRef]
- Gray, J.; Oehrle, K.; Worthen, G.; Alenghat, T.; Whitsett, J.; Deshmukh, H. Intestinal commensal bacteria mediate lung mucosal immunity and promote resistance of newborn mice to infection. Sci. Transl. Med. 2017, 9, eaaf9412. [Google Scholar] [CrossRef]
- Novitsky, A.; Tuttle, D.; Locke, R.G.; Saiman, L.; Mackley, A.; Paul, D.A. Prolonged Early Antibiotic Use and Bronchopulmonary Dysplasia in Very Low Birth Weight Infants. Am. J. Perinatol. 2015, 32, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Singer, B.H.; Newstead, M.W.; Falkowski, N.R.; Erb-Downward, J.R.; Standiford, T.J.; Huffnagle, G.B. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat. Microbiol. 2016, 1, 16113. [Google Scholar] [CrossRef] [PubMed]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.A.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging pathogenic links between microbiota and the gut–lung axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Fischäder, G.; Röder-Stolinski, C.; Wichmann, G.; Nieber, K.; Lehmann, I. Release of MCP-1 and IL-8 from lung epithelial cells exposed to volatile organic compounds. Toxicol. Vitr. 2008, 22, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.I.; Hong, Y.-C.; Cho, S.-H.; Kim, H.; Kim, Y.; Sohn, J.R.; Kwon, M.; Park, S.-H.; Cho, M.-H.; Cheong, H.-K. Exposure to volatile organic compounds and loss of pulmonary function in the elderly. Eur. Respir. J. 2010, 36, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Romijn, M.; van Kaam, A.H.; Fenn, D.; Bos, L.D.; Akker, C.H.v.D.; Finken, M.J.; Rotteveel, J.; Cerullo, J.; Brinkman, P.; Onland, W. Exhaled Volatile Organic Compounds for Early Prediction of Bronchopulmonary Dysplasia in Infants Born Preterm. J. Pediatr. 2023, 257, 113368. [Google Scholar] [CrossRef]
- Davidson, S. Energy Intake, Growth, and Development in Ventilated Very-Low-Birth-Weight Infants with and Without Bronchopulmonary Dysplasia. Arch. Pediatr. Adolesc. Med. 1990, 144, 553–559. [Google Scholar] [CrossRef]
- Underwood, M.A.; Lakshminrusimha, S.; Steinhorn, R.H.; Wedgwood, S. Malnutrition, poor post-natal growth, intestinal dysbiosis and the developing lung. J. Perinatol. 2021, 41, 1797–1810. [Google Scholar] [CrossRef]
- Milanesi, B.G.; Lima, P.A.; Villela, L.D.; Martins, A.S.; Gomes-Junior, S.C.S.; Moreira, M.E.L.; Méio, M.D.B.B. Assessment of early nutritional intake in preterm infants with bronchopulmonary dysplasia: A cohort study. Eur. J. Pediatr. 2021, 180, 1423–1430. [Google Scholar] [CrossRef]
- Stephens, B.E.; Gargus, R.A.; Walden, R.V.; Mance, M.; Nye, J.; McKinley, L.; Tucker, R.; Vohr, B.R. Fluid regimens in the first week of life may increase risk of patent ductus arteriosus in extremely low birth weight infants. J. Perinatol. 2008, 28, 123–128. [Google Scholar] [CrossRef]
- Massaro, D.; Massaro, G.D. Retinoids, alveolus formation, and alveolar deficiency: Clinical implications. Am. J. Respir. Cell Mol. Biol. 2003, 28, 271–274. [Google Scholar] [CrossRef]
- Shenai, J.P.; Chytil, F.; Stahlman, M.T. Liver Vitamin A Reserves of Very Low Birth Weight Neonates. Pediatr. Res. 1985, 19, 892–893. [Google Scholar] [CrossRef]
- Araki, S.; Kato, S.; Namba, F.; Ota, E. Vitamin A to prevent bronchopulmonary dysplasia in extremely low birth weight infants: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0207730. [Google Scholar] [CrossRef]
- Rakshasbhuvankar, A.A.; Simmer, K.; Patole, S.K.; Stoecklin, B.; Nathan, E.A.; Clarke, M.W.; Pillow, J.J. Enteral Vitamin A for Reducing Severity of Bronchopulmonary Dysplasia: A Randomized Trial. Pediatrics 2021, 147, e2020009985. [Google Scholar] [CrossRef] [PubMed]
- Lykkedegn, S.; Sorensen, G.L.; Beck-Nielsen, S.S.; Christesen, H.T. The impact of vitamin D on fetal and neonatal lung maturation. A systematic review. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L587–L602. [Google Scholar] [CrossRef] [PubMed]
- Mandell, E.; Seedorf, G.; Gien, J.; Abman, S.H. Vitamin D treatment improves survival and infant lung structure after intra-amniotic endotoxin exposure in rats: Potential role for the prevention of bronchopulmonary dysplasia. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L420–L428. [Google Scholar] [CrossRef]
- Ge, H.; Qiao, Y.; Ge, J.; Li, J.; Hu, K.; Chen, X.; Cao, X.; Xu, X.; Wang, W. Effects of early vitamin D supplementation on the prevention of bronchopulmonary dysplasia in preterm infants. Pediatr. Pulmonol. 2022, 57, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Aristizabal, N.; Holder, M.P.; Durham, L.; Ashraf, A.P.; Taylor, S.; Salas, A.A. Safety and Efficacy of Early Vitamin D Supplementation in Critically Ill Extremely Preterm Infants: An Ancillary Study of a Randomized Trial. J. Acad. Nutr. Diet. 2023, 123, 87–94. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dankhara, N.; Holla, I.; Ramarao, S.; Kalikkot Thekkeveedu, R. Bronchopulmonary Dysplasia: Pathogenesis and Pathophysiology. J. Clin. Med. 2023, 12, 4207. https://doi.org/10.3390/jcm12134207
Dankhara N, Holla I, Ramarao S, Kalikkot Thekkeveedu R. Bronchopulmonary Dysplasia: Pathogenesis and Pathophysiology. Journal of Clinical Medicine. 2023; 12(13):4207. https://doi.org/10.3390/jcm12134207
Chicago/Turabian StyleDankhara, Nilesh, Ira Holla, Sumana Ramarao, and Renjithkumar Kalikkot Thekkeveedu. 2023. "Bronchopulmonary Dysplasia: Pathogenesis and Pathophysiology" Journal of Clinical Medicine 12, no. 13: 4207. https://doi.org/10.3390/jcm12134207
APA StyleDankhara, N., Holla, I., Ramarao, S., & Kalikkot Thekkeveedu, R. (2023). Bronchopulmonary Dysplasia: Pathogenesis and Pathophysiology. Journal of Clinical Medicine, 12(13), 4207. https://doi.org/10.3390/jcm12134207