

Rationale and Design of a Randomized Controlled Clinical Trial on the Safety and Efficacy of Flecainide versus Amiodarone in the Cardioversion of Atrial Fibrillation at the Emergency Department in Patients with Coronary Artery Disease (FLECA-ED)
 ,
,  , , , , ,
, , , , , 

Abstract
:1. Introduction
2. Objectives
3. Materials and Methods
3.1. Trial Design Overview
3.2. Eligibility Criteria
3.3. Randomization
3.4. Sample Size
3.5. Study Phases and Assessments
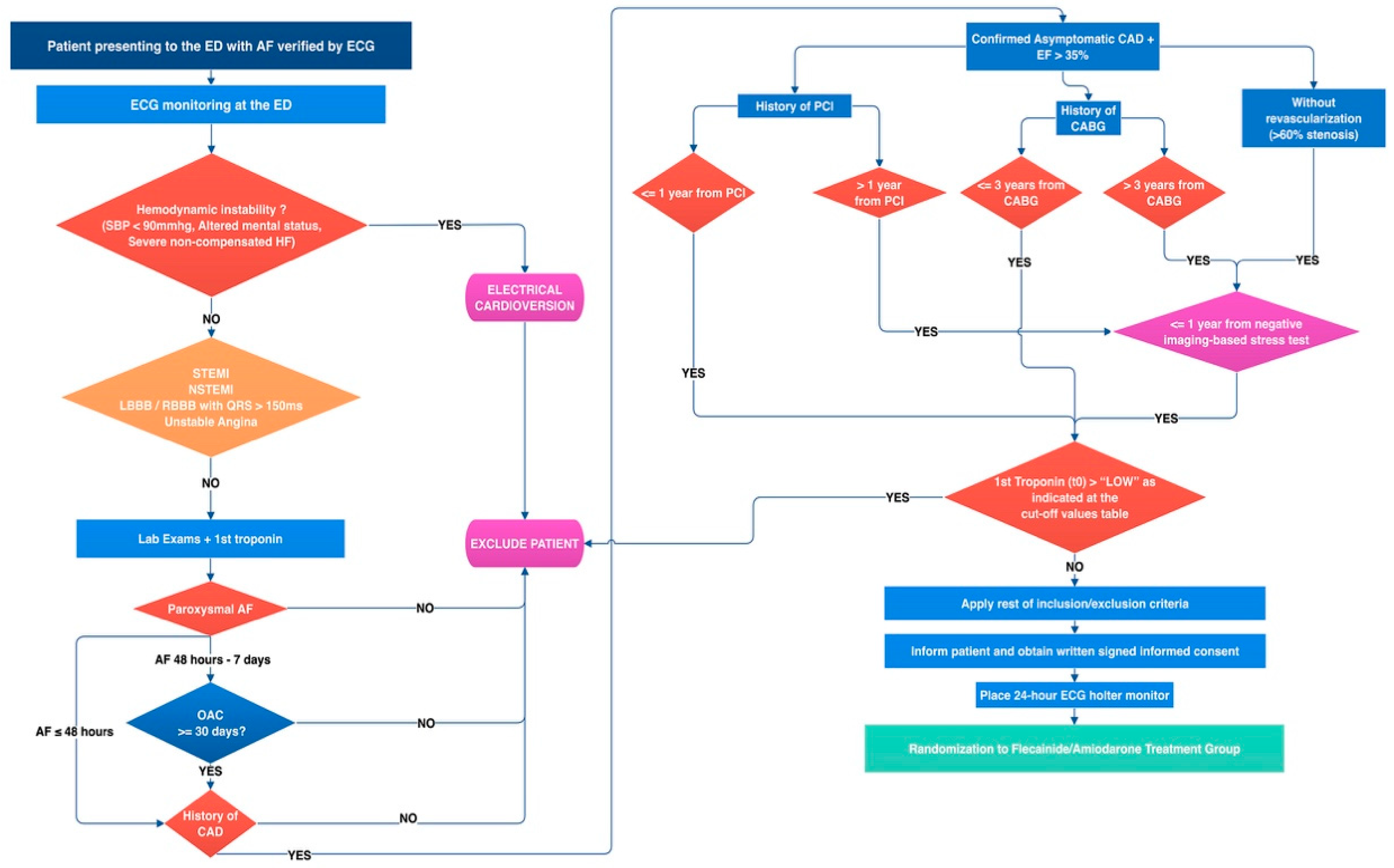
3.5.1. Screening Phase (Vscr, Emergency Department)
3.5.2. Intervention Visit (V0)
3.5.3. Follow-Up Phase (from Intervention Visit to 1 Month)
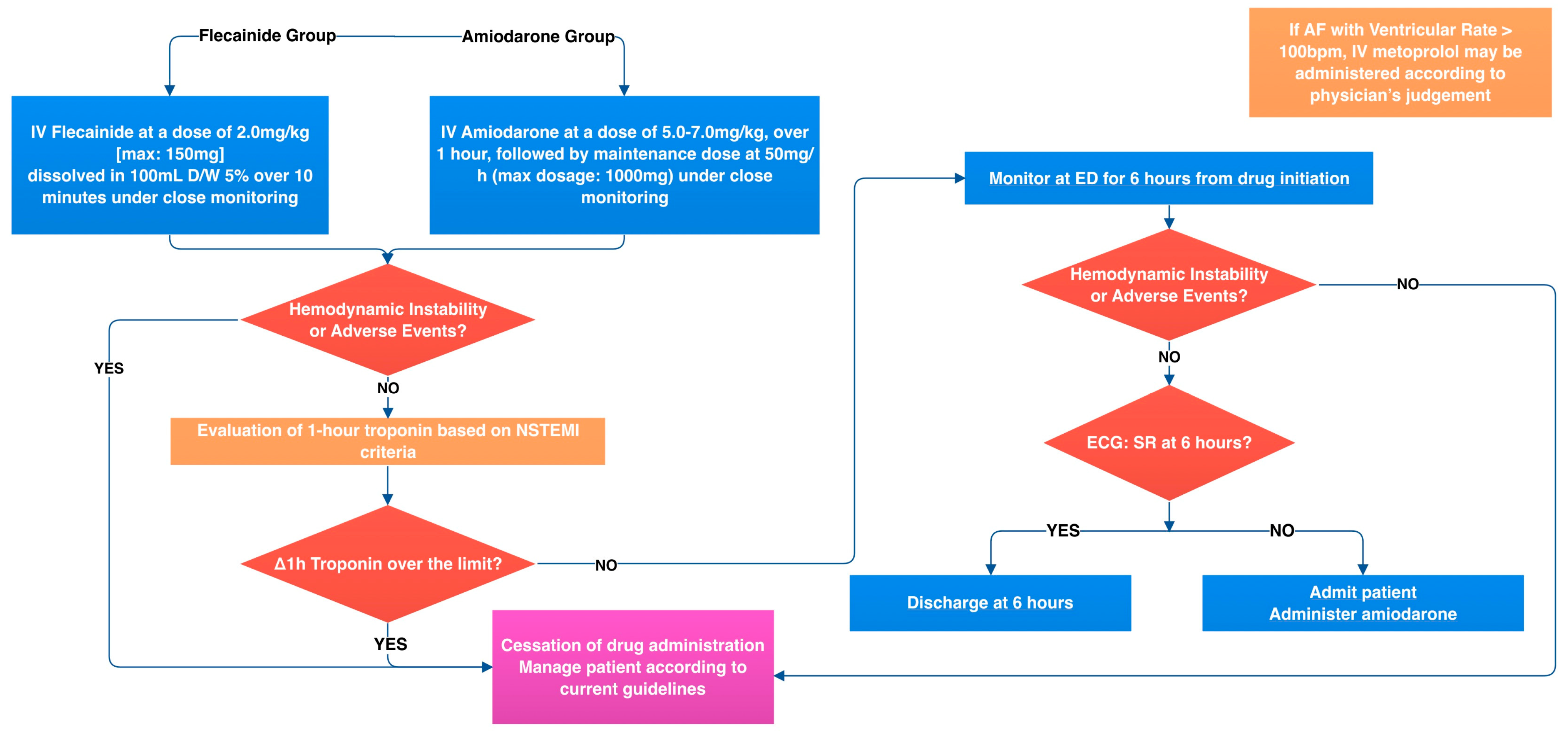
3.6. Interventions
3.7. Outcomes
3.8. Statistical Analysis
4. Discussion
5. Conclusions
Current Status
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.-H.; McAnulty, J.H., Jr.; Zheng, Z.-J.; et al. Worldwide Epidemiology of Atrial Fibrillation. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.P.; Ma, J.; Weissman, J.S.; Bernard, K.R.; Schuur, J.D. Hospital-Level Variation and Predictors of Admission after ED Visits for Atrial Fibrillation: 2006 to 2011. Am. J. Emerg. Med. 2016, 34, 2094–2100. [Google Scholar] [CrossRef]
- Jackson, S.L.; Tong, X.; Yin, X.; George, M.G.; Ritchey, M.D. Emergency Department, Hospital Inpatient, and Mortality Burden of Atrial Fibrillation in the United States, 2006 to 2014. Am. J. Cardiol. 2017, 120, 1966–1973. [Google Scholar] [CrossRef]
- Rozen, G.; Hosseini, S.M.; Kaadan, M.I.; Biton, Y.; Heist, E.K.; Vangel, M.; Mansour, M.C.; Ruskin, J.N. Emergency Department Visits for Atrial Fibrillation in the United States: Trends in Admission Rates and Economic Burden From 2007 to 2014. J. Am. Heart Assoc. 2018, 7, e009024. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.J.; Deshmukh, A.; Pant, S.; Singh, V.; Patel, N.; Arora, S.; Shah, N.; Chothani, A.; Savani, G.T.; Mehta, K.; et al. Contemporary Trends of Hospitalization for Atrial Fibrillation in the United States, 2000 through 2010. Circulation 2014, 129, 2371–2379. [Google Scholar] [CrossRef] [Green Version]
- Bellew, S.D.; Bremer, M.L.; Kopecky, S.L.; Lohse, C.M.; Munger, T.M.; Robelia, P.M.; Smars, P.A. Impact of an Emergency Department Observation Unit Management Algorithm for Atrial Fibrillation. J. Am. Heart Assoc. 2016, 5, e002984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchetti, A.; Williams, J.; Levi, S.; Akula, D. Impact of Emergency Department Management of Atrial Fibrillation on Hospital Charges. West. J. Emerg. Med. 2013, 14, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Besser, K.; Mills, A.M. Is Discharge to Home After Emergency Department Cardioversion Safe for the Treatment of Recent-Onset Atrial Fibrillation? Ann. Emerg. Med. 2011, 58, 517–520. [Google Scholar] [CrossRef]
- Martín, A.; Coll-Vinent, B.; Suero, C.; Fernández-Simón, A.; Sánchez, J.; Varona, M.; Cancio, M.; Sánchez, S.; Carbajosa, J.; Malagón, F.; et al. Benefits of Rhythm Control and Rate Control in Recent-onset Atrial Fibrillation: The HERMES-AF Study. Acad. Emerg. Med. 2019, 26, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Y.; Qiu, C.; Davis, P.J.; Jhaveri, M.; Prystowsky, E.N.; Kowey, P.; Weintraub, W.S. Predictors of Progression of Recently Diagnosed Atrial Fibrillation in REgistry on Cardiac Rhythm DisORDers Assessing the Control of Atrial Fibrillation (RecordAF)–United States Cohort. Am. J. Cardiol. 2013, 112, 79–84. [Google Scholar] [CrossRef]
- Ganesan, A.N.; Chew, D.P.; Hartshorne, T.; Selvanayagam, J.B.; Aylward, P.E.; Sanders, P.; McGavigan, A.D. The Impact of Atrial Fibrillation Type on the Risk of Thromboembolism, Mortality, and Bleeding: A Systematic Review and Meta-Analysis. Eur. Heart J. 2015, 37, 1591–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulizia, M.M.; Cemin, R.; Colivicchi, F.; Luca, L.D.; Lenarda, A.D.; Boriani, G.; Pasquale, G.D.; Nardi, F.; Scherillo, M.; Lucci, D.; et al. Management of Atrial Fibrillation in the Emergency Room and in the Cardiology Ward: The BLITZ AF Study. EP Eur. 2018, 21, 230–238. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the Diagnosis and Management of Atrial Fibrillation Developed in Collaboration with the European Association of Cardio-Thoracic Surgery (EACTS): The Task Force for the Diagnosis and Management of Atrial Fibrillation of the European Society of Cardiology (ESC) Developed with the Special Contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2020, 42, ehaa612. [Google Scholar] [CrossRef]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.-C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the Management of Atrial Fibrillation Developed in Collaboration with EACTS. EP Eur. 2016, 18, 1609–1678. [Google Scholar] [CrossRef]
- January, C.T.; Wann, L.S.; Calkins, H.; Chen, L.Y.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; Furie, K.L.; et al. 2019 AHA/ACC/HRS Focused Update of the 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation. Circulation 2019, 140, e125–e151. [Google Scholar] [CrossRef]
- Bager, J.-E.; Martín, A.; Dalmau, J.C.; Simon, A.; Merino, J.L.; Ritz, B.; Hartikainen, J.E.K. Vernakalant for Cardioversion of Recent-Onset Atrial Fibrillation in the Emergency Department: The SPECTRUM Study. Cardiology 2022, 147, 566–577. [Google Scholar] [CrossRef]
- Tsiachris, D.; Doundoulakis, I.; Tsioufis, P.; Pagkalidou, E.; Antoniou, C.-K.; Zafeiropoulos, S.M.; Gatzoulis, K.A.; Tsioufis, K.; Stefanadis, C. Reappraising the Role of Class Ic Antiarrhythmics in Atrial Fibrillation. Eur. J. Clin. Pharmacol. 2022, 78, 1039–1045. [Google Scholar] [CrossRef]
- Ibrahim, O.A.; Belley-Côté, E.P.; Um, K.J.; Baranchuk, A.; Benz, A.P.; Dalmia, S.; Wang, C.N.; Alhazzani, W.; Conen, D.; Devereaux, P.J.; et al. Single-Dose Oral Anti-Arrhythmic Drugs for Cardioversion of Recent-Onset Atrial Fibrillation: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. EP Eur. 2020, 23, 1200–1210. [Google Scholar] [CrossRef]
- Tsiachris, D.; Doundoulakis, I.; Pagkalidou, E.; Kordalis, A.; Deftereos, S.; Gatzoulis, K.A.; Tsioufis, K.; Stefanadis, C. Pharmacologic Cardioversion in Patients with Paroxysmal Atrial Fibrillation: A Network Meta-Analysis. Cardiovasc. Drug. Ther. 2021, 35, 293–308. [Google Scholar] [CrossRef] [PubMed]
- deSouza, I.S.; Tadrous, M.; Sexton, T.; Benabbas, R.; Carmelli, G.; Sinert, R. Pharmacologic Cardioversion of Recent-Onset Atrial Fibrillation and Flutter in the Emergency Department: A Systematic Review and Network Meta-Analysis. Ann. Emerg. Med. 2019, 76, 14–30. [Google Scholar] [CrossRef]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L.; et al. Mortality and Morbidity in Patients Receiving Encainide, Flecainide, or Placebo. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Platia, E.V.; Hallstrom, A.; Henthorn, R.W.; Buckingham, T.A.; Carlson, M.D.; Carson, P.E. Interaction of Baseline Characteristics with the Hazard of Encainide, Flecainide, and Moricizine Therapy in Patients with Myocardial Infarction. A Possible Explanation for Increased Mortality in the Cardiac Arrhythmia Suppression Trial (CAST). Circulation 2018, 90, 2843–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, T.; Pawitan, Y.; Greenberg, H.; Kuo, C.-S.; Reynolds-Haertle, R.A.; The CAST Investigators. Increased Risk of Death and Cardiac Arrest from Encainide and Flecainide in Patients after Non-Q-Wave Acute Myocardial Infarction in the Cardiac Arrhythmia Suppression Trial. Am. J. Cardiol. 1991, 68, 1551–1555. [Google Scholar] [CrossRef]
- Meinertz, T.; Lip, G.Y.H.; Lombardi, F.; Sadowski, Z.P.; Kalsch, B.; Camez, A.; Hewkin, A.; Eberle, S.; Investigators, E. Efficacy and Safety of Propafenone Sustained Release in the Prophylaxis of Symptomatic Paroxysmal Atrial Fibrillation (The European Rythmol/Rytmonorm Atrial Fibrillation Trial [ERAFT] Study). Am. J. Cardiol. 2002, 90, 1300–1306. [Google Scholar] [CrossRef]
- Kiani, S.; Sayegh, M.N.; Ibraham, R.; Bhatia, N.K.; Merchant, F.M.; Shah, A.D.; Westerman, S.B.; Lurgio, D.B.D.; Patel, A.M.; Thompkins, C.M.; et al. The Feasibility and Safety of Flecainide Use Among Patients with Varying Degrees of Coronary Disease. JACC Clin. Electrophysiol. 2023, in press. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Inclusion Criteria |
|---|
|
| Exclusion Criteria |
|
| Event | Screening Visit Vscr | Intervention Visit V0 | Visit at 24-h (V1) | Visit at 30 Days (V2) |
|---|---|---|---|---|
| Type of contact | On site | On site | On site | Phone |
| Study phase | Screening | Intervention | Follow-up | Follow-up |
| Inclusion/Exclusion criteria | x | |||
| Informed consent | x | |||
| Demographics and medical history | x | |||
| Physical Examination | x | x | x | |
| Electrocardiogram | x | x | x | |
| Echocardiography | x (if not within last year) | x | ||
| Chest X-ray | x | |||
| Laboratory Exams | x | x (1 h hs-TnI only) | ||
| 24-h ECG holter monitoring | x | |||
| Medication profile | x | x | ||
| Adverse event assessment | x | x | x | |
| Endpoints assessment | x | x | x | |
| Primary Outcomes |
|---|
|
| Secondary Outcomes |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsioufis, P.; Tsiachris, D.; Doundoulakis, I.; Kordalis, A.; Antoniou, C.-K.; Vlachakis, P.K.; Theofilis, P.; Manta, E.; Gatzoulis, K.A.; Parissis, J.; et al. Rationale and Design of a Randomized Controlled Clinical Trial on the Safety and Efficacy of Flecainide versus Amiodarone in the Cardioversion of Atrial Fibrillation at the Emergency Department in Patients with Coronary Artery Disease (FLECA-ED). J. Clin. Med. 2023, 12, 3961. https://doi.org/10.3390/jcm12123961
Tsioufis P, Tsiachris D, Doundoulakis I, Kordalis A, Antoniou C-K, Vlachakis PK, Theofilis P, Manta E, Gatzoulis KA, Parissis J, et al. Rationale and Design of a Randomized Controlled Clinical Trial on the Safety and Efficacy of Flecainide versus Amiodarone in the Cardioversion of Atrial Fibrillation at the Emergency Department in Patients with Coronary Artery Disease (FLECA-ED). Journal of Clinical Medicine. 2023; 12(12):3961. https://doi.org/10.3390/jcm12123961
Chicago/Turabian StyleTsioufis, Panagiotis, Dimitris Tsiachris, Ioannis Doundoulakis, Athanasios Kordalis, Christos-Konstantinos Antoniou, Panayotis K. Vlachakis, Panagiotis Theofilis, Eleni Manta, Konstantinos A. Gatzoulis, John Parissis, and et al. 2023. "Rationale and Design of a Randomized Controlled Clinical Trial on the Safety and Efficacy of Flecainide versus Amiodarone in the Cardioversion of Atrial Fibrillation at the Emergency Department in Patients with Coronary Artery Disease (FLECA-ED)" Journal of Clinical Medicine 12, no. 12: 3961. https://doi.org/10.3390/jcm12123961
APA StyleTsioufis, P., Tsiachris, D., Doundoulakis, I., Kordalis, A., Antoniou, C.-K., Vlachakis, P. K., Theofilis, P., Manta, E., Gatzoulis, K. A., Parissis, J., & Tsioufis, K. (2023). Rationale and Design of a Randomized Controlled Clinical Trial on the Safety and Efficacy of Flecainide versus Amiodarone in the Cardioversion of Atrial Fibrillation at the Emergency Department in Patients with Coronary Artery Disease (FLECA-ED). Journal of Clinical Medicine, 12(12), 3961. https://doi.org/10.3390/jcm12123961