

Cellular and Molecular Mechanisms Underlying Tricuspid Valve Development and Disease
 ,
,  ,
,  and
and 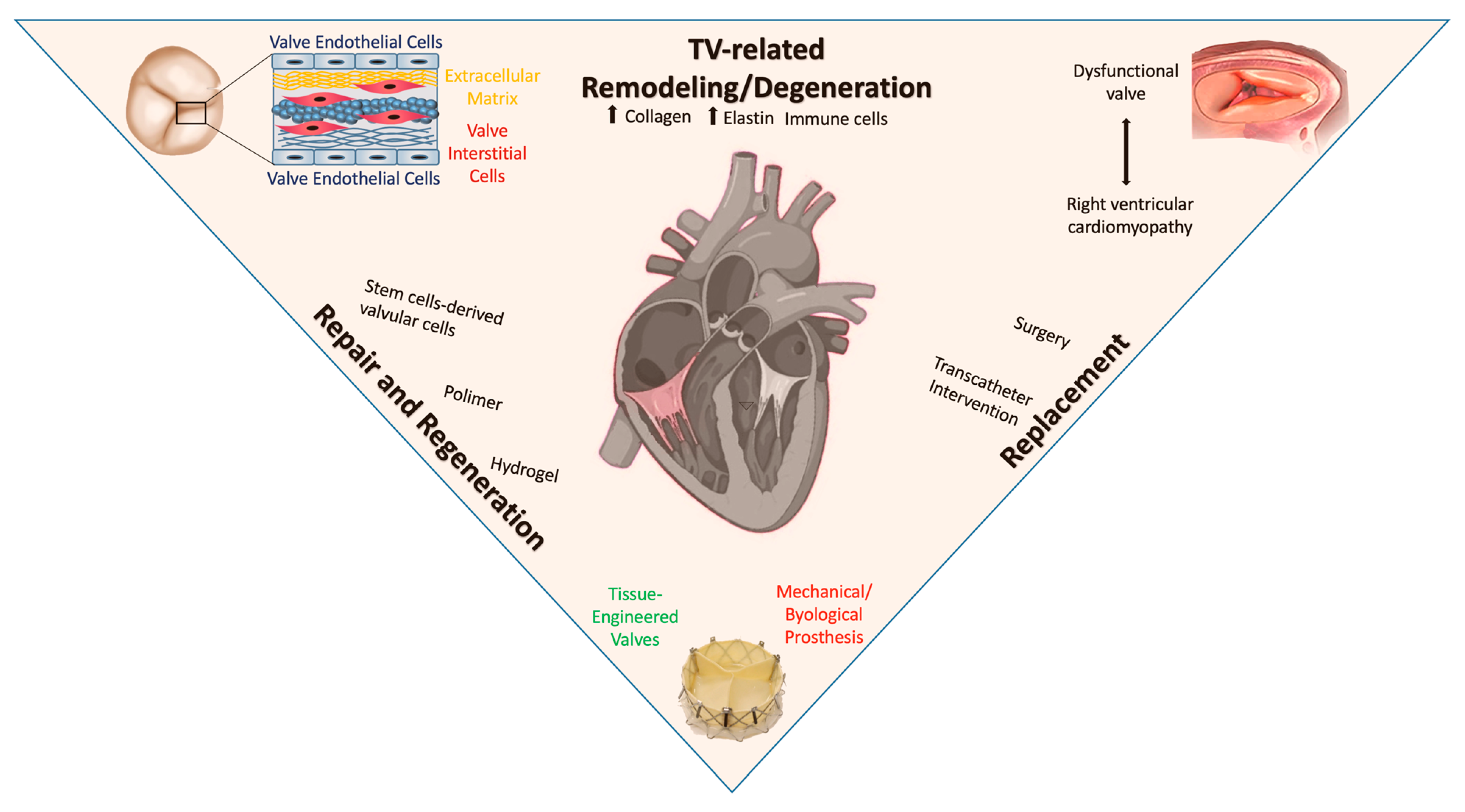{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tricuspid Valve Development: Embryology and Anatomy
3. Developmental Mechanisms Implicated in Tricuspid Valve Disease
4. Postnatal Valve Remodeling
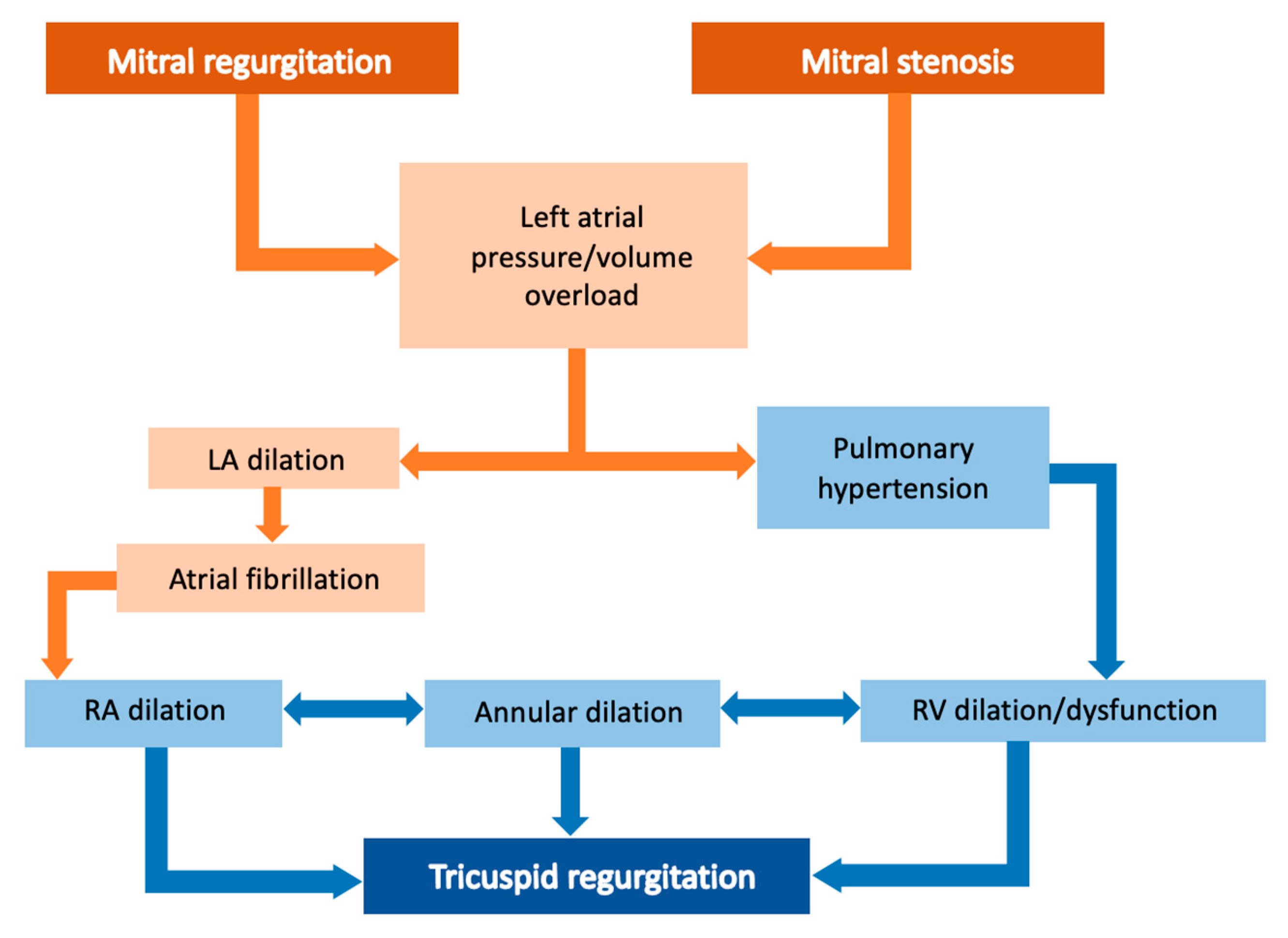
5. Right Ventricular Cardiomyopathy Induced by Tricuspid Valve Disease
6. Valvular Repair and Regeneration
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nkomo, V.T.; Gardin, J.M.; Skelton, T.N.; Gottdiener, J.S.; Scott, C.G.; Enriquez-Sarano, M. Burden of valvular heart diseases: A population-based study. Lancet 2006, 368, 1005–1011. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech. Ageing Dev. 2022, 208, 111740. [Google Scholar] [CrossRef]
- Topilsky, Y.; Maltais, S.; Medina Inojosa, J.; Oguz, D.; Michelena, H.; Maalouf, J.; Mahoney, D.W.; Enriquez-Sarano, M. Burden of Tricuspid Regurgitation in Patients Diagnosed in the Community Setting. JACC Cardiovasc. Imaging 2019, 12, 433–442. [Google Scholar] [CrossRef]
- Topilsky, Y.; Nkomo, V.T.; Vatury, O.; Michelena, H.I.; Letourneau, T.; Suri, R.M.; Pislaru, S.; Park, S.; Mahoney, D.W.; Biner, S.; et al. Clinical outcome of isolated tricuspid regurgitation. JACC Cardiovasc. Imaging 2014, 7, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Sorajja, P.; Whisenant, B.; Hamid, N.; Naik, H.; Makkar, R.; Tadros, P.; Price, M.J.; Singh, G.; Fam, N.; Kar, S.; et al. Transcatheter Repair for Patients with Tricuspid Regurgitation. N. Engl. J. Med. 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef]
- Sabatino, J.; Bassareo, P.P.; Ciliberti, P.; Cazzoli, I.; Oreto, L.; Secinaro, A.; Guccione, P.; Indolfi, C.; DI Salvo, G.; on behalf of the Congenital Heart Disease Working Group of the Italian Society of Cardiol.ogy (SIC). Tricuspid valve in congenital heart disease: Multimodality imaging and electrophysiological considerations. Minerva. Cardiol. Angiol. 2022, 70, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Hahn, R.T.; Weckbach, L.T.; Noack, T.; Hamid, N.; Kitamura, M.; Bae, R.; Lurz, P.; Kodali, S.K.; Sorajja, P.; Hausleiter, J.; et al. Proposal for a Standard Echocardiographic Tricuspid Valve Nomenclature. JACC Cardiovasc. Imaging 2021, 14, 1299–1305. [Google Scholar] [CrossRef]
- Shah, S.; Jenkins, T.; Markowitz, A.; Gilkeson, R.; Rajiah, P. Multimodal imaging of the tricuspid valve: Normal appearance and pathological entities. Insights Imaging 2016, 7, 649–667. [Google Scholar] [CrossRef]
- Volpato, V.; Lang, R.M.; Yamat, M.; Veronesi, F.; Weinert, L.; Tamborini, G.; Muratori, M.; Fusini, L.; Pepi, M.; Genovese, D.; et al. Echocardiographic Assessment of the Tricuspid Annulus: The Effects of the Third Dimension and Measurement Methodology. J. Am. Soc. Echocardiogr. 2019, 32, 238–247. [Google Scholar] [CrossRef]
- Bouchardy, J.; Therrien, J.; Pilote, L.; Ionescu-Ittu, R.; Martucci, G.; Bottega, N.; Maretlli, A.J. Atrial arrhythmias in adults with congenital heart disease. Circulation 2009, 120, 1679–1686. [Google Scholar] [CrossRef]
- Hebe, J. Ebstein’s anomaly in adults. Arrhythmias: Diagnosis and therapeutic approach. Thorac. Cardiovasc. Surg. 2000, 48, 214–219. [Google Scholar] [CrossRef]
- Hager, A.; Zrenner, B.; Brodherr-Heberlein, S.; Steinbauer-Rosenthal, I.; Schreieck, J.; Hess, J. Congenital and surgically acquired Wolff-Parkinson-White syndrome in patients with tricuspid atresia. J. Thorac. Cardiovasc. Surg. 2005, 130, 48–53. [Google Scholar] [CrossRef]
- Sylva, M.; van den Hoff, M.J.B.; Moorman, A.F.M. Development of the human heart. Am. J. Med. Genet. A 2014, 164, 1347–1371. [Google Scholar] [CrossRef]
- Walsh, E.P. Ebstein’s Anomaly of the Tricuspid Valve. JACC Clin. Electrophysiol. 2018, 4, 1271–1288. [Google Scholar] [CrossRef]
- Attenhofer Jost, C.H.; Connolly, H.M.; Dearani, J.A.; Edwards, W.D.; Danielson, G.K. Ebstein’s Anomaly. Circulation 2007, 115, 277–285. [Google Scholar] [CrossRef]
- Qureshi, M.Y.; Sommer, R.J.; Cabalka, A.K. Tricuspid Valve Imaging and Intervention in Pediatric and Adult Patients with Congenital Heart Disease. JACC Cardiovasc. Imaging 2019, 12, 637–651. [Google Scholar] [CrossRef]
- Lamers, W.H.; Virágh, S.; Wessels, A.; Moorman, A.F.M.; Anderson, R.H. Formation of the Tricuspid Valve in the Human Heart. Circulation 1995, 91, 111–121. [Google Scholar] [CrossRef]
- Gaussin, V.; Morley, G.E.; Cox, L.; Zwijsen, A.; Vance, K.M.; Emile, L.; Tian, Y.; Liu, J.; Hong, C.; Myers, D.; et al. Alk3/Bmpr1a receptor is required for development of the atrioventricular canal into valves and annulus fibrosus. Circ. Res. 2005, 97, 219–226. [Google Scholar] [CrossRef]
- Sicko, R.J.; Browne, M.L.; Rigler, S.L.; Druschel, C.M.; Liu, G.; Fan, R.; Romitti, P.A.; Caggana, M.; Kay, D.M.; Brody, L.C.; et al. Genetic Variants in Isolated Ebstein Anomaly Implicated in Myocardial Development Pathways. PLoS ONE 2016, 11, e0165174. [Google Scholar] [CrossRef]
- Digilio, M.C.; Bernardini, L.; Lepri, F.R.; Giuffrida, M.G.; Guida, V.; Baban, A.; Versacci, P.; Capolino, R.; Torres, B.; De Luca, A.; et al. Ebstein anomaly: Genetic heterogeneity and association with microdeletions 1p36 and 8p23.1. Am. J. Med. Genet. A 2011, 155, 2196–2202. [Google Scholar] [CrossRef] [PubMed]
- Postma, A.V.; van Engelen, K.; van de Meerakker, J.; Rahman, T.; Probst, S.; Baars, M.J.H.; Bauer, U.; Pickardt, T.; Sperling, S.R.; Berger, F.; et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ. Cardiovasc. Genet. 2011, 4, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Thareja, S.K.; Frommelt, M.A.; Lincoln, J.; Lough, J.W.; Mitchell, M.E.; Tomita-Mitchell, A. A Systematic Review of Ebstein’s Anomaly with Left Ventricular Noncompaction. J. Cardiovasc. Dev. Dis. 2022, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Patorno, E.; Huybrechts, K.F.; Hernandez-Diaz, S. Lithium Use in Pregnancy and the Risk of Cardiac Malformations. N. Engl. J. Med. 2017, 377, 893–894. [Google Scholar]
- Correa-Villaseñor, A.; Ferencz, C.; Neill, C.A.; Wilson, P.D.; Boughman, J.A. Ebstein’s malformation of the tricuspid valve: Genetic and environmental factors. Teratology 1994, 50, 137–147. [Google Scholar] [CrossRef]
- Lang, D.; Oberhoffer, R.; Cook, A.; Sharland, G.; Allan, L.; Fagg, N.; Andetrson, R.H. Pathologic spectrum of malformations of the tricuspid valve in prenatal and neonatal life. J. Am. Coll. Cardiol. 1991, 17, 1161–1167. [Google Scholar] [CrossRef]
- Shikata, F.; Nagashima, M.; Nishimura, K.; Suetsugu, F.; Kawachi, K. Repair of Congenitally Absent Chordae in a Tricuspid Valve Leaflet with Hypoplastic Papillary Muscle Using Artificial Chordae. J. Card. Surg. 2010, 25, 737–739. [Google Scholar] [CrossRef]
- Rao, P.S. A unified classification for tricuspid atresia. Am. Heart. J. 1980, 99, 799–804. [Google Scholar] [CrossRef]
- Floriani, M.A.; Glaeser, A.B.; Dorfman, L.E.; Agnes, G.; Rosa, R.F.M.; Zen, P.R.G. GATA 4 Deletions Associated with Congenital Heart Diseases in South Brazil. J. Pediatr. Genet. 2021, 10, 92–97. [Google Scholar]
- Svensson, E.C.; Huggins, G.S.; Lin, H.; Clendenin, C.; Jiang, F.; Tufts, R.; Dardik, F.B.; Letiden, J.M. A syndrome of tricuspid atresia in mice with a targeted mutation of the gene encoding Fog-2. Nat. Genet. 2000, 25, 353–356. [Google Scholar] [CrossRef]
- Lu, J.R.; McKinsey, T.A.; Xu, H.; Wang, D.Z.; Richardson, J.A.; Olson, E.N. FOG-2, a heart- and brain-enriched cofactor for GATA transcription factors. Mol. Cell. Biol. 1999, 19, 4495–4502. [Google Scholar] [CrossRef]
- Pugnaloni, F.; Digilio, M.C.; Putotto, C.; De Luca, E.; Marino, B.; Versacci, P. Genetics of atrioventricular canal defects. Ital. J. Pediatr. 2020, 46, 61. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Markwald, R.R.; Lin, A.E. Genetic aspects of atrioventricular septal defects. Am. J. Med. Genet. 2000, 97, 289–296. [Google Scholar] [CrossRef]
- Rajagopal, S.K.; Ma, Q.; Obler, D.; Shen, J.; Manichaikul, A.; Tomita-Mitchell, A.; Boardman, K.; Briggs, C.; Garg, V.; Srivastava, D.; et al. Spectrum of Heart Disease Associated with Murine and Human GATA4 Mutation. J. Mol. Cell. Cardiol. 2007, 43, 677–685. [Google Scholar] [CrossRef]
- Wessels, A.; Anderson, R.H.; Markwald, R.R.; Webb, S.; Brown, N.A.; Viragh, S.; Moorman, A.; Lamers, W. Atrial development in the human heart: An immunohistochemical study with emphasis on the role of mesenchymal tissues. Anat. Rec. 2000, 259, 288–300. [Google Scholar] [CrossRef]
- Burns, T.; Yang, Y.; Hiriart, E.; Wessels, A. The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects. J. Cardiovasc. Dev. Dis. 2016, 3, 29. [Google Scholar] [CrossRef]
- Posch, M.G.; Perrot, A.; Schmitt, K.; Mittelhaus, S.; Esenwein, E.M.; Stiller, B.; Geier, C.; Dietz, R.; Gessner, R.; Ozcelik, C.; et al. Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am. J. Med. Genet. A 2008, 146, 251–253. [Google Scholar] [CrossRef]
- Reamon-Buettner, S.M.; Borlak, J. TBX5 mutations in non-Holt-Oram syndrome (HOS) malformed hearts. Hum. Mutat. 2004, 24, 104. [Google Scholar] [CrossRef]
- Inga, A.; Reamon-Buettner, S.M.; Borlak, J.; Resnick, M.A. Functional dissection of sequence-specific NKX2-5 DNA binding domain mutations associated with human heart septation defects using a yeast-based system. Hum. Mol. Genet. 2005, 14, 1965–1975. [Google Scholar] [CrossRef]
- Zatyka, M.; Priestley, M.; Ladusans, E.J.; E Fryer, A.E.; Mason, J.; Latif, F.; Mahr, E.R. Analysis of CRELD1 as a candidate 3p25 atrioventicular septal defect locus (AVSD2). Clin. Genet. 2005, 67, 526–528. [Google Scholar] [CrossRef]
- Ai, D.; Fu, X.; Wang, J.; Lu, M.F.; Chen, L.; Baldini, A.; Klein, W.H.; Martina, J.F. Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc. Natl. Acad. Sci. USA 2007, 104, 9319–9324. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.E.; Burns, T.A.; Lockhart, M.M.; Phelps, A.L.; Van den Hoff, M.J.B.; Wessels, A. Wnt/β-catenin and sonic hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2016, 245, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Brida, M.; Diller, G.P.; Gatzoulis, M.A. Systemic Right Ventricle in Adults with Congenital Heart Disease. Circulation 2018, 137, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L.; Waldo, K.L. Neural crest and cardiovascular patterning. Circ. Res. 1995, 77, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Yelbuz, T.M.; Waldo, K.L.; Kumiski, D.H.; Stadt, H.A.; Wolfe, R.R.; Leatherbury, L.; Kirby, M.L. Shortened outflow tract leads to altered cardiac looping after neural crest ablation. Circulation 2002, 106, 504–510. [Google Scholar] [CrossRef]
- Williams, K.; Carson, J.; Lo, C. Genetics of Congenital Heart Disease. Biomolecules 2019, 9, 879. [Google Scholar] [CrossRef]
- Gao, W.; Higaki, T.; Eguchi-Ishimae, M.; Iwabuki, H.; Wu, Z.; Yamamoto, E.; Takata, H.; Ohta, M.; Imoto, I.; Ishii, E.; et al. DGCR6 at the proximal part of the DiGeorge critical region is involved in conotruncal heart defects. Hum. Genome. Var. 2015, 2, 15004. [Google Scholar] [CrossRef]
- Warburton, D.; Ronemus, M.; Kline, J.; Jobanputra, V.; Williams, I.; Anyane-Yeboa, K.; Chung, W.; Yu, L.; Wong, N.; Awad, D.; et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum. Genet. 2014, 133, 11–27. [Google Scholar] [CrossRef]
- Kerstjens-Frederikse, W.S.; van de Laar, I.M.B.H.; Vos, Y.J.; Verhagen, J.M.A.; Berger, R.M.F.; Lichtenbelt, K.D.; Wassink-Ruiter, J.S.K.; van der Zwaag, P.A.; Sarvaas, G.J.D.M.; Bergman, K.A.; et al. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: Data on 428 probands with left-sided CHD and their families. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 914–923. [Google Scholar] [CrossRef]
- Rabkin-Aikawa, E.; Mayer, J.E.; Schoen, F.J. Heart valve regeneration. Adv. Biochem. Eng. Biotechnol. 2005, 94, 141–179. [Google Scholar]
- Schoen, F.J. Cardiac valves and valvular pathology: Update on function, disease, repair, and replacement. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2005, 14, 189–194. [Google Scholar] [CrossRef]
- Hinton, R.B.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzy, K.E. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef]
- Amofa, D.; Hulin, A.; Nakada, Y.; Sadek, H.A.; Yutzey, K.E. Hypoxia promotes primitive glycosaminoglycan-rich extracellular matrix composition in developing heart valves. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1143–H1154. [Google Scholar] [CrossRef]
- Hulin, A.; Hortells, L.; Gomez-Stallons, M.V.; O’Donnell, A.; Chetal, K.; Adam, M.; Lancellotti, P.; Oury, C.; Potter, S.S.; Salomonis, N.; et al. Maturation of heart valve cell populations during postnatal remodeling. Dev. Camb. Engl. 2019, 146, dev173047. [Google Scholar] [CrossRef]
- Mjaatvedt, C.H.; Kern, C.B.; Norris, R.A.; Fairey, S.; Cave, C.L. Normal distribution of melanocytes in the mouse heart. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol. 2005, 285, 748–757. [Google Scholar] [CrossRef]
- Yajima, I.; Larue, L. The location of heart melanocytes is specified and the level of pigmentation in the heart may correlate with coat color. Pigment. Cell Melanoma. Res. 2008, 21, 471–476. [Google Scholar] [CrossRef]
- Lincoln, J.; Lange, A.W.; Yutzey, K.E. Hearts and bones: Shared regulatory mechanisms in heart valve, cartilage, tendon, and bone development. Dev. Biol. 2006, 294, 292–302. [Google Scholar] [CrossRef]
- Lei, T.C.; Vieira, W.D.; Hearing, V.J. In vitro migration of melanoblasts requires matrix metalloproteinase-2: Implications to vitiligo therapy by photochemotherapy. Pigment. Cell Res. 2002, 15, 426–432. [Google Scholar] [CrossRef]
- Brito, F.C.; Kos, L. Timeline and distribution of melanocyte precursors in the mouse heart. Pigment. Cell Melanoma. Res. 2008, 21, 464–470. [Google Scholar] [CrossRef]
- Balani, K.; Brito, F.C.; Kos, L.; Agarwal, A. Melanocyte pigmentation stiffens murine cardiac tricuspid valve leaflet. J. R. Soc. Interface 2009, 6, 1097–1102. [Google Scholar] [CrossRef]
- Braunwald, N.S.; Ross, J.; Morrow, A.G. Conservative management of tricuspid regurgitation in patients undergoing mitral valve replacement. Circulation 1967, 5 (Suppl. S4), I63–I69. [Google Scholar] [CrossRef] [PubMed]
- Nath, J.; Foster, E.; Heidenreich, P.A. Impact of tricuspid regurgitation on long-term survival. J. Am. Coll. Cardiol. 2004, 43, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.R.; Sell, J.E.; McIntosh, C.L.; Clark, R.E. Tricuspid regurgitation in patients with acquired, chronic, pure mitral regurgitation. I. Prevalence, diagnosis, and comparison of preoperative clinical and hemodynamic features in patients with and without tricuspid regurgitation. J. Thorac. Cardiovasc. Surg. 1987, 94, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Shiran, A.; Sagie, A. Tricuspid regurgitation in mitral valve disease incidence, prognostic implications, mechanism, and management. J. Am. Coll. Cardiol. 2009, 53, 401–408. [Google Scholar] [CrossRef]
- Aydın, A.; Demircin, M.; Doğan, R.; Yılmaz, M.; Paşaoğlu, İ. Predictors for Progression of Tricuspid Insufficiency following Left-Sided Valvular Surgery: A Retrospective Cohort Study. Heart Surg. Forum. 2019, 22, E262–E268. [Google Scholar] [CrossRef]
- Ton-Nu, T.T.; Levine, R.A.; Handschumacher, M.D.; Dorer, D.J.; Yosefy, C.; Fan, D.; Hua, L.; Jiang, L.; Hung, J. Geometric determinants of functional tricuspid regurgitation: Insights from 3-dimensional echocardiography. Circulation 2006, 114, 143–149. [Google Scholar] [CrossRef]
- Fukuda, S.; Saracino, G.; Matsumura, Y.; Daimon, M.; Tran, H.; Greenberg, N.L.; Hozumi, T.; Yoshikawa, J.; Thomas, J.D.; Shiota, T. Three-dimensional geometry of the tricuspid annulus in healthy subjects and in patients with functional tricuspid regurgitation: A real-time, 3-dimensional echocardiographic study. Circulation 2006, 114 (Suppl. S1), I492–I498. [Google Scholar] [CrossRef]
- Taramasso, M.; Pozzoli, A.; Guidotti, A.; Nietlispach, F.; Inderbitzin, D.T.; Benussi, S.; Alfieri, O.; Maisano, F. Percutaneous tricuspid valve therapies: The new frontier. Eur. Heart J. 2017, 38, 639–647. [Google Scholar] [CrossRef]
- Dietz, M.F.; Prihadi, E.A.; van der Bijl, P.; Goedemans, L.; Mertens, B.J.A.; Gursoy, E.; van Getnderen, O.S.; Marsan, N.A.; Delgado, V.; Bax, J.J. Prognostic Implications of Right Ventricular Remodeling and Function in Patients with Significant Secondary Tricuspid Regurgitation. Circulation 2019, 140, 836–845. [Google Scholar] [CrossRef]
- Mutlak, D.; Lessick, J.; Reisner, S.A.; Aronson, D.; Dabbah, S.; Agmon, Y. Echocardiography-based spectrum of severe tricuspid regurgitation: The frequency of apparently idiopathic tricuspid regurgitation. J. Am. Soc. Echocardiogr. 2007, 20, 405–408. [Google Scholar] [CrossRef]
- Topilsky, Y.; Khanna, A.; Le Tourneau, T.; Park, S.; Michelena, H.; Suri, R.; Mahoney, D.W.; Enriquez-Sarano, M. Clinical context and mechanism of functional tricuspid regurgitation in patients with and without pulmonary hypertension. Circ. Cardiovasc. Imaging 2012, 5, 314–323. [Google Scholar] [CrossRef]
- Dandel, M.; Hetzer, R. Echocardiographic assessment of the right ventricle: Impact of the distinctly load dependency of its size, geometry and performance. Int. J. Cardiol. 2016, 221, 1132–1142. [Google Scholar] [CrossRef]
- Tian, C.; Yang, Y.; Ke, Y.; Yang, L.; Zhong, L.; Wang, Z.; Huang, H. Integrative Analyses of Genes Associated with Right Ventricular Cardiomyopathy Induced by Tricuspid Regurgitation. Front. Genet. 2021, 12, 708275. [Google Scholar] [CrossRef]
- Polimeni, A.; Albanese, M.; Salerno, N.; Aquila, I.; Sabatino, J.; Sorrentino, S.; Leo, I.; Cacia, M.; Signorile, V.; Mongiardo, A. Predictors of outcomes in patients with mitral regurgitation undergoing percutaneous valve repair. Sci. Rep. 2020, 10, 17144. [Google Scholar] [CrossRef]
- Harken, D.E. Heart valves: Ten commandments and still counting. Ann. Thorac. Surg. 1989, 48 (Suppl. S3), S18–S19. [Google Scholar] [CrossRef]
- Mancuso, A.; Cianflone, E.; Cristiano, M.C.; Salerno, N.; Tarsitano, M.; Marino, F.; Molinaro, C.; Fresta, M.; Torella, D.; Paolino, D. Lyotropic Liquid Crystals: A Biocompatible and Safe Material for Local Cardiac Application. Pharmaceutics 2022, 14, 452. [Google Scholar] [CrossRef]
- Nachlas, A.L.Y.; Li, S.; Jha, R.; Singh, M.; Xu, C.; Davis, M.E. Human iPSC-derived mesenchymal stem cells encapsulated in PEGDA hydrogels mature into valve interstitial-like cells. Acta Biomater. 2018, 71, 235–246. [Google Scholar] [CrossRef]
- Wissing, T.B.; Bonito, V.; Bouten, C.V.C.; Smits, A.I.P.M. Biomaterial-driven in situ cardiovascular tissue engineering-a multi-disciplinary perspective. NPJ. Regen. Med. 2017, 2, 18. [Google Scholar] [CrossRef]
- Gerdisch, M.; Stelly, T.; Slaughter, M.; Rodriguez, V. Early Results from the FDA Extracellular Matrix (ECM) Cylinder Valve Clinical Feasibility Trial. Struct. Heart 2020, 4, 102. [Google Scholar] [CrossRef]
- Salerno, N.; Salerno, L.; Marino, F.; Scalise, M.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Cianflone, E.; Urbanek, K.; Torella, D. Myocardial regeneration protocols towards the routine clinical scenario: An unseemly path from bench to bedside. Eclinicalmedicine 2022, 50, 101530. [Google Scholar] [CrossRef]
- Cheng, L.; Xie, M.; Qiao, W.; Song, Y.; Zhang, Y.; Geng, Y.; Xu, W.; Wang, L.; Wang, Z.; Huang, K.; et al. Generation and characterization of cardiac valve endothelial-like cells from human pluripotent stem cells. Commun. Biol. 2021, 4, 1039. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Marino, F.; Salerno, L.; Cianflone, E.; Molinaro, C.; Salerno, N.; De Angelis, A.; Viglietto, G.; Urbanek, K.; Torella, D. From Spheroids to Organoids: The Next Generation of Model Systems of Human Cardiac Regeneration in a Dish. Int. J. Mol. Sci. 2021, 22, 13180. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, C.; Salerno, L.; Marino, F.; Scalise, M.; Salerno, N.; Pagano, L.; De Angelis, A.; Cianflone, E.; Torella, D.; Urbanek, K. Unraveling and Targeting Myocardial Regeneration Deficit in Diabetes. Antioxid 2022, 11, 208. [Google Scholar] [CrossRef] [PubMed]
- Fioretta, E.S.; Motta, S.E.; Lintas, V.; Loerakker, S.; Parker, K.K.; Baaijens, F.P.T.; Falk, V.; Hoerstrup, S.P.; Emmert, M.Y. Next-generation tissue-engineered heart valves with repair, remodelling and regeneration capacity. Nat. Rev. Cardiol. 2021, 18, 92–116. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salerno, N.; Panuccio, G.; Sabatino, J.; Leo, I.; Torella, M.; Sorrentino, S.; De Rosa, S.; Torella, D. Cellular and Molecular Mechanisms Underlying Tricuspid Valve Development and Disease. J. Clin. Med. 2023, 12, 3454. https://doi.org/10.3390/jcm12103454
Salerno N, Panuccio G, Sabatino J, Leo I, Torella M, Sorrentino S, De Rosa S, Torella D. Cellular and Molecular Mechanisms Underlying Tricuspid Valve Development and Disease. Journal of Clinical Medicine. 2023; 12(10):3454. https://doi.org/10.3390/jcm12103454
Chicago/Turabian StyleSalerno, Nadia, Giuseppe Panuccio, Jolanda Sabatino, Isabella Leo, Michele Torella, Sabato Sorrentino, Salvatore De Rosa, and Daniele Torella. 2023. "Cellular and Molecular Mechanisms Underlying Tricuspid Valve Development and Disease" Journal of Clinical Medicine 12, no. 10: 3454. https://doi.org/10.3390/jcm12103454
APA StyleSalerno, N., Panuccio, G., Sabatino, J., Leo, I., Torella, M., Sorrentino, S., De Rosa, S., & Torella, D. (2023). Cellular and Molecular Mechanisms Underlying Tricuspid Valve Development and Disease. Journal of Clinical Medicine, 12(10), 3454. https://doi.org/10.3390/jcm12103454