
Hereditary Hemorrhagic Telangiectasia and Arterio-Venous Malformations—From Diagnosis to Therapeutic Challenges

 ,
,  , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hereditary Hemorrhagic Telangiectasia—Diagnosis
- Arterial hypoxemia caused by right-to-left shunts;
- A paradoxical embolism with transient ischemic attack or stroke, and brain abscess caused by the absence of normally filtering capillary bed;
- Chronic bleeding like hemoptysis or hemothorax due to the rupture of the thin wall of the AVMs.
2.1. Classification
- Type 1 is associated with an ENG gene mutation, encoding endoglin, and involves a higher number of pulmonary and central nervous system vascular malformations.
- Type 2 is associated with activin A receptor type II—like 1 (ACVRL1) gene mutations and with a higher incidence of primary pulmonary hypertension and hepatic manifestations of the disease. These two types are found in approximately 85% of cases referred to genetic testing for clinical suspicion of HHT [2].
- Type 3 is a subset linked to chromosome 5q31, but the gene has not been identified [6].
- simple (one artery supplying an aneurismal communication with a single draining vein) and
- complex (two or more arterial branches with two or more draining veins).
2.2. Clinical Diagnosis
- Telangiectasia: these are pink to red lesions, usually 0.5–1.0 mm in diameter, which may appear at any site, especially on the face, nose, fingertips, lips, tongue, oral, and gastrointestinal mucosa. These lesions blanch when pressure is applied and refill immediately after release. Given their thin walls and close situation to the surface of the skin and mucosa, they are predisposed to rupture and bleeding [13].
- Epistaxis: the most prevalent symptom in HHT is epistaxis related to nasal telangiectasia, which can be spontaneous or recurrent and in severe cases can lead to anemia.
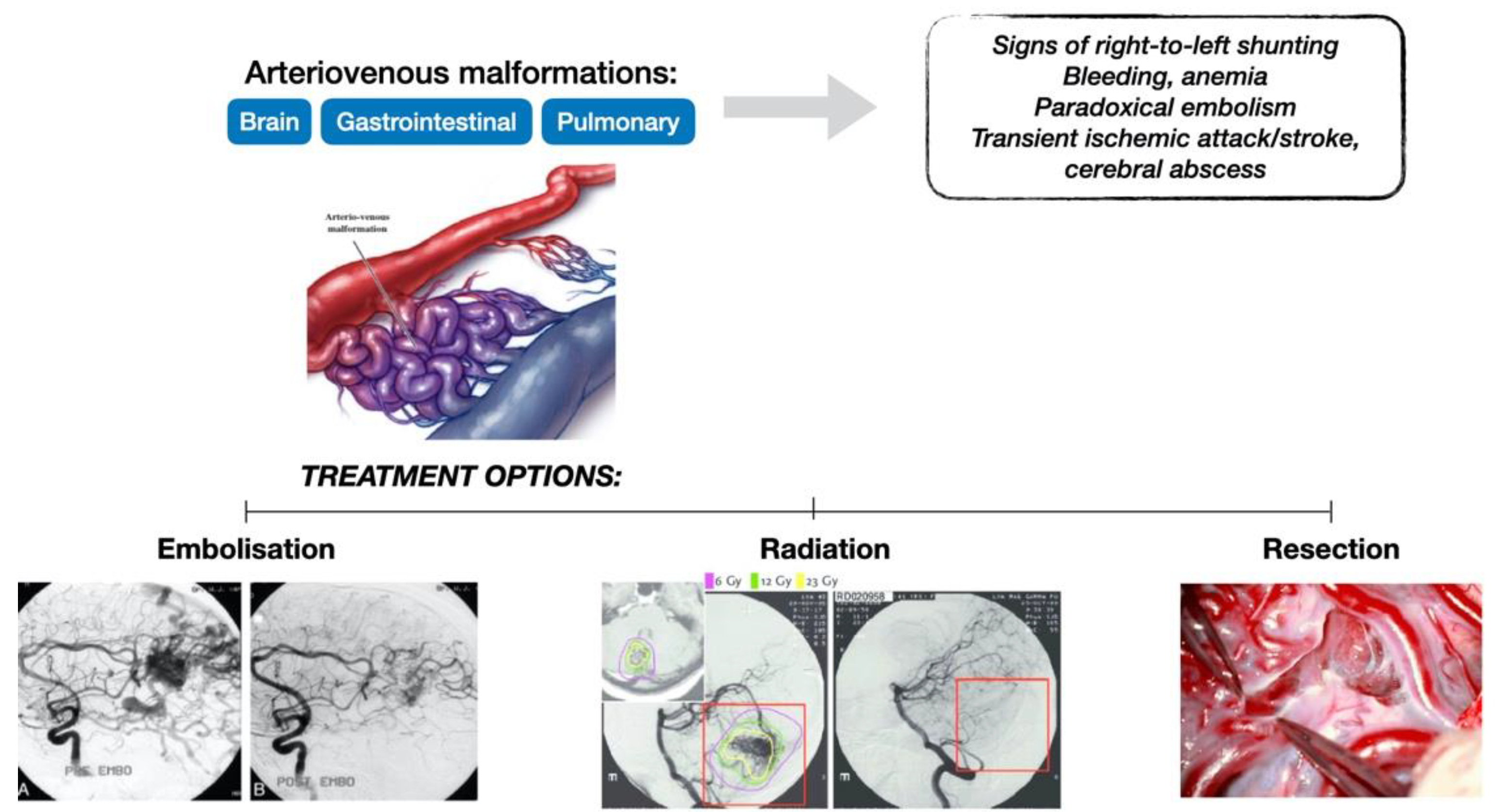
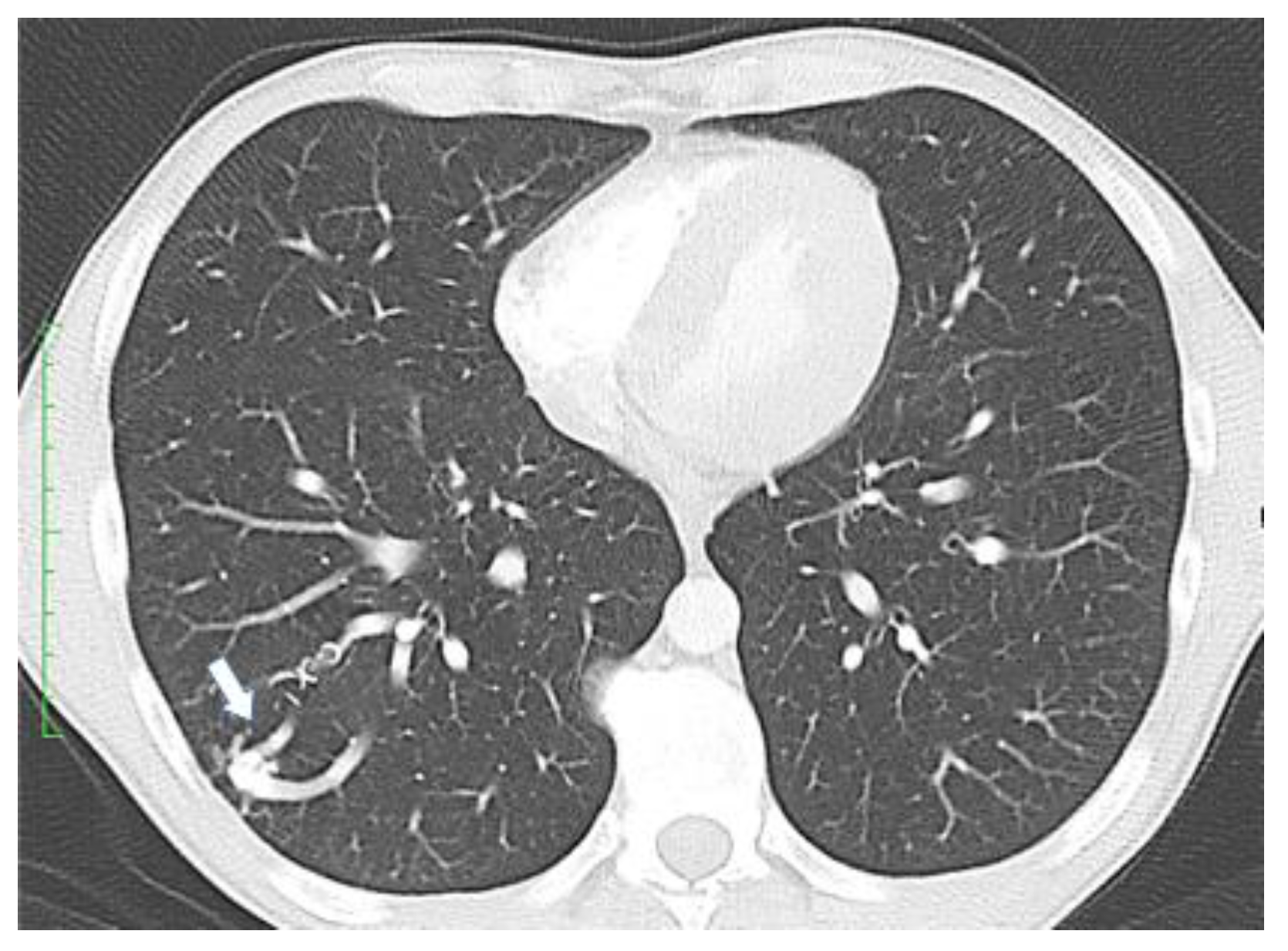
- Visceral lesions: represented by AMVs that may arise in various organs, with characteristic clinical presentations. Gastrointestinal lesions are usually evaluated by upper or lower digestive endoscopies and may become symptomatic through massive hemorrhages or anemia [7]. Pulmonary AVMs can be asymptomatic or may present with bleeding (hemoptysis, hemothorax) or signs of right-to-left shunting such as hypoxemia, cyanosis, poor exercise tolerance, dyspnea, orthodeoxia (decrease in oxygen saturation in an upright posture), nail clubbing, polycythemia, pulmonary embolism, heart failure, and pulmonary hypertension [14]. Neurological symptoms resulting from right-to-left shunting with embolic or infected material through pulmonary AVMs are migraines, transient ischemic attacks, strokes, seizures, and cerebral abscesses. The annual rate for stroke calculated by a meta-analysis is 0.92% and 0.32% for brain abscess [2]. Lesions in the liver may lead to congestive heart failure, portal hypertension, liver failure, and subsequent encephalopathy, and can be evaluated either by Doppler US or CT scan [7]. In the central nervous system, AVMs may be present in the brain or spinal cord and may determine cerebral abscesses, transient ischemic attacks, or even ischemic strokes [7]. These lesions can be examined by angiography or MRI scan.
- Family history: first degree relatives affected by the disease.
- Stage I (latency or inactivity): localized hyperemia. Pediatric patients can remain stable for long time in this stage if AVMs are small.
- Stage II (expansion): increasing of arterio-venous shunting.
- Stage III (progression): onset of symptoms and signs, such as pain, bleeding, or ulceration.
- Stage IV: large AVMs with a risk of cardiac involvement and decompensation.
2.3. Paraclinical Diagnosis
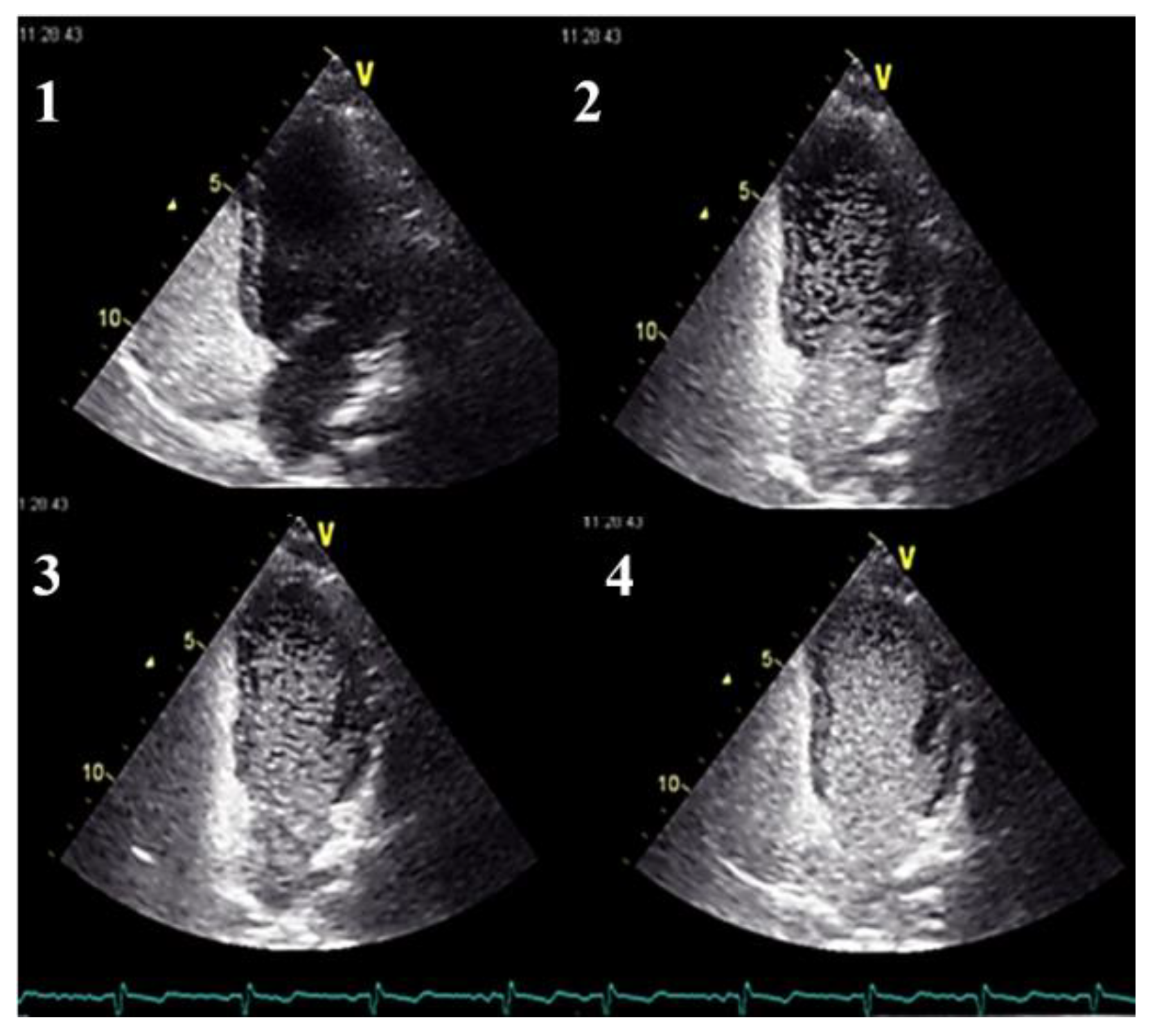
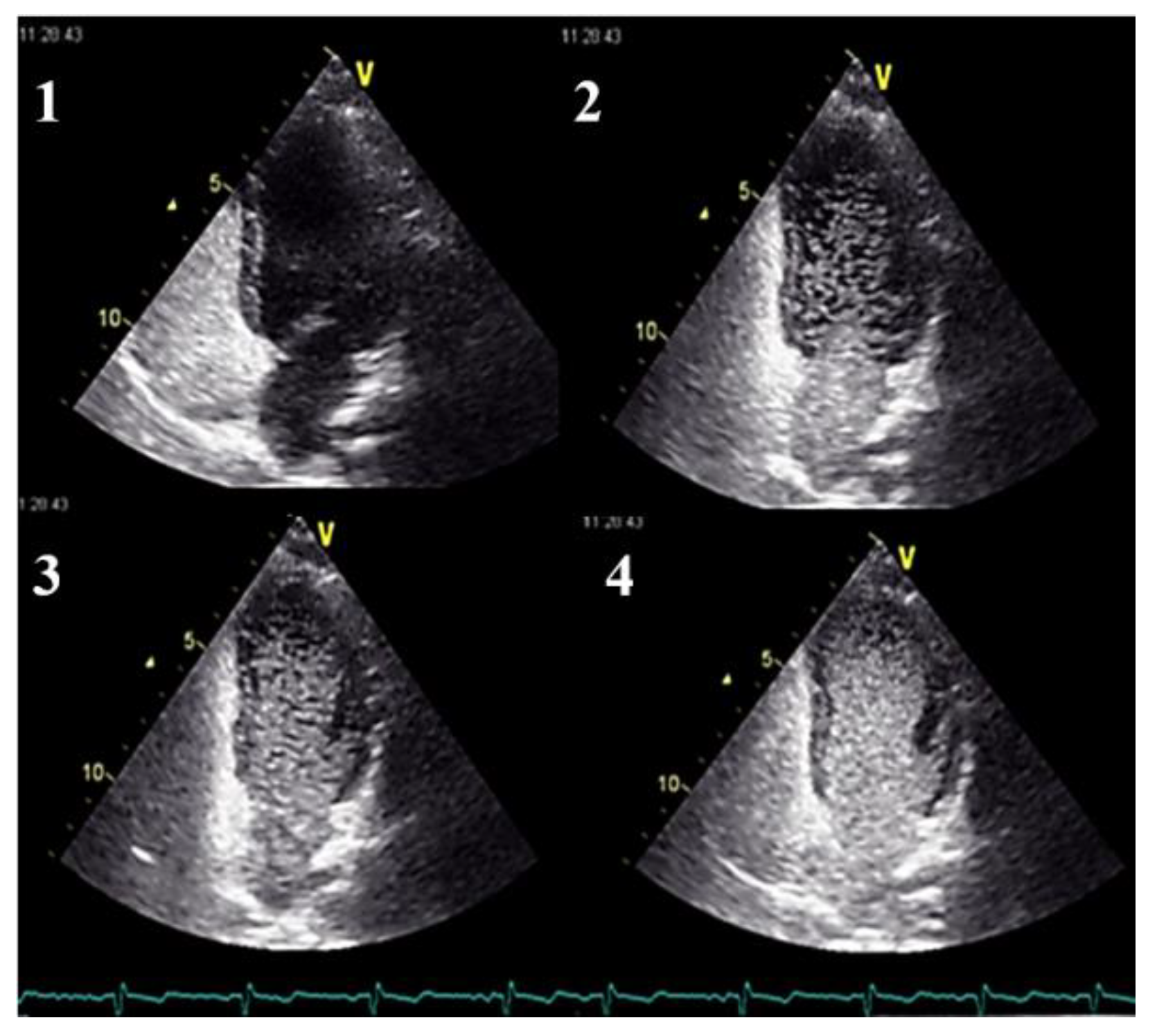
- in the absence of symptoms, pulmonary AVMs screening should be performed initially, either by an agitated saline transthoracic contrast echocardiography or by a low-dose chest CT without contrast, according to local availability and expertise. When opting for a chest CT examination, it should ideally be performed early in the second trimester.
- in cases presenting with symptoms indicative of pulmonary AVMs, diagnostic testing by a low-dose non-contrast chest CT scan should be performed. This examination can be carried out at any gestational age, as clinically indicated.
- treatment of pulmonary AVMs is recommended starting with the second trimester, in the absence of other clinical contraindications.
3. Hereditary Hemorrhagic Telangiectasia—Therapeutic Challenges
- Screening for liver AVMs should be performed in adults with definite or suspected HHT.
- In HHT patients with signs or symptoms suggestive of complicated liver AVMs, such as abnormal liver function tests, abdominal pain, portal hypertension, encephalopathy, pulmonary hypertension, heart failure, or abnormal levels of cardiac biomarkers, diagnostic testing should be performed using a Doppler ultrasound, a multiphase contrast CT scan, or contrast abdominal magnetic resonance imaging.
- An intensive first-line approach should only be instated in cases presenting with complicated or symptomatic liver AVMs, adapted to the specific type of complication.
- HHT cases that exhibit high-output cardiac failure and pulmonary hypertension should be managed multidisciplinarily by the Center of Excellence for HHT and a HHT cardiologist or a pulmonary hypertension specialty clinic.
- Clinicians should evaluate liver AVMs prognosis using available predictors in an effort to select patients in which closer monitoring may be advised.
- Intravenous administration of bevacizumab should be taken into consideration in cases with symptomatic high-output cardiac failure caused by liver AVMs with an insufficient response to first-line treatment.
- A referral to evaluation for liver transplantation is recommended in cases of refractory high-output cardiac failure, biliary ischemia, or complicated portal hypertension that arise as complications of liver AVMs.
- In patients with a proven or suspected HHT diagnosis, liver biopsy should be avoided.
- Given the fact that hepatic artery embolization is a temporising intervention with significant morbidity and mortality, it should not be performed in cases with liver AVMs.
- Clinicians should screen all patients with a suspected or confirmed HHT diagnosis for pulmonary AVMs.
- Clinicians should use transthoracic contrast echocardiography as the initial screening test for pulmonary AVMs.
- Clinicians should treat pulmonary AVMs with transcatheter embolotherapy.
- Clinicians should provide the following long-term recommendations to patients with diagnosed pulmonary AVMs (irrespective of previous treatment):
- In the event of procedures with risk of bacteremia, antibiotic prophylaxis should be done;
- When intravenous access is in place, special care should be taken in order to avoid intravenous air;
- SCUBA diving should be avoided.
- Long-term follow-up should be provided in cases with pulmonary AVMs, in an effort to diagnose the possible growth of untreated AVMs or the reperfusion of AVMs that have undegone treatment.
- Magnetic resonance imaging as a screening method for brain AVMs in patients with suspected or definite HHT should be performed using a protocol both with and without contrast administration and implementing sequences that detect blood products, for sensitivity optimization.
- In cases of acute hemorrhage arrising as a complication of brain AVMs, definitive treatment should be recommended in a specialized center with neurovascular expertise.
- In all other patients diagnosed with brain AVMs, invasive testing and individualized management should be recommended in a center with neurovascular expertise.
- In pregnant women with possible or confirmed HHT with concurrent asymptomatic brain AVMs, definitive treatment of the brain AVMs should ideally be postponed until after delivery (which should follow obstetrical indications).
- In HHT-related epistaxis, moisturizing topical therapies that humidify the nasal mucosa should be used.
- Oral administration of tranexamic acid should be considered in cases of epistaxis that do not resolve with the use of moisturizing topical therapies.
- Ablative therapies such as laser treatment, radiofrequency ablation, electrosurgery, and sclerotherapy should be considered for the treatment for nasal telangiectasias, in cases that are non-responsive to moisturizing topical therapies.
- Systemic antiangiogenic agents should be considered for the management of epistaxis that has failed to resolve with any of the aforementioned therapies.
- Septodermoplasty or nasal closure should be taken into consideration in cases of epistaxis with insufficient improvement after topical application of moisturising therapies, ablative procedures and/or administration of tranexamic acid.
- Physicians should recommend the topical use of nasal mucosa humidifying agents for HHT-related epistaxis prophylaxis.
- Patients with HHT-related epistaxis who are willing to undergo treatment should be referred to otorhinolaryngologists with HHT expertise.
- If nasal surgery is considered for reasons other than epistaxis, an evaluation should be performed by an otorhinolaryngologist with expertise in HHT-related epistaxis.
- Interventions performed for acute epistaxis should involve packing with material or products that are associated with a low likelihood of rebleeding upon removal, such as lubricated low-pressure pneumatic packing.
- Esophagogastroduodenoscopy should be used as the first-line diagnostic investigation in patients with possible HHT-related bleeding. If colorectal cancer screening criteria are fullfilled and in cases with either genetically proven or suspected SMAD4-HHT syndrome, a colonoscopy should also be performed.
- If the esophagogastroduodenoscopy does not identify significant HHT-related telangiectasia, a videocapsule endoscopy should be taken into consideration.
- HHT-related gastrointestinal bleeding severity should be assessed using the following framework:
- Mild: oral iron replacement is sufficient to achieve the target hemoglobin level;
- Moderate: hemoglobin goals are met with intravenous iron treatment;
- Severe: target hemoglobin levels are not met in spite of adequate iron replacement or blood transfusions are necessary.
- The use of endoscopic argon plasma coagulation during endoscopy should be limited.
- In mild HHT-related gastrointestinal bleeding, the administration of oral antifibrinolytics should be taken into consideration.
- In moderate to severe HHT-related gastrointestinal bleeding, the administration of intravenous systemic antiangiogenic therapies, such as bevacizumab, should be taken into consideration.
- Diagnostic genetic testing should be performed in asymptomatic children of an affected parent.
- Asymptomatic children with HHT or at risk for HHT at the time of presentation should be screened for brain and pulmonary AVMs.
- Large pulmonary AVMs and those associated with decreased oxygen saturation levels should undergo treatment in an effort to avoid major complications.
- Pulmonary AVM screening in asymptomatic children with definite HHT or at risk for HHT should typically be repeated at 5-year intervals.
- Brain AVMs that harbour high-risk features should be treated.
- Iron deficiency and anemia should be investigated in the following HHT patients:
- Adults, irrespective of symptoms;
- Children presenting with symptoms of anemia and/or recurrent bleeding episodes.
- Iron replacement therapy is recommended as follows:
- Oral iron administration constitutes the first therapeutic step;
- In cases in which oral administration of iron proves ineffective, cannot be absorbed or tolerated, and in patients presenting with severe anemia, intravenous iron replacement should be performed.
- Red blood cell transfusion is recommended as follows:
- Hemodynamic instability or shock;
- Comorbidities in which a higher hemoglobin target is needed;
- Clinical settings in which an urgent increase in hemoglobin level is necessary (e.g., preoperatively, or during pregnancy);
- Inadequate hemoglobin levels in spite of frequent intravenous iron infusions.
- Additional causes of anemia should be investigated in case of an inadequate response to iron replacement.
- Prophylactic or therapeutic anticoagulation or antiplatelets should be administered in the presence of an appropriate indication, taking into account the individualized bleeding risks of HHT patients; HHT-related bleeding does not constitute an absolute contraindication for these medications.
- Dual antiplatelet therapy and/or a combination of antiplatelet medication and anticoagulation should be avoided in HHT when possible.
- Clinicians should address preconception and prenatal diagnostic options in patients with HHT.
- Testing by unenhanced magnetic resonance imaging should be done in pregnant women presenting with symptoms indicative of brain AVMs.
- HHT-affected pregnant women without recent screening and/or treatment for pulmonary AVMs should be managed as follows:
- In the absence of symptoms, pulmonary AVMs screening should be performed initially, either by an agitated saline transthoracic contrast echocardiography or by a low-dose chest CT without contrast, according to local availability and expertise. When opting for a chest CT examination, it should ideally be performed early in the second trimester.
- In cases presenting with symptoms indicative of pulmonary AVMs, diagnostic testing by a low-dose non-contrast chest CT scan should be performed. This examination can be carried out at any gestational age, as clinically indicated.
- The treatment of pulmonary AVMs is recommended starting with the second trimester, in the absence of other clinical contraindications.
- HHT-affected pregnant women with previously untreated pulmonary and/or brain AVMs or those who have not undergone pulmonary AVMs screening recently should be managed by a multidisciplinary team, ideally in a tertiary care center.
- Epidural anesthesia should not be withheld on the basis of a HHT diagnosis and spinal vascular malformations screening is not necessary.
- Labor and vaginal delivery are not contraindicated in pregnant HHT women who harbour objectively defined, non-high-risk brain AVMs. An assisted second stage may be necessary on a case-by-case basis.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grosse, S.D.; Boulet, S.L.; Grant, A.M.; Hulihan, M.M.; Faughnan, M.E. The Use of US Health Insurance Data for Surveillance of Rare Disorders: Hereditary Hemorrhagic Telangiectasia. Genet. Med. 2014, 16, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Brady, A.P.; Murphy, M.M.; O’Connor, T.M. Hereditary Haemorrhagic Telangiectasia: A Cause of Preventable Morbidity and Mortality. Ir. J. Med. Sci. 2009, 178, 135–146. [Google Scholar] [CrossRef]
- McDonald, J.; Wooderchak-Donahue, W.; VanSant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary Hemorrhagic Telangiectasia: Genetics and Molecular Diagnostics in a New Era. Front. Genet. 2015, 6, 1. [Google Scholar] [CrossRef]
- Mallet, C.; Lamribet, K.; Giraud, S.; Dupuis-Girod, S.; Feige, J.-J.; Bailly, S.; Tillet, E. Functional Analysis of Endoglin Mutations from Hereditary Hemorrhagic Telangiectasia Type 1 Patients Reveals Different Mechanisms for Endoglin Loss of Function. Hum. Mol. Genet. 2015, 24, 1142–1154. [Google Scholar] [CrossRef]
- Gallione, C.J.; Klaus, D.J.; Yeh, E.Y.; Stenzel, T.T.; Xue, Y.; Anthony, K.B.; McAllister, K.A.; Baldwin, M.A.; Berg, J.N.; Lux, A.; et al. Mutation and Expression Analysis of the Endoglin Gene in Hereditary Hemorrhagic Telangiectasia Reveals Null Alleles. Hum. Mutat. 1998, 11, 286–294. [Google Scholar] [CrossRef]
- Gallione, C.J.; Richards, J.A.; Letteboer, T.G.W.; Rushlow, D.; Prigoda, N.L.; Leedom, T.P.; Ganguly, A.; Castells, A.; van Amstel, J.K.P.; Westermann, C.J.J.; et al. SMAD4 Mutations Found in Unselected HHT Patients. J. Med. Genet. 2006, 43, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Rössler, J.; Baumgartner, I.; Kremer Hovinga, J.A. EHA Endorsement of the Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. HemaSphere 2021, 5, e647. [Google Scholar] [CrossRef]
- Hernandez, F.; Huether, R.; Carter, L.; Johnston, T.; Thompson, J.; Gossage, J.R.; Chao, E.; Elliott, A.M. Mutations in RASA1 and GDF2 Identified in Patients with Clinical Features of Hereditary Hemorrhagic Telangiectasia. Hum. Genome Var. 2015, 2, 15040. [Google Scholar] [CrossRef]
- Begbie, M.; Wallace, G.; Shovlin, C. Hereditary Haemorrhagic Telangiectasia (Osler-Weber-Rendu Syndrome): A View from the 21st Century. Postgrad. Med. J. 2003, 79, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Morgan, T.; McDonald, J.; Anderson, C.; Ismail, M.; Miller, F.; Mao, R.; Madan, A.; Barnes, P.; Hudgins, L.; Manning, M. Intracranial Hemorrhage in Infants and Children with Hereditary Hemorrhagic Telangiectasia (Osler-Weber-Rendu Syndrome). PEDIATRICS 2002, 109, e12. [Google Scholar] [CrossRef] [Green Version]
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic Criteria for Hereditary Hemorrhagic Telangiectasia (Rendu-Osler-Weber Syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Van Gent, M.W.F.; Velthuis, S.; Post, M.C.; Snijder, R.J.; Westermann, C.J.J.; Letteboer, T.G.W.; Mager, J.J. Hereditary Hemorrhagic Telangiectasia: How Accurate Are the Clinical Criteria? Am. J. Med. Genet. A 2013, 161, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Khunger, M.; Gupta, A.; Kumar, N. Optimal Management of Hereditary Hemorrhagic Telangiectasia. J. Blood Med. 2014, 5, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Faughnan, M.E.; Lui, Y.W.; Wirth, J.A.; Pugash, R.A.; Redelmeier, D.A.; Hyland, R.H.; White, R.I. Diffuse Pulmonary Arteriovenous Malformations: Characteristics and Prognosis. CHEST 2000, 117, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacombe, P.; Lacout, A.; Marcy, P.-Y.; Binsse, S.; Sellier, J.; Bensalah, M.; Chinet, T.; Bourgault-Villada, I.; Blivet, S.; Roume, J.; et al. Diagnosis and Treatment of Pulmonary Arteriovenous Malformations in Hereditary Hemorrhagic Telangiectasia: An Overview. Diagn. Interv. Imaging 2013, 94, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Andorfer, K.E.C.; Seebauer, C.T.; Dienemann, C.; Marcrum, S.C.; Fischer, R.; Bohr, C.; Kühnel, T.S. HHT-Related Epistaxis and Pregnancy—A Retrospective Survey and Recommendations for Management from an Otorhinolaryngology Perspective. J. Clin. Med. 2022, 11, 2178. [Google Scholar] [CrossRef]
- Thompson, K.P.; Nelson, J.; Kim, H.; Pawlikowska, L.; Marchuk, D.A.; Lawton, M.T.; Faughnan, M.E. Brain Vascular Malformation Consortium HHT Investigator Group Predictors of Mortality in Patients with Hereditary Hemorrhagic Telangiectasia. Orphanet J. Rare Dis. 2021, 16, 12. [Google Scholar] [CrossRef]
- Schimmel, K.; Ali, M.K.; Tan, S.Y.; Teng, J.; Do, H.M.; Steinberg, G.K.; Stevenson, D.A.; Spiekerkoetter, E. Arteriovenous Malformations—Current Understanding of the Pathogenesis with Implications for Treatment. Int. J. Mol. Sci. 2021, 22, 9037. [Google Scholar] [CrossRef]
- Faughnan, M.E.; Mager, J.J.; Hetts, S.W.; Palda, V.A.; Lang-Robertson, K.; Buscarini, E.; Deslandres, E.; Kasthuri, R.S.; Lausman, A.; Poetker, D.; et al. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann. Intern. Med. 2020, 173, 989–1001. [Google Scholar] [CrossRef]
- Grand’Maison, A. Hereditary Hemorrhagic Telangiectasia. CMAJ 2009, 180, 833–835. [Google Scholar] [CrossRef] [Green Version]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S.; et al. International Guidelines for the Diagnosis and Management of Hereditary Haemorrhagic Telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef]
- Tau, N.; Atar, E.; Mei-Zahav, M.; Bachar, G.N.; Dagan, T.; Birk, E.; Bruckheimer, E. Amplatzer Vascular Plugs Versus Coils for Embolization of Pulmonary Arteriovenous Malformations in Patients with Hereditary Hemorrhagic Telangiectasia. Cardiovasc. Intervent. Radiol. 2016, 39, 1110–1114. [Google Scholar] [CrossRef]
- Fish, A.; Henderson, K.; Moushey, A.; Pollak, J.; Schlachter, T. Incidence of Spontaneous Pulmonary AVM Rupture in HHT Patients. J. Clin. Med. 2021, 10, 4714. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jie, B.; Yu, D.; Li, L.-L.; Jiang, S. Massive Haemorrhagic Complications of Ruptured Pulmonary Arteriovenous Malformations: Outcomes from a 12 Years’ Retrospective Study. BMC Pulm. Med. 2021, 21, 230. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.D.; Li, Q.; Hester, S.T.; Leitner, O.; Smith, K.L.; Kasthuri, R.S. Integration of Clinical Parameters, Genotype and Epistaxis Severity Score to Guide Treatment for Hereditary Hemorrhagic Telangiectasia Associated Bleeding. Orphanet J. Rare Dis. 2020, 15, 185. [Google Scholar] [CrossRef]
- Marcos, S.; Botella, L.M.; Albiñana, V.; Arbia, A.; de Rosales, A.M. Sclerotherapy on Demand with Polidocanol to Treat HHT Nosebleeds. J. Clin. Med. 2021, 10, 3845. [Google Scholar] [CrossRef]
- Tortora, A.; Riccioni, M.E.; Gaetani, E.; Ojetti, V.; Holleran, G.; Gasbarrini, A. Rendu-Osler-Weber Disease: A Gastroenterologist’s Perspective. Orphanet J. Rare Dis. 2019, 14, 130. [Google Scholar] [CrossRef] [Green Version]
- Al-Samkari, H.; Kasthuri, R.S.; Parambil, J.G.; Albitar, H.A.; Almodallal, Y.A.; Vázquez, C.; Serra, M.M.; Dupuis-Girod, S.; Wilsen, C.B.; McWilliams, J.P.; et al. An International, Multicenter Study of Intravenous Bevacizumab for Bleeding in Hereditary Hemorrhagic Telangiectasia: The InHIBIT-Bleed Study. Haematologica 2021, 106, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; McWilliams, J.P. Approach to Pulmonary Arteriovenous Malformations: A Comprehensive Update. J. Clin. Med. 2020, 9, 1927. [Google Scholar] [CrossRef] [PubMed]
- Kiliç, K.; Konya, D.; Kurtkaya, Ö.; Sav, A.; Pamir, M.N.; Kiliç, T. Inhibition of Angiogenesis Induced by Cerebral Arteriovenous Malformations Using Gamma Knife Irradiation. J. Neurosurg. 2007, 106, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, S.; Bell, J.-A.; Brooker, J. Tamoxifen Therapy for Recurrent Mucosal Bleeding in Hereditary Hemorrhagic Telangiectasia. J. Hematol. 2021, 10, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Kroon, S.; Snijder, R.J.; Mager, J.J.; Post, M.C.; Tenthof van Noorden, J.; van Geenen, E.J.M.; Drenth, J.P.H.; Grooteman, K.V. Octreotide for Gastrointestinal Bleeding in Hereditary Hemorrhagic Telangiectasia: A Prospective Case Series. Am. J. Hematol. 2019, 94, E247–E249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, K.D.; Umar, B.; Schairer, J. Successful Treatment of Hereditary Hemorrhagic Telangiectasia with Octreotide. ACG Case Rep. J. 2019, 6, e00088. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Floria, M.; Năfureanu, E.D.; Iov, D.-E.; Sîrbu, O.; Dranga, M.; Ouatu, A.; Tănase, D.M.; Bărboi, O.B.; Drug, V.L.; Cobzeanu, M.D. Hereditary Hemorrhagic Telangiectasia and Arterio-Venous Malformations—From Diagnosis to Therapeutic Challenges. J. Clin. Med. 2022, 11, 2634. https://doi.org/10.3390/jcm11092634
Floria M, Năfureanu ED, Iov D-E, Sîrbu O, Dranga M, Ouatu A, Tănase DM, Bărboi OB, Drug VL, Cobzeanu MD. Hereditary Hemorrhagic Telangiectasia and Arterio-Venous Malformations—From Diagnosis to Therapeutic Challenges. Journal of Clinical Medicine. 2022; 11(9):2634. https://doi.org/10.3390/jcm11092634
Chicago/Turabian StyleFloria, Mariana, Elena Diana Năfureanu, Diana-Elena Iov, Oana Sîrbu, Mihaela Dranga, Anca Ouatu, Daniela Maria Tănase, Oana Bogdana Bărboi, Vasile Liviu Drug, and Mihail Dan Cobzeanu. 2022. "Hereditary Hemorrhagic Telangiectasia and Arterio-Venous Malformations—From Diagnosis to Therapeutic Challenges" Journal of Clinical Medicine 11, no. 9: 2634. https://doi.org/10.3390/jcm11092634
APA StyleFloria, M., Năfureanu, E. D., Iov, D.-E., Sîrbu, O., Dranga, M., Ouatu, A., Tănase, D. M., Bărboi, O. B., Drug, V. L., & Cobzeanu, M. D. (2022). Hereditary Hemorrhagic Telangiectasia and Arterio-Venous Malformations—From Diagnosis to Therapeutic Challenges. Journal of Clinical Medicine, 11(9), 2634. https://doi.org/10.3390/jcm11092634