
Strategy and Challenges of Paraclinical Examinations in Adult-Onset Still’s Disease


Abstract
1. Introduction
2. Paraclinical Examinations to Support the Diagnosis
2.1. Serological
2.2. Radiological

{kind=link}
| Marker | Description in AOSD | Usefulness for AOSD | References |
|---|---|---|---|
| CRP | >5 mg/L usually very high >50 mg/L | Not specific but essential for diagnosis | [4] |
| Polymorphonuclear neutrophils | >80% neutrophils among leukocytes | Cardinal criteria | [4] |
| Ferritin | >ULN often > 5 × ULN | High sensitivity if >ULN but high specificity (80%) only if >5 × ULN | [1,25,26] |
| Glycosylated ferritin | Low (<20%) | Sensitivity 79.5% Specificity 66.4% | [1,10] |
| IL-1β | Elevated but no standard and not different in sepsis | Not routinely used | [27,28,29] |
| IL-6 | Elevated but no standard and not different in sepsis | Not routinely used | [28,29] |
| IL-18 | No standard but levels >150 or 366 ng/L | Not routinely used Sensitivity 91.7% and specificity 99.1% when >366 ng/L | [30,31] |
| TNFα | Elevated but no standard and not different in sepsis | Not routinely used | [28,29] |
| Histology on skin biopsy | Broad histologic spectrum Interstitial dermal neutrophils aligned between the collagen bundles | Allow to exclude differential diagnosis in atypical forms | [13,32] |
| Joint X-ray | Peri-capitate carpal destruction/fusion with metacarpophalangeal joints sparing | Useful in advanced and articular forms of the disease/late-onset abnormality | [18] |
| Joint US | Active synovitis of large and medium joints | Useful in articular forms of the disease | [16] |
| CT scan/PET-CT scan | Lymphadenopathy, HSM/hypermetabolism in lymph node, spleen, and bone marrow | Not essential for diagnosis, useful for differential diagnosis | [33] |
3. Paraclinical Tests to Exclude Other Diagnoses
4. Paraclinical Examinations to Detect Complications of AoSD
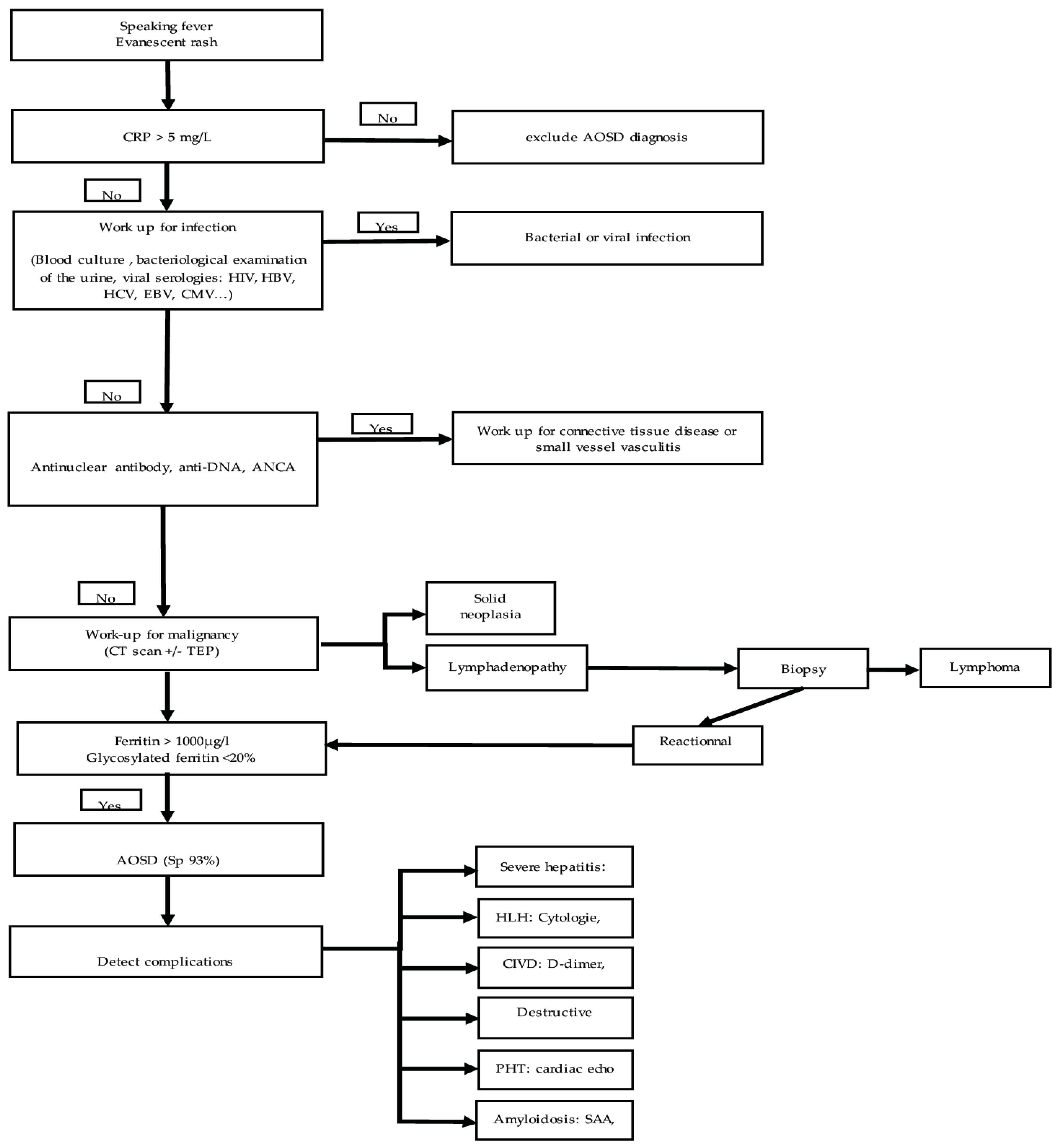
5. Proposal for Stratification of These Tests
6. Future and Perspectives of Paraclinical Examinations for AOSD
Author Contributions
Funding
Conflicts of Interest
References
- Feist, E.; Mitrovic, S.; Fautrel, B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat. Rev. Rheumatol. 2018, 14, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Gerfaud-Valentin, M.; Maucort-Boulch, D.; Hot, A.; Iwaz, J.; Ninet, J.; Durieu, I.; Broussolle, C.; Sève, P. Adult-onset still disease: Manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine 2014, 93, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Kalyoncu, U.; Solmaz, D.; Emmungil, H.; Yazici, A.; Kasifoglu, T.; Kimyon, G.; Balkarli, A.; Bes, C.; Ozmen, M.; Alibaz-Oner, F.; et al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still’s disease: Data from a large multicenter cohort. J. Autoimmun. 2016, 69, 59–63. [Google Scholar] [CrossRef]
- Mitrovic, S.; Fautrel, B. New Markers for Adult-Onset Still’s Disease. Jt. Bone Spine 2018, 85, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T. Preliminary criteria for classification of adult Still’s disease. J. Rheumatol. 1992, 19, 424–430. [Google Scholar] [PubMed]
- Fautrel, B.; Zing, E.; Golmard, J.L.; Le Moel, G.; Bissery, A.; Rioux, C.; Rozenberg, S.; Piette, J.; Bourgeois, P. Proposal for a new set of classification criteria for adult-onset still disease. Medicine 2002, 81, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, D.; Mestrallet, S.; Dehoux, M.; Golmard, J.L.; Granger, B.; Georgin-Lavialle, S.; Arnaud, L.; Grateau, G.; Pouchot, J.; Fautrel, B. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin. Arthritis Rheum. 2018, 47, 578–585. [Google Scholar] [CrossRef]
- Fautrel, B.; Le Moel, G.; Saint-Marcoux, B.; Taupin, P.; Vignes, S.; Rozenberg, S.; Koeger, A.C.; Meyer, O.; Guillevin, L.; Piette, J.C.; et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J. Rheumatol. 2001, 28, 322–329. [Google Scholar]
- Dayer, E.; Dayer, J.M.; Roux-Lombard, P. Primer: The practical use of biological markers of rheumatic and systemic inflammatory diseases. Nat. Clin. Pract. Rheumatol. 2007, 3, 512–520. [Google Scholar] [CrossRef]
- Vignes, S.; Le Moel, G.; Fautrel, B.; Wechsler, B.; Godeau, P.; Piette, J.C. Percentage of glycosylated serum ferritin remains low throughout the course of adult onset Still’s disease. Ann. Rheum. Dis. 2000, 59, 347–350. [Google Scholar] [CrossRef]
- Seo, J.Y.; Suh, C.H.; Jung, J.Y.; Kim, A.R.; Yang, J.W.; Kim, H.A. The neutrophil-to-lymphocyte ratio could be a good diagnostic marker and predictor of relapse in patients with adult-onset Still’s disease: A STROBE-compliant retrospective observational analysis. Medicine 2017, 96, e7546. [Google Scholar] [CrossRef] [PubMed]
- Wouters, J.M.; Reekers, P.; van de Putte, L.B. Adult-onset Still’s disease. Disease course and HLA associations. Arthritis Rheum. 1986, 29, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Fortna, R.R.; Gudjonsson, J.E.; Seidel, G.; Dicostanzo, D.; Jacobson, M.; Kopelman, M.; Patel, R.M. Persistent pruritic papules and plaques: A characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still’s disease. J. Cutan. Pathol. 2010, 37, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, A.; Papagrigoraki, A.; Biasi, D.; Colato, C.; Girolomoni, G. Cutaneous manifestations of adult-onset Still’s disease: A case report and review of literature. Clin. Rheumatol. 2016, 35, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Medsger, T.A., Jr.; Christy, W.C. Carpal arthritis with ankylosis in late onset Still’s disease. Arthritis Rheum. 1976, 19, 232–242. [Google Scholar] [CrossRef]
- Jacques, T.; Sudol-Szopinska, I.; Larkman, N.; O’Connor, P.; Cotton, A. Musculoskeletal Manifestations of Non-RA Connective Tissue Diseases: Scleroderma, Systemic Lupus Erythematosus, Still’s Disease, Dermatomyositis/Polymyositis, Sjogren’s Syndrome, and Mixed Connective Tissue Disease. Semin. Musculoskelet. Radiol. 2018, 22, 166–179. [Google Scholar]
- Belghali, S.; El Amri, N.; Baccouche, K.; Laataoui, S.; Bouzaoueche, M.; Zeglaoui, H.; Bouajina, E. Atypical form of Adult-onset Still’s Disease with Distal Interphalangeal Joints Involvement. Curr. Rheumatol. Rev. 2018, 14, 284–288. [Google Scholar] [CrossRef]
- Ziegeler, K.; Eshed, I.; Diekhoff, T.; Hermann, K.G. Imaging of Joints and Bones in Autoinflammation. J. Clin. Med. 2020, 9, 4074. [Google Scholar] [CrossRef]
- Ruscitti, P.; Barile, A.; Berardicurti, O.; Iafrate, S.; Di Benedetto, P.; Vitale, A.; Caso, F.; Costa, L.; Bruno, F.; Ursini, F.; et al. The joint involvement in adult onset Still’s disease is characterised by a peculiar magnetic resonance imaging and a specific transcriptomic profile. Sci. Rep. 2021, 11, 12455. [Google Scholar] [CrossRef]
- Ruscitti, P.; Berardicurti, O.; Iacono, D.; Pantano, I.; Liakouli, V.; Caso, F.; Emmi, G.; Grembiale, R.D.; Cantatore, F.P.; Atzeni, F.; et al. Parenchymal lung disease in adult onset Still’s disease: An emergent marker of disease severity-characterisation and predictive factors from Gruppo Italiano di Ricerca in Reumatologia Clinica e Sperimentale (GIRRCS) cohort of patients. Arthritis Res. Ther. 2020, 22, 151. [Google Scholar] [CrossRef]
- Gerfaud-Valentin, M.; Cottin, V.; Jamilloux, Y.; Hot, A.; Gaillard-Coadon, A.; Durieu, I.; Broussolle, C.; Iwaz, J.; Sève, P. Parenchymal lung involvement in adult-onset Still disease: A STROBE-compliant case series and literature review. Medicine 2016, 95, e4258. [Google Scholar] [CrossRef] [PubMed]
- Koplay, M.; Sivri, M.; Kendir, I.C.; Erdogan, H.; Yilmaz, S. Education and Imaging. Hepatobiliary and Pancreatic: Magnetic resonance imaging (MRI) features of liver involvement in adult-onset Still’s disease. J. Gastroenterol. Hepatol. 2015, 30, 1229. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Nhan, P.; Cholet, C.; Jachiet, V.; Ederhy, S.; Mekinian, A.; Boccara, F.; Fain, O.; Cohen, A. Acute Myocarditis Revealing Adult-Onset Still’s Disease. JACC Case Rep. 2021, 3, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Bozek, M.; Konopko, M.; Wierzba-Bobrowicz, T.; Witkowski, G.; Makowicz, G.; Sienkiewicz-Jarosz, H. Autoimmune meningitis and encephalitis in adult-onset still disease—Case report. Neurol. Neurochir. Pol. 2017, 51, 421–426. [Google Scholar] [CrossRef][Green Version]
- Ota, T.; Higashi, S.; Suzuki, H.; Eto, S. Increased serum ferritin levels in adult Still’s disease. Lancet 1987, 1, 562–563. [Google Scholar] [CrossRef]
- Rosario, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’Cruz, D.P.; Shoenfeld, Y. The hyperferritinemic syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185. [Google Scholar] [CrossRef]
- Rau, M.; Schiller, M.; Krienke, S.; Heyder, P.; Lorenz, H.; Blank, N. Clinical manifestations but not cytokine profiles differentiate adult-onset Still’s disease and sepsis. J. Rheumatol. 2010, 37, 2369–2376. [Google Scholar] [CrossRef]
- Choi, J.H.; Suh, C.H.; Lee, Y.M.; Suh, Y.J.; Lee, S.K.; Kim, S.S.; Nahm, D.; Park, H. Serum cytokine profiles in patients with adult onset Still’s disease. J. Rheumatol. 2003, 30, 2422–2427. [Google Scholar]
- Chen, D.Y.; Lan, J.L.; Lin, F.J.; Hsieh, T.Y. Proinflammatory cytokine profiles in sera and pathological tissues of patients with active untreated adult onset Still’s disease. J. Rheumatol. 2004, 31, 2189–2198. [Google Scholar]
- Chen, P.K.; Lan, J.L.; Huang, P.H.; Hsu, J.L.; Chang, C.K.; Tien, N.; Lin, H.-J.; Chen, D.-Y. Interleukin-18 Is a Potential Biomarker to Discriminate Active Adult-Onset Still’s Disease From COVID-19. Front. Immunol. 2021, 12, 719544. [Google Scholar] [CrossRef]
- Priori, R.; Colafrancesco, S.; Alessandri, C.; Minniti, A.; Perricone, C.; Iaiani, G.; Palazzo, D.; Valesini, G. Interleukin 18: A biomarker for differential diagnosis between adult-onset Still’s disease and sepsis. J. Rheumatol. 2014, 41, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.R.; Laga, A.C.; Granter, S.R. The spectrum of histopathologic findings in cutaneous lesions in patients with Still disease. Am. J. Clin. Pathol. 2015, 144, 945–951. [Google Scholar] [CrossRef]
- Schonau, V.; Vogel, K.; Englbrecht, M.; Wacker, J.; Schmidt, D.; Manger, B.; Kuwert, T.; Schett, G. The value of (18)F-FDG-PET/CT in identifying the cause of fever of unknown origin (FUO) and inflammation of unknown origin (IUO): Data from a prospective study. Ann. Rheum. Dis. 2018, 77, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Vanderschueren, S.; Knockaert, D.; Adriaenssens, T.; Demey, W.; Durnez, A.; Blockmans, D.; Bobbaers, H. From prolonged febrile illness to fever of unknown origin: The challenge continues. Arch. Intern. Med. 2003, 163, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Brisset, J.; Jamilloux, Y.; Dumonteil, S.; Lades, G.; Killian, M.; Gerfaud-Valentin, M.; Lemaire, A.; Chroboczek, T.; Liozon, E.; Gondran, G.; et al. Characteristics and Clinical Value of 18F-FDG PET/CT in the Management of Adult-Onset Still’s Disease: 35 Cases. J. Clin. Med. 2021, 10, 2489. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dong, C.; Ma, X.; Wang, Y. (18)F-FDG PET/CT Associates With Disease Activity and Clinical Recurrence of AOSD Patients. Front. Med. 2021, 8, 668323. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, R.; Ruscitti, P.; Shoenfeld, Y. A comprehensive review on adult onset Still’s disease. J. Autoimmun. 2018, 93, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Abdirakhmanova, A.; Sazonov, V.; Mukusheva, Z.; Assylbekova, M.; Abdukhakimova, D.; Poddighe, D. Macrophage Activation Syndrome in Pediatric Systemic Lupus Erythematosus: A Systematic Review of the Diagnostic Aspects. Front. Med. 2021, 8, 681875. [Google Scholar] [CrossRef]
- Fauter, M.; Gerfaud-Valentin, M.; Delplanque, M.; Georgin-Lavialle, S.; Seve, P.; Jamilloux, Y. Adult-onset Still’s disease complications. Rev. Med. Interne 2020, 41, 168–179. [Google Scholar] [CrossRef]
- Muller, R.; Briantais, A.; Faucher, B.; Borentain, P.; Nafati, C.; Blasco, V.; Gregoire, E.; Bernit, E.; Seguier, J.; Meunier, B.; et al. Acute severe hepatitis in adult-onset Still’s disease: Case report and comprehensive review of a life-threatening manifestation. Clin. Rheumatol. 2021, 40, 2467–2476. [Google Scholar] [CrossRef]
- Tomaras, S.; Goetzke, C.C.; Kallinich, T.; Feist, E. Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach. J. Clin. Med. 2021, 10, 733. [Google Scholar] [CrossRef] [PubMed]
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014, 66, 2613–2620. [Google Scholar] [CrossRef] [PubMed]
- Efthimiou, P.; Kadavath, S.; Mehta, B. Life-threatening complications of adult-onset Still’s disease. Clin. Rheumatol. 2014, 33, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.V.; Manson, D.K.; Horn, E.M.; Haythe, J. An atypical presentation of adult-onset Still’s disease complicated by pulmonary hypertension and macrophage activation syndrome treated with immunosuppression: A case-based review of the literature. Pulm. Circ. 2016, 6, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Humbert, P.G.; Billerey, C.; Fest, T.; Dupond, J.L. Adult onset Still’s disease and related renal amyloidosis. Ann. Rheum. Dis. 1991, 50, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, T.; Zhang, H.; Xu, Y.; Yang, Q.; Liu, Q.; Gao, Y.; Wu, J.; Shao, L.; Zhang, W. Biomarker screening and validation for the differentiation of bloodstream infection from adult-onset Still’s disease: A prospective cohort study. Cytokine 2021, 146, 155642. [Google Scholar] [CrossRef]
- Koga, T.; Sumiyoshi, R.; Furukawa, K.; Sato, S.; Migita, K.; Shimizu, T.; Umeda, M.; Endo, Y.; Fukui, S.; Kawashiri, S.-Y.; et al. Interleukin-18 and fibroblast growth factor 2 in combination is a useful diagnostic biomarker to distinguish adult-onset Still’s disease from sepsis. Arthritis Res. Ther. 2020, 22, 108. [Google Scholar] [CrossRef]
- Jung, K.H.; Kim, J.J.; Lee, J.S.; Park, W.; Kim, T.H.; Jun, J.B.; Yoo, D.H. Interleukin-18 as an efficient marker for remission and follow-up in patients with inactive adult-onset Still’s disease. Scand. J. Rheumatol. 2014, 43, 162–169. [Google Scholar] [CrossRef]
- Chi, H.; Liu, D.; Sun, Y.; Hu, Q.; Liu, H.; Cheng, X.; Ye, J.; Shi, H.; Yin, Y.; Liu, M.; et al. Interleukin-37 is increased in adult-onset Still’s disease and associated with disease activity. Arthritis Res. Ther. 2018, 20, 54. [Google Scholar] [CrossRef]
- Nam, S.W.; Kang, S.; Lee, J.H.; Yoo, D.H. Different Features of Interleukin-37 and Interleukin-18 as Disase Activity Markers of Adult-Onset Still’s Disease. J. Clin. Med. 2021, 10, 910. [Google Scholar] [CrossRef]
- Kim, H.A.; An, J.M.; Nam, J.Y.; Jeon, J.Y.; Suh, C.H. Serum S100A8/A9, but not follistatin-like protein 1 and interleukin 18, may be a useful biomarker of disease activity in adult-onset Still’s disease. J. Rheumatol. 2012, 39, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zha, X.; Li, C.; Jia, Y.; Zhu, L.; Guo, J.; Su, Y. Serum calprotectin—A promising diagnostic marker for adult-onset Still’s disease. Clin. Rheumatol. 2016, 35, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Segu-Verges, C.; Coma, M.; Kessel, C.; Smeets, S.; Foell, D.; Aldea, A. Application of systems biology-based in silico tools to optimize treatment strategy identification in Still’s disease. Arthritis Res. Ther. 2021, 23, 126. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poursac, N.; Odriozola, I.; Truchetet, M.-E. Strategy and Challenges of Paraclinical Examinations in Adult-Onset Still’s Disease. J. Clin. Med. 2022, 11, 2232. https://doi.org/10.3390/jcm11082232
Poursac N, Odriozola I, Truchetet M-E. Strategy and Challenges of Paraclinical Examinations in Adult-Onset Still’s Disease. Journal of Clinical Medicine. 2022; 11(8):2232. https://doi.org/10.3390/jcm11082232
Chicago/Turabian StylePoursac, Nicolas, Itsaso Odriozola, and Marie-Elise Truchetet. 2022. "Strategy and Challenges of Paraclinical Examinations in Adult-Onset Still’s Disease" Journal of Clinical Medicine 11, no. 8: 2232. https://doi.org/10.3390/jcm11082232
APA StylePoursac, N., Odriozola, I., & Truchetet, M.-E. (2022). Strategy and Challenges of Paraclinical Examinations in Adult-Onset Still’s Disease. Journal of Clinical Medicine, 11(8), 2232. https://doi.org/10.3390/jcm11082232