
Advances in the Treatment of Giant Cell Arteritis



 , , and
, , and 
Abstract
:1. Introduction
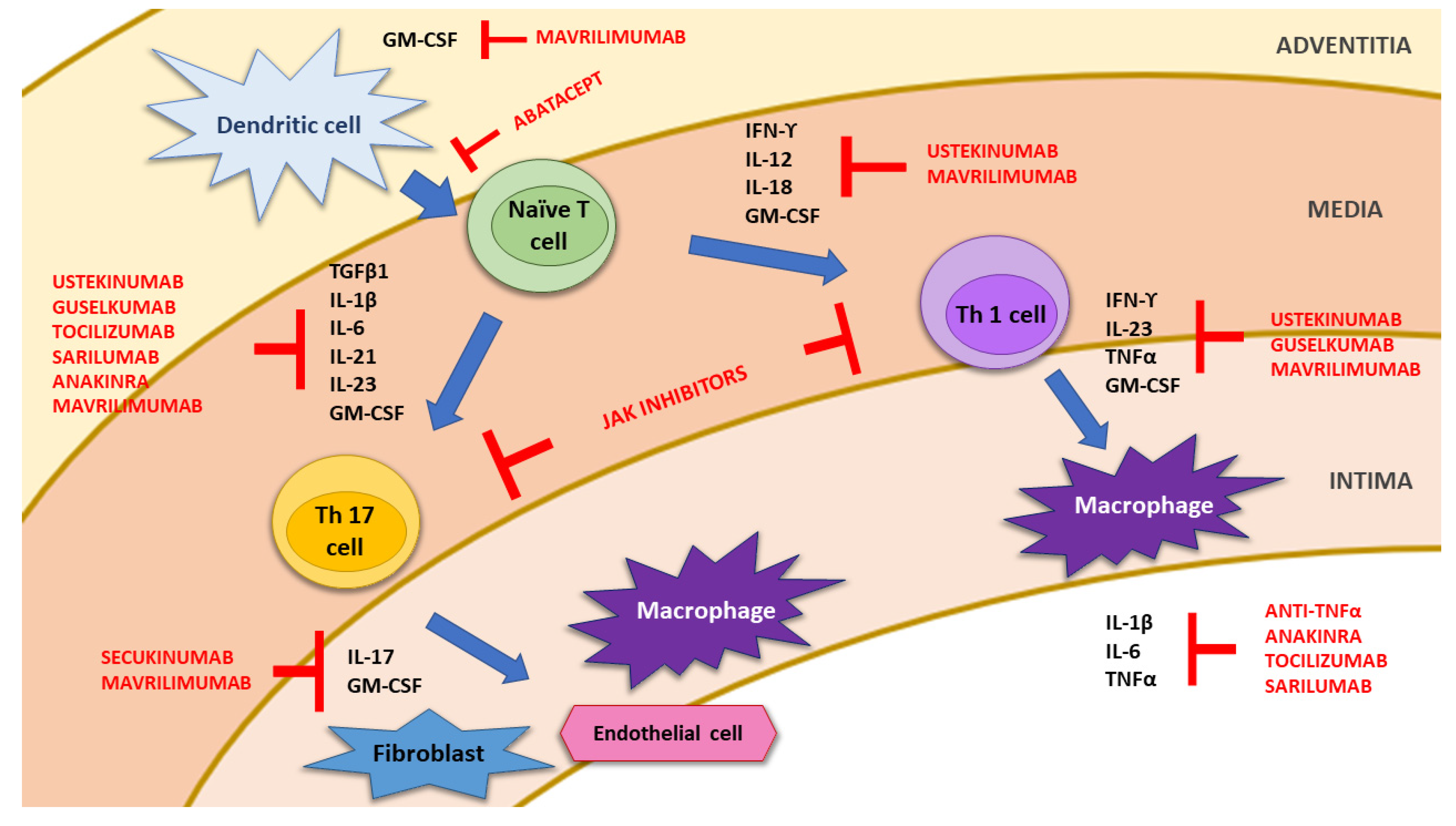
2. Pathophysiology of Giant Cell Arteritis
3. Treatment of Giant Cell Arteritis
3.1. Drugs Currently Used in the Management of GCA
3.1.1. Glucocorticoids
3.1.2. Methotrexate
3.1.3. Other Conventional Synthetic Disease-Modifying Anti-Rheumatic Drugs (csDMARD)
3.1.4. IL-6 Blockade: Tocilizumab
3.2. Role of Non-Tocilizumab Biologic Disease-Modifying Anti-Rheumatic Drugs
3.2.1. TNF-α Antagonists
3.2.2. IL-12/IL-23 Pathway Inhibition

{kind=link}
| Author, Year [Ref.] | Type of Study | n | Sex F (%) | Population/Median Follow-Up | Therapeutic Protocol | Prior DMARD n (%) | Main Efficacy Outcomes | Serious Adverse Events |
|---|---|---|---|---|---|---|---|---|
| TNF-α Antagonists | ||||||||
| Hoffman et al., 2007 [80] | Multicenter, randomized, DB, PBO-controlled trial | 44 (IFX 28, PBO 16) 2:1 ratio | IFX 24 (86) PBO 11 (69) | Newly diagnosed GCA in GC-induced remission. FU: 54 weeks (early termination after interim analysis at week 22) | i.v. IFX 5mg/kg vs. PBO | Prior DMARD not allowed | Differences in relapse-free patients (43% IFX vs. 50% PBO) and % of patients with GC tapering without relapse (61% IFX vs. 75% PBO) at 22w between groups were NS | 29% IFX vs. 25% PBO, NS Serious infections 11% IFX vs. 6% PBO, NS |
| Martinez-Taboada et al., 2008 [82] | Multicenter, randomized, DB, PBO-controlled trial | 17 (ETN 8, PBO 9) 1:1 ratio | ETN 6 (75) PBO 8 (88.9) | Clinically asymptomatic biopsy-proven GCA with GC-related comorbidity. FU: 52 weeks | s.c. ETN 25 mg twice weekly vs. PBO | Prior DMARD not allowed | Clinical disease control without GC at 52 w: 50% ETN vs. 22.2% PBO (NS). ETN cumulative GC dose was significantly lower (p = 0.03) | 12.5% ETN vs. 11.1% PBO, NS |
| Seror et al., 2018 [81] | Multicenter, randomized, DB, PBO-controlled trial (HECTHOR trial) | 70 (ADA 34, PBO 36) | ADA 24 (71) PBO 28 (78) | Newly diagnosed GCA (GCA-related visual symptoms were an exclusion criteria). FU: 10 weeks | s.c. ADA 45 mg q2w vs. PBO | Prior DMARD not allowed | Remission on less than 0.1 mg/kg of prednisone at week 26: 59% ADA vs. 50% PBO (NS). NS differences in prednisone dose reduction or % of relapse-free patients | 14.5% ADA vs. 47.2% PBO. Serious infections: 3 ADA vs. 5 PBO. Deaths: 1 ADA (pneumonia) vs. 2 PBO (septic shock and cancer) |
| Ustekinumab | ||||||||
| Conway et al., 2016 [29] | Single center, prospective open-label registry | 14 | 11 (79) | Refractory GCA (inability to taper GC to <10 mg/d due to symptoms of active GCA with a minimum of two relapses). FU: 13.5 months | s.c. UST 90 mg every 3 months | 12 (86) | No relapses. Significant reduction of GC dose (p = 0.001). 29% stopped GC and 92% stopped other DMARD. Image improvement in LVV (n = 5), without new stenoses or aneurysm | 3 Discontinuations due to AE |
| Conway et al., 2018 [86] | Multicenter, open-label prospective registry | 25 | 20 (80) | Refractory GCA (inability to taper GC due to recurrence of symptoms consistent with active GCA, after an initial treatment response to high-dose GC). FU: 52 weeks | s.c. UST 90 mg every 3 months | 17 (68) | No relapses. Significant reduction of GC dose (p < 0.001) and CRP decrease (p = 0.006). 24% stopped GC and 76% stopped other DMARD. Image improvement in LVV (n = 8), without new stenoses or aneurysm | 3 Discontinuations due to AE: 1 recurrent respiratory tract infections, 1 alopecia and 1 non-dermatomal limb paresthesia |
| Matza et al., 2021 [87] | Single center, single-arm prospective open-label trial | 13 | 11 (85) | Active new-onset (39%) or relapsing GCA. FU: 52 weeks (enrollment closed prematurely due to relapse of 7/10 initial patients) | s.c. UST 90 mg every 3 months | 2 (15) | 23% achieved prednisone-free remission (absence of relapse through week 52 and normalization of ESR and CRP). 7 Patients relapsed after a mean period of 23 w | 1 SAE: mild diverticulitis that required hospitalization |
| Abatacept | ||||||||
| Langford et al., 2017 [90] | Multicenter, randomized DB, PBO-controlled study | 41 (20 ABA, 21 PBO) 1:1 ratio | ABA 16 (80) PBO 21 (100) | Newly-diagnosed or relapsing GCA with active disease within the prior 2 m. FU: 12 months | Initially (n = 49): i.v. ABA 10 mg/kg/m At 12 w (n = 41): DB randomization to ABA vs. PBO of cases in remission | NR Prior bDMARD was not allowed within established time schedule | Relapse-free survival at 12 m: 48% ABA vs. 31% PBO (p = 0.049). Longer duration of remission with ABA (9.9 m) vs. PBO (3.9 m), p = 0.023 | 23 SAE in 15 patients. NS difference in frequency or severity of AE between treatment arms, including the rate of infection or SAE. No deaths |
| Rossi et al., 2021 [91] | Single center, two-arm prospective open-label study | 33 (17 TCZ, 16 ABA) 1:1 ratio | 21 (63.6) | Consecutive biopsy-proven newly diagnosed or relapsing GCA. FU: 12 months | TCZ: i.v. 8mg/kg/m (n = 8), s.c. 162 mg/w (n = 9) ABA: s.c. 125 mg/w Combination with other DMARD was not allowed | 22 (66.6) | i.v. TCZ, s.c. TCZ and ABA clinical response was complete in 57%, 67% and 31%, and partial in 43%, 16% and 31%, respectively 100% TCZ group and 43% ABA group reduced prednisone to ≤ 7.5 mg/d at 12 m, p = 0.0003. Switch due to inefficacy more frequent with ABA (0.0445) | No significant side effects |
| Sirukumab | ||||||||
| Schmidt et al., 2020 [79] | Multicenter, randomized DB, PBO-controlled parallel-group study + open-label extension | 161 (SIR 107, PBO 54) | 124 (77) | Newly diagnosed or relapsing GCA. FU of DB phase: 52 weeks FU of OL phase: 104 weeks (early termination by sponsor decision) | DB phase: s.c. SIR 100 mg q2w + 6 m or 3 m of GC taper; s.c. SIR 50 mg q4w + 6 m GC taper;PBO q2w + 6 m or 12 m GC taper OL phase: SIR 100 mg q2w at investigator discretion | Prior cs- and bDMARD was not allowed within established time schedule | At 52 w: Sustained remission not achieved by 82.4–88.9% patients in SIR arms and 100% in PBO arms; Lower % of flares with SIR than PBO (18.4–30.8% vs. 37–40%); Highest % of flares (23.1%) and withdrawals (61.5%) with SIR 100 mg q2w + 3 m GC taper. OL phase: 60% maintained remission at 4w without SIR administration; No flares | At 52 w: 19.3% SAE; NS differences across arms; No deaths. OL phase: No SAE; No deaths |
| Meta-analysis | ||||||||
| Berti et al., 2018 [75] | 10 RCT (9 phase 2 and 1 phase 3) | 645 | TCZ, i.v. GC and MTX significantly improved the likelihood of being relapse free with relative risks and 95% CI of 3.54 (2.28, 5.51), 5.11 (1.39, 18.81) and 1.54 (1.02, 2.30) | |||||
| Song et al., 2020 [92] | 6 RCT (2 TCZ, 3 TNF antagonists 1 and ABA) | 260 patients 193 controls | Remission rate higher for TCZ than PBO (OR 7.009, 95% CI 3.854–12.75, p < 0.001). Relapse rate lower for TCZ than PBO (OR 0.222, 95% CI 0.129–0.381, p < 0.001). NS difference in remission and relapse between groups with TNF antagonists, ABA and PBO | Number of SAE lower for TCZ than PBO (OR 0.539, 95% CI 0.296–0.982, p = 0.044). NS difference in SAE among patients treated with TNF antagonists, ABA and PBO. Infection rate higher for TNF antagonists than PBO (OR 2.407, 95% CI 1.168-4.960, p = 0.017), but with NS differences between TCZ, ABA and PBO | ||||
3.2.3. T Cell Modulation: Abatacept
3.2.4. IL-1 Inhibition
3.3. Therapeutic Lines under Research
3.3.1. IL-17 Inhibition
3.3.2. Janus Kinase Inhibitors
3.3.3. Granulocyte-Macrophage Colony-Stimulating Factor
| Drug [Therapeutic Regimen ] [Ref.] | Trial Name and Identifier | Target | Duration | Type of Trial and Phase | Control | Population | Target n | Primary Outcome | Status (January 2022) |
|---|---|---|---|---|---|---|---|---|---|
| IL-6R antagonists | |||||||||
| Tocilizumab [s.c. TCZ 162 mg/w 52 w + 8 w GC taper] | NCT03726749 | IL-6 | 52 weeks | Phase 4, open-label | None | New-onset and relapsing GCA | 30 | Sustained remission at w52 | Recruiting |
| Tocilizumab [s.c. TCZ 162 mg/w 4 w + GC taper 18 m (1g iv MP/d 3 d + oral GC) + ASA 75 mg/d vs. GC + ASA 75 mg/d] | TOCIAION NCT04239196 | IL-6 | 18 months | Phase 2, randomized, parallel assignment, open-label, non-comparative | None | AION due to GCA | 58 | Ocular change at w8 | Unknown |
| Tocilizumab [s.c. TCZ 162 mg/w 52 w + GC versus escalating s.c. MTX up to 0.3 mg/Kg/w 52 w + GC] | METOGiA NCT03892785 | IL-6 | 78 weeks | Phase 3, randomized, parallel assignment, open-label | MTX (≤20 mg/w) + GC | Active GCA within 6 weeks before randomization | 200 | Percentage of patients alive without relapse after initial remission or deviation from the scheduled regimen of prednisone at w78 | Recruiting |
| Tocilizumab [s.c. TCZ 162 mg/w 24 w versus s.c. PBO/w 24 w] | TOGIAC NCT04888221 | IL-6 | 52 weeks | Phase 3, randomized, parallel assignment, quadruple blind | PBO | GCA with cerebrovascular involvement | 66 | Percentage of patients in complete remission of GCA with absence of ischemic stroke recurrence at w24 | Not yet recruiting |
| Tocilizumab [s.c. TCZ 162 mg/w 156 w or commercial availability of TCZ] | NCT03202368 | IL-6 | 156 weeks | Phase 3, open-label extension of WA28119 (NCT01791153) | None | GCA flare or persistent disease activity | 3 | Percentage of subjects with adverse events at w160 | Completed (not published) |
| Tocilizumab TCZ 8 mg/kg on day 3 and thereafter weekly s.c. TCZ injections (162 mg) over 52 w | GUSTO NCT03745586 | IL-6 | 52 weeks | Open-label Phase 1 and Phase 2 | GC | New-onset GCA | 18 | Analyze the effect of ultra-short GCs followed by TCZ monotherapy. Proportion of patients achieving remission within 31 days and without relapse until w24 | Completed (final results not published yet) |
| Sarilumab | NCT03600805 | IL-6 | 52 weeks | Randomized, parallel assignment, quadruple blind Phase 3 | PBO | New-onset and refractory GCA | 83 | Proportion of patients with sustained remission at w52 | Terminated (Protracted recruitment timeline exacerbated by COVID-19 pandemic) |
| JAK inhibitors | |||||||||
| Baricitinib [4 mg/d 52 w] | NCT03026504 | JAK1+JAK2 | 52 weeks | Phase 2, open-label | None | Relapsing GCA | 15 | Percentage of subjects experiencing AE at w52 | Completed (not published) |
| Upadacitinib [UPA dose A or dose B + 26 w GC taper versus PBO + 52 w GC taper] | SELECT-GCA NCT03725202 | JAK1 | 52 weeks | Phase 3, randomized, parallel assignment, quadruple blind | PBO | New-onset and relapsing GCA | 420 | Percentage of patients achieving sustained remission at w52 | Recruiting |
| IL-17 inhibitors | |||||||||
| Secukinumab [s.c. SEC 300 mg/4 w to w 48 + 26 w GC taper versus s.c. PBO to w 48 + 26 w GC taper] | TitAIN* NCT03765788 | IL-17A | 52 weeks | Randomized, parallel assignment, double-blind Phase 2 | PBO | New-onset or relapsing GCA | 52 | Percentage of patients in sustained remission until w28 | Completed (not published) |
| Secukinumab [s.c. SEC 300 mg/4 w 52 w + 26 w GC taper versus s.c. PBO + 52 w GC taper] | NCT04930094 | IL-17A | 52 weeks | Phase 3, randomized, parallel assignment, double-blind | PBO | New-onset or relapsing GCA | 240 | Number of participants with sustained remission at w52 | Recruiting |
| Other drugs | |||||||||
| Anakinra [s.c. ANK 100 mg/d 16 w versus s.c. PBO/d 16 w] | GiAnT NCT02902731 | IL-1 | 52 weeks | Phase 3, randomized, parallel assignment, double-blind | PBO | New-onset and relapsing GCA | 70 | Global relapse rate at w26 | Recruiting |
| Abatacept [s.c. ABA 125 mg/w 12 m versus s.c. PBO/w 12 m] | ABAGART NCT04474847 | CTLA-4 CD80/CD86 | 12 months | Phase 3, randomized, parallel assignment, double-blind | PBO | Newly diagnosed or relapsing GCA | 78 | Proportion of participants in remission of those randomized to ABA as compared to PBO at m12 | Recruiting |
| Ustekinumab [s.c. UST 90 mg at w0, w4, w12 and w28 + GC taper versus GC taper] | ULTRA NCT03711448 | IL-12/IL-23 | 52 weeks | Phase 2, randomized, parallel assignment, open-label | None | Relapsing GCA | 38 | Percentage of patients in remission, without a new relapse or deviation from the GC tapering protocol planned at w52 | Recruiting |
| Guselkumab [i.v. GUS dose 1 at w0, w4 and w8 and s.c. GUS dose 2 q4w from w12 to w48 versus PBO, with 26 w GC taper in both arms] | THEIA NCT04633447 | IL-23 | 52 weeks | Phase 2, randomized, parallel assignment, double-blind | PBO | New-onset or relapsing GCA | 60 | Percentage of participants achieving GC-free remission at w28 | Recruiting |
| Mavrilimumab [s.c. MAV 150 mg q2w versus s.c. PBO, with 26 w GC taper in both arms] [105] | NCT03827018 | GM-CSF | 26 weeks | Phase 2, randomized, parallel assignment, quadruple blind | PBO | New-onset or relapsing GCA | 70 | Time to flare by w26 | Completed (not published) |
| Bosentan [145 mg/d 14 d] | CECIBO NCT03841734 | Endothelin receptors A and B | 3 months | Phase 3, open-label | None | Sudden blindness due to GCA | 8 | Visual acuity calculated according to the Early Treatment Diabetic Retinopathy Study at m3 | Unknown |
| Glucocorticoids [28 w GC taper versus 52 w GC taper] | CORTODOSE NCT04012905 | 52 weeks | Phase 3, randomized, parallel assignment, open-label | GC | New-onset GCA | 150 | Patients in complete remission throughout 52 w, without relapse | Not yet recruiting | |
3.3.4. Endothelin Receptor Antagonists
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- González-Gay, M.A.; García-Porrúa, C. Epidemiology of the vasculitides. Rheum. Dis. Clin. N. Am. 2001, 27, 729–749. [Google Scholar] [CrossRef]
- Salvarani, C.; Cantini, F.; Boiardi, L.; Hunder, G.G. Polymyalgia rheumatica and giant-cell arteritis. N. Engl. J. Med. 2002, 347, 261–271. [Google Scholar] [CrossRef] [PubMed]
- De Smit, E.; Palmer, A.J.; Hewitt, A.W. Projected worldwide disease burden from giant cell arteritis by 2050. J. Rheumatol. 2015, 42, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Gay, M.A.; Barros, S.; Lopez-Diaz, M.J.; García-Porrua, C.; Sanchez-Andrade, A.; Llorca, J. Giant cell arteritis: Disease patterns of clinical presentation in a series of 240 patients. Medicine 2005, 84, 269–276. [Google Scholar] [CrossRef]
- Hoffman, G.S. Giant Cell Arteritis. Ann. Intern. Med. 2016, 165, ITC65–ITC80. [Google Scholar] [CrossRef]
- De Boysson, H.; Liozon, E.; Ly, K.H.; Dumont, A.; Delmas, C.; Aouba, A. The different clinical patterns of giant cell arteritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S117), 57–60. [Google Scholar]
- De Boysson, H.; Liozon, E.; Espitia, O.; Daumas, A.; Vautier, M.; Lambert, M.; Parienti, J.J.; Granel, B.; Dumont, A.; Sultan, A.; et al. Different patterns and specific outcomes of large-vessel involvements in giant cell arteritis. J. Autoimmun. 2019, 103, 102283. [Google Scholar] [CrossRef]
- González-Gay, M.A.; Prieto-Peña, D.; Calderón-Goercke, M.; Atienza-Mateo, B.; Castañeda, S. Giant cell arteritis: More than a cranial disease. Clin. Exp. Rheumatol. 2020, 38 (Suppl. S124), 15–17. [Google Scholar]
- Hunder, G.G.; Bloch, D.A.; Michel, B.A.; Stevens, M.B.; Arend, W.P.; Calabrese, L.H.; Edworthy, S.M.; Fauci, A.S.; Leavitt, R.Y.; Lie, J.T.; et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990, 33, 1122–1128. [Google Scholar] [CrossRef]
- Dejaco, C.; Ramiro, S.; Duftner, C.; Besson, F.L.; Bley, T.A.; Blockmans, D.; Brouwer, E.; Cimmino, M.A.; Clark, E.; Dasgupta, B.; et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann. Rheum. Dis. 2018, 77, 636–643. [Google Scholar] [CrossRef]
- Martínez-Lado, L.; Calviño-Díaz, C.; Piñeiro, A.; Dierssen, T.; Vazquez-Rodriguez, T.R.; Miranda-Filloy, J.A.; Lopez-Diaz, M.J.; Blanco, R.; Llorca, J.; Gonzalez-Gay, M.A. Relapses and recurrences in giant cell arteritis: A population-based study of patients with biopsy-proven disease from northwestern Spain. Medicine 2011, 90, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Villiger, P.M.; Adler, S.; Kuchen, S.; Wermelinger, F.; Dan, D.; Fiege, V.; Bütikofer, L.; Seitz, M.; Reichenbach, S. Tocilizumab for induction and maintenance of remission in giant cell arteritis: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1921–1927. [Google Scholar] [CrossRef]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. Immune mechanisms in medium and large-vessel vasculitis. Nat. Rev. Rheumatol. 2013, 9, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, M.; Audia, S.; Fraszczak, J.; Trad, M.; Orneti, P.; Lakomy, D.; Ciudad, M.; Leguy, V.; Berthier, S.; Vinit, J.; et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. 2012, 64, 3788–3798. [Google Scholar] [CrossRef]
- Samson, M.; Corbera-Bellalta, M.; Audia, S.; Planas-Rigol, E.; Martin, L.; Cid, M.C.; Bonnotte, B. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmun. Rev. 2017, 16, 833–844. [Google Scholar] [CrossRef]
- Akiyama, M.; Ohtsuki, S.; Berry, G.J.; Liang, D.H.; Goronzy, J.J.; Weyand, C.M. Innate and Adaptive Immunity in Giant Cell Arteritis. Front. Immunol. 2021, 11, 621098. [Google Scholar] [CrossRef]
- Carmona, F.D.; Martín, J.; González-Gay, M.A. Genetics of vasculitis. Curr. Opin. Rheumatol. 2015, 27, 10–17. [Google Scholar] [CrossRef]
- Carmona, F.D.; Mackie, S.L.; Martín, J.E.; Taylor, J.C.; Vaglio, A.; Eyre, S.; Bossini-Castillo, L.; Castañeda, S.; Cid, M.C.; Hernández-Rodríguez, J.; et al. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am. J. Hum. Genet. 2015, 96, 565–580. [Google Scholar] [CrossRef] [Green Version]
- Dammacco, R.; Alessio, G.; Giancipoli, E.; Leone, P.; Cirulli, A.; Resta, L.; Vacca, A.; Dammacco, F. Giant Cell Arteritis: The Experience of Two Collaborative Referral Centers and an Overview of Disease Pathogenesis and Therapeutic Advancements. Clin. Ophthalmol. 2020, 14, 775–793. [Google Scholar] [CrossRef] [Green Version]
- Harrington, R.; Al Nokhatha, S.A.; Conway, R. Biologic Therapies for Giant Cell Arteritis. Biologics 2021, 15, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Ríos-Garcés, R.; Terrades-García, N.; Espígol-Frigolé, G. Treatment of giant-cell arteritis: From broad spectrum immunosuppressive agents to targeted therapies. Rheumatology 2020, 59 (Suppl. S3), iii17–iii27. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Audia, S.; Martin, L.; Janikashvili, N.; Bonnotte, B. Pathogenesis of giant cell arteritis: New insight into the implication of CD161+ T cells. Clin. Exp. Rheumatol. 2013, 31 (Suppl. S75), S65–S73. [Google Scholar] [PubMed]
- Rittner, H.L.; Kaiser, M.; Brack, A.; Szweda, L.I.; Goronzy, J.J.; Weyand, C.M. Tissue-destructive macrophages in giant cell arteritis. Circ. Res. 1999, 84, 1050–1058. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Medium- and large-vessel vasculitis. N. Engl. J. Med. 2003, 349, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Weyand, C.M.; Björnsson, J.; Goronzy, J.J. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum. 1998, 41, 623–633. [Google Scholar] [CrossRef]
- Luo, J.; Wu, S.J.; Lacy, E.R.; Orlovsky, Y.; Baker, A.; Teplyakov, A.; Obmolova, G.; Heavner, G.A.; Richter, H.T.; Benson, J. Structural basis for the dual recognition of IL-12 and IL-23 by ustekinumab. J. Mol. Biol. 2010, 402, 797–812. [Google Scholar] [CrossRef]
- Conway, R.; O’Neill, L.; O’Flynn, E.; Gallagher, P.; McCarthy, G.M.; Murphy, C.C.; Veale, D.J.; Fearon, U.; Molloy, E.S. Ustekinumab for the treatment of refractory giant cell arteritis. Ann. Rheum. Dis. 2016, 75, 1578–1579. [Google Scholar] [CrossRef]
- Choy, E.H. Clinical significance of Janus Kinase inhibitor selectivity. Rheumatology 2019, 58, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Weyand, C.M.; Goronzy, J.J. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N. Engl. J. Med. 2014, 371, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koster, M.J.; Warrington, K.J. Giant cell arteritis: Pathogenic mechanisms and new potential therapeutic targets. BMC Rheumatol. 2017, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, M.; Ly, K.H.; Tournier, B.; Janikashvili, N.; Trad, M.; Ciudad, M.; Gautheron, A.; Devilliers, H.; Quipourt, V.; Maurieret, F.; et al. Involvement and prognosis value of CD8(+) T cells in giant cell arteritis. J. Autoimmun. 2016, 72, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Parreau, S.; Warrington, K.; Berry, G.J.; Goronzy, J.; Weyand, C.M. Regulatory T cells in Autoimmune Vasculitis. Front. Immunol. 2022, in press. [Google Scholar] [CrossRef]
- Harkins, P.; Conway, R. Giant cell arteritis: What is new in the preclinical and early clinical development pipeline? Expert Opin. Investig. Drugs 2021, 30, 1–12. [Google Scholar] [CrossRef]
- Hellmich, B.; Agueda, A.; Monti, S.; Buttgereit, F.; de Boysson, H.; Brouwer, E.; Cassie, R.; Cid, M.C.; Dasgupta, B.; Dejaco, C.; et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann. Rheum. Dis. 2020, 79, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Mackie, S.L.; Dejaco, C.; Appenzeller, S.; Camellino, D.; Duftner, C.; Gonzalez-Chiappe, S.; Mahr, A.; Mukhtyar, C.; Reynolds, G.; de Souza, A.W.S.; et al. British Society for Rheumatology guideline on diagnosis and treatment of giant cell arteritis. Rheumatology 2020, 59, e1–e23. [Google Scholar] [CrossRef] [Green Version]
- Mainbourg, S.; Addario, A.; Samson, M.; Puéchal, X.; François, M.; Durupt, S.; Gueyffier, F.; Cucherat, M.; Durieu, I.; Reynaud, Q.; et al. Prevalence of Giant Cell Arteritis Relapse in Patients Treated with Glucocorticoids: A Meta-Analysis. Arthritis Care Res. 2020, 72, 838–849. [Google Scholar] [CrossRef]
- Samson, M.; Devilliers, H.; Ly, K.H.; Maurier, F.; Bienvenu, B.; Terrier, B.; Charles, P.; Guillevin, L.; Besancenot, J.F.; Liozon, E.; et al. Tocilizumab as an add-on therapy to glucocorticoids during the first 3 months of treatment of Giant cell arteritis: A prospective study. Eur. J. Intern. Med. 2018, 57, 96–104. [Google Scholar] [CrossRef]
- Proven, A.; Gabriel, S.E.; Orces, C.; O’Fallon, W.M.; Hunder, G.G. Glucocorticoid therapy in giant cell arteritis: Duration and adverse outcomes. Arthritis Rheum. 2003, 49, 703–708. [Google Scholar] [CrossRef]
- Mukhtyar, C.; Guillevin, L.; Cid, M.C.; Dasgupta, B.; de Groot, K.; Gross, W.; Hauser, T.; Hellmich, B.; Jayne, D.; Kallenberg, C.G.M.; et al. EULAR recommendations for the management of large vessel vasculitis. Ann. Rheum. Dis. 2009, 68, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrineau, S.; Ghesquière, T.; Charles, P.; Paule, R.; Samson, M.; Gayraud, M.; Chauvin, A.; Terrier, B.; Guillevin, L.; Bonnotte, B.; et al. A French cohort of patients with giant cell arteritis: Glucocorticoid treatment and its associated side effects. Clin. Exp. Rheumatol. 2021, 39 (Suppl. S129), 155–160. [Google Scholar] [PubMed]
- Spiera, R.F.; Mitnick, H.J.; Kupersmith, M.; Richmond, M.; Spiera, H.; Peterson, M.G.; Paget, S.A. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin. Exp. Rheumatol. 2001, 19, 495–501. [Google Scholar] [PubMed]
- Jover, J.A.; Hernandez-Garcia, C.; Morado, I.C.; Vargas, E.; Bañares, A.; Fernández-Gutiérrez, B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2001, 134, 106–114. [Google Scholar] [CrossRef]
- Hoffman, G.S.; Cid, M.C.; Hellmann, D.B.; Guillevin, L.; Stone, J.H.; Schousboe, J.; Cohen, P.; Calabrese, L.H.; Dickler, H.; Merkel, P.A.; et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002, 46, 1309–1318. [Google Scholar] [CrossRef]
- Mahr, A.D.; Jover, J.A.; Spiera, R.F.; Hernández-García, C.; Fernández-Gutiérrez, B.; Lavalley, M.P.; Merkel, P.A. Adjunctive methotrexate for treatment of giant cell arteritis: An individual patient data meta-analysis. Arthritis Rheum. 2007, 56, 2789–2797. [Google Scholar] [CrossRef]
- Maz, M.; Chung, S.A.; Abril, A.; Langford, C.A.; Gorelik, M.; Guyatt, G.; Archer, A.M.; Conn, D.L.; Full, K.A.; Grayson, P.C.; et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Giant Cell Arteritis and Takayasu Arteritis. Arthritis Rheumatol. 2021, 73, 1349–1365. [Google Scholar] [CrossRef]
- De Silva, M.; Hazleman, B.L. Azathioprine in giant cell arteritis/polymyalgia rheumatica: A double-blind study. Ann. Rheum. Dis. 1986, 45, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Diamantopoulos, A.P.; Hetland, H.; Myklebust, G. Leflunomide as a corticosteroid-sparing agent in giant cell arteritis and polymyalgia rheumatica: A case series. Biomed. Res. Int. 2013, 2013, 120638. [Google Scholar] [CrossRef] [Green Version]
- Adizie, T.; Christidis, D.; Dharmapaliah, C.; Borg, F.; Dasgupta, B. Efficacy and tolerability of leflunomide in difficult-to-treat polymyalgia rheumatica and giant cell arteritis: A case series. Int. J. Clin. Pract. 2012, 66, 906–909. [Google Scholar] [CrossRef]
- Yates, M.; Loke, Y.K.; Watts, R.A.; MacGregor, A.J. Prednisolone combined with adjunctive immunosuppression is not superior to prednisolone alone in terms of efficacy and safety in giant cell arteritis: Meta-analysis. Clin. Rheumatol. 2014, 33, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Ly, K.H.; Dalmay, F.; Gondran, G.; Palat, S.; Bezanahary, H.; Cypierre, A.; Fauchais, A.L.; Liozon, E. Steroid-sparing effect and toxicity of dapsone treatment in giant cell arteritis: A single-center, retrospective study of 70 patients. Medicine 2016, 95, e4974. [Google Scholar] [CrossRef] [PubMed]
- Quartuccio, L.; Maset, M.; De Maglio, G.; Pontarini, E.; Fabris, M.; Mansutti, E.; Mariuzzi, L.; Pizzolitto, S.; Beltrami, C.A.; De Vita, S. Role of oral cyclophosphamide in the treatment of giant cell arteritis. Rheumatology 2012, 51, 1677–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loock, J.; Henes, J.; Kötter, I.; Witte, T.; Lamprecht, P.; Schirmer, M.; Gross, W.L. Treatment of refractory giant cell arteritis with cyclophosphamide: A retrospective analysis of 35 patients from three centres. Clin. Exp. Rheumatol. 2012, 30 (Suppl. S70), S70–S76. [Google Scholar]
- De Boysson, H.; Boutemy, J.; Creveuil, C.; Ollivier, Y.; Letellier, P.; Pagnoux, C.; Bienvenu, B. Is there a place for cyclophosphamide in the treatment of giant-cell arteritis? A case series and systematic review. Semin. Arthritis Rheum. 2013, 43, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Karabayas, M.; Dospinescu, P.; Fluck, N.; Kidder, D.; Fordyce, G.; Hollick, R.J.; De Bari, C.; Basu, N. Evaluation of adjunctive mycophenolate for large vessel giant cell arteritis. Rheumatol. Adv. Pract. 2020, 4, rkaa069. [Google Scholar] [CrossRef]
- Dasgupta, B.; Panayi, G.S. Interleukin-6 in serum of patients with polymyalgia rheumatica and giant cell arteritis. Br. J. Rheumatol. 1990, 29, 456–458. [Google Scholar] [CrossRef]
- Seitz, M.; Reichenbach, S.; Bonel, H.M.; Adler, S.; Wermelinger, F.; Villiger, P.M. Rapid induction of remission in large vessel vasculitis by IL-6 blockade. A case series. Swiss Med. Wkly. 2011, 141, w13156. [Google Scholar]
- Salvarani, C.; Magnani, L.; Catanoso, M.; Pipitone, N.; Versari, A.; Dardani, L.; Pulsatelli, L.; Meliconi, R.; Boiardi, L. Tocilizumab: A novel therapy for patients with large-vessel vasculitis. Rheumatology 2012, 51, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Loricera, J.; Blanco, R.; Hernández, J.L.; Castañeda, S.; Mera, A.; Pérez-Pampín, E.; Peiró, E.; Humbría, A.; Calvo-Alén, J.; Aurrecoechea, E.; et al. Tocilizumab in giant cell arteritis: Multicenter open-label study of 22 patients. Semin. Arthritis Rheum. 2015, 44, 717–723. [Google Scholar] [CrossRef]
- Régent, A.; Redeker, S.; Deroux, A.; Kieffer, P.; Ly, K.H.; Dougados, M.; Liozon, E.; Larroche, C.; Guillevin, L.; Bouillet, L.; et al. Tocilizumab in Giant Cell Arteritis: A Multicenter Retrospective Study of 34 Patients. J. Rheumatol. 2016, 43, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Adler, S.; Reichenbach, S.; Gloor, A.; Yerly, D.; Cullmann, J.L.; Villiger, P.M. Risk of relapse after discontinuation of tocilizumab therapy in giant cell arteritis. Rheumatology 2019, 58, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Nannini, C.; Niccoli, L.; Sestini, S.; Laghai, I.; Coppola, A.; Cantini, F. Remission maintenance after tocilizumab dose-tapering and interruption in patients with giant cell arteritis: An open-label, 18-month, prospective, pilot study. Ann. Rheum. Dis. 2019, 78, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Goercke, M.; Loricera, J.; Aldasoro, V.; Castañeda, S.; Villa, I.; Humbría, A.; Moriano, C.; Romero-Yuste, S.; Narváez, J.; Gómez-Arango, C.; et al. Tocilizumab in giant cell arteritis. Observational, open-label multicenter study of 134 patients in clinical practice. Semin. Arthritis Rheum. 2019, 49, 126–135. [Google Scholar] [CrossRef]
- Amsler, J.; Kysela, I.; Tappeiner, C.; Seitz, L.; Christ, L.; Scholz, G.; Stalder, O.; Kollert, F.; Reichenbach, S.; Villiger, P.M. Vision loss in patients with giant cell arteritis treated with tocilizumab. Arthritis Res Ther. 2021, 23, 92. [Google Scholar] [CrossRef]
- Tuckwell, K.; Collinson, N.; Dimonaco, S.; Klearman, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; Salvarani, C.; et al. GiACTA Investigators. Newly diagnosed vs. relapsing giant cell arteritis: Baseline data from the GiACTA trial. Semin. Arthritis Rheum. 2017, 46, 657–664. [Google Scholar] [CrossRef]
- Loricera, J.; Blanco, R.; Castañeda, S.; Humbría, A.; Ortego-Centeno, N.; Narváez, J.; Mata, C.; Melchor, S.; Aurrecoechea, E.; Calvo-Alén, J.; et al. Tocilizumab in refractory aortitis: Study on 16 patients and literature review. Clin. Exp. Rheumatol. 2014, 32 (Suppl. S82), S79–S89. [Google Scholar] [CrossRef]
- Loricera, J.; Blanco, R.; Hernández, J.L.; Castañeda, S.; Humbría, A.; Ortego-Centeno, N.; Bravo, B.; Freire, M.; Melchor, S.; Mínguez, M.; et al. Tocilizumab in patients with Takayasu arteritis: A retrospective study and literature review. Clin. Exp. Rheumatol. 2016, 34 (Suppl. S97), S44–S53. [Google Scholar]
- Unizony, S.; McCulley, T.J.; Spiera, R.; Pei, J.; Sidiropoulos, P.N.; Best, J.H.; Birchwood, C.; Pavlov, A.; Stone, J.H. Clinical outcomes of patients with giant cell arteritis treated with tocilizumab in real-world clinical practice: Decreased incidence of new visual manifestations. Arthritis Res. Ther. 2021, 23, 8. [Google Scholar] [CrossRef]
- Saito, S.; Okuyama, A.; Okada, Y.; Shibata, A.; Sakai, R.; Kurasawa, T.; Kondo, T.; Takei, H.; Amano, K. Tocilizumab monotherapy for large vessel vasculitis: Results of 104-week treatment of a prospective, single-centre, open study. Rheumatology 2020, 59, 1617–1621. [Google Scholar] [CrossRef]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Glucocorticoid Dosages and Acute-Phase Reactant Levels at Giant Cell Arteritis Flare in a Randomized Trial of Tocilizumab. Arthritis Rheumatol. 2019, 71, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unizony, S.H.; Bao, M.; Han, J.; Luder, Y.; Pavlov, A.; Stone, J.H. Treatment failure in giant cell arteritis. Ann. Rheum. Dis. 2021, 80, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Han, J.; Mohan, S. Efficacy of Adjunctive Methotrexate in Patients with Giant Cell Arteritis Treated with Tocilizumab Plus Prednisone Tapering: Subanalysis of a Phase 3 Trial [abstract 1926]. Arthritis Rheumatol. 2020, 72 (Suppl. S10), 3863–3865. [Google Scholar]
- Stone, J.H.; Bao, M.; Han, J.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; Salvarani, C.; et al. Long-Term Outcome of Tocilizumab for Patients with Giant Cell Arteritis: Results from Part 2 of a Randomized Controlled Phase 3 Trial [abstract 0808]. Arthritis Rheumatol. 2019, 71 (Suppl. S10), 1389–1390. [Google Scholar]
- Berti, A.; Cornec, D.; Medina Inojosa, J.R.; Matteson, E.L.; Murad, M.H. Treatments for giant cell arteritis: Meta-analysis and assessment of estimates reliability using the fragility index. Semin. Arthritis Rheum. 2018, 48, 77–82. [Google Scholar] [CrossRef]
- Unizony, S.; Mohan, S.; Han, J.; Stone, J. Characteristics of Giant Cell Arteritis Flares After Successful Treatment with Tocilizumab: Results from the Long-Term Extension of a Randomized Controlled Phase 3 Trial [abstract 0516]. Arthritis Rheumatol. 2020, 72 (Suppl. S10), 1050–1052. [Google Scholar]
- Garvey, T.D.; Koster, M.J.; Warrington, K.J. My Treatment Approach to Giant Cell Arteritis. Mayo Clin. Proc. 2021, 96, 1530–1545. [Google Scholar] [CrossRef]
- Regola, F.; Cerudelli, E.; Bosio, G.; Andreoli, L.; Tincani, A.; Franceschini, F.; Toniati, P. Long-term treatment with tocilizumab in giant cell arteritis: Efficacy and safety in a monocentric cohort of patients. Rheumatol. Adv. Pract. 2020, 4, rkaa017. [Google Scholar] [CrossRef]
- Schmidt, W.A.; Dasgupta, B.; Luqmani, R.; Unizony, S.H.; Blockmans, D.; Lai, Z.; Kurrasch, R.H.; Lazic, I.; Brown, K.; Rao, R. A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy and Safety of Sirukumab in the Treatment of Giant Cell Arteritis. Rheumatol. Ther. 2020, 7, 793–810. [Google Scholar] [CrossRef]
- Hoffman, G.S.; Cid, M.C.; Rendt-Zagar, K.E.; Merkel, P.A.; Weyand, C.M.; Stone, J.H.; Salvarani, C.; Xu, W.; Visvanathan, S.; Rahman, M.H.; et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: A randomized trial. Ann. Intern. Med. 2007, 146, 621–630. [Google Scholar] [CrossRef]
- Seror, R.; Baron, G.; Hachulla, E.; Debandt, M.; Larroche, C.; Puéchal, X.; Maurier, F.; de Wazieres, B.; Quéméneur, T.; Ravaud, P.; et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: Results of a multicentre randomized controlled trial. Ann. Rheum. Dis. 2014, 73, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Taboada, V.M.; Rodriguez-Valverde, V.; Carreno, L.; Lopez-Longo, J.; Figueroa, M.; Belzunegui, J.; Mola, E.M.; Bonilla, G. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann. Rheum. Dis. 2008, 67, 625–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, R.; O’Neill, L.; McCarthy, G.M.; Murphy, C.C.; Fabre, A.; Kennedy, S.; Veale, D.J.; Wade, S.M.; Fearon, U.; Molloy, E.S. Interleukin 12 and interleukin 23 play key pathogenic roles in inflammatory and proliferative pathways in giant cell arteritis. Ann. Rheum. Dis. 2018, 77, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Espígol-Frigolé, G.; Planas-Rigol, E.; Lozano, E.; Corbera-Bellalta, M.; Terrades-García, N.; Prieto-González, S.; García-Martínez, A.; Hernández-Rodríguez, J.; Grau, J.M.; Cid, M.C. Expression and Function of IL12/23 Related Cytokine Subunits (p35, p40, and p19) in Giant-Cell Arteritis Lesions: Contribution of p40 to Th1- and Th17-Mediated Inflammatory Pathways. Front. Immunol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, J.M.; Peritt, D.; Scallon, B.J.; Heavner, G.A.; Shealy, D.J.; Giles-Komar, J.M.; Mascelli, M.A. Discovery and mechanism of ustekinumab: A human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs 2011, 3, 535–545. [Google Scholar] [CrossRef]
- Conway, R.; O’Neill, L.; Gallagher, P.; McCarthy, G.M.; Murphy, C.C.; Veale, D.J.; Fearon, U.; Molloy, E.S. Ustekinumab for refractory giant cell arteritis: A prospective 52-week trial. Semin. Arthritis Rheum. 2018, 48, 523–528. [Google Scholar] [CrossRef]
- Matza, M.A.; Fernandes, A.D.; Stone, J.H.; Unizony, S.H. Ustekinumab for the treatment of giant cell arteritis. Arthritis Care Res. 2021, 73, 893–897. [Google Scholar] [CrossRef]
- Samson, M.; Bonnotte, B. Ustekinumab for the Treatment of Giant Cell Arteritis: Comment on the Article by Matza et al. Arthritis Care Res. 2021, 73, 1058–1059. [Google Scholar] [CrossRef]
- Conway, R.; Molloy, E.S. Ustekinumab in Giant Cell Arteritis: Comment on the Article by Matza et al. Arthritis Care Res. 2021, 73, 1056–1057. [Google Scholar] [CrossRef]
- Langford, C.A.; Cuthbertson, D.; Ytterberg, S.R.; Khalidi, N.; Monach, P.A.; Carette, S.; Seo, P.; Moreland, L.W.; Weisman, M.; Koening, C.L.; et al. Vasculitis Clinical Research Consortium. A Randomized, Double-Blind Trial of Abatacept (CTLA-4Ig) for the Treatment of Giant Cell Arteritis. Arthritis Rheumatol. 2017, 69, 837–845. [Google Scholar] [CrossRef]
- Rossi, D.; Cecchi, I.; Sciascia, S.; Naretto, C.; Alpa, M.; Roccatello, D. An agent-to-agent real life comparison study of tocilizumab versus abatacept in giant cell arteritis. Clin. Exp. Rheumatol. 2021, 39 (Suppl. S129), 125–128. [Google Scholar] [PubMed]
- Song, G.G.; Lee, Y.H. Efficacy and safety of biological agents in patients with giant cell arteritis: A meta-analysis of randomized trials. Int. J. Clin. Pharmacol. Ther. 2020, 58, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Pountain, G.; Hazleman, B.; Cawston, T. Circulating levels of IL-1beta, IL-6 and soluble IL-2 receptor in polymyalgia rheumatica and giant cell arteritis and rheumatoid arthritis. Br. J. Rheumatol. 1998, 37, 797–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Rodríguez, J.; Segarra, M.; Vilardell, C.; Sánchez, M.; García-Martínez, A.; Esteban, M.J.; Queralt, C.; Grau, J.M.; Urbano-Márquez, A.; Palacín, A.; et al. Tissue production of pro-inflammatory cytokines (IL-1β, TNFα and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology 2004, 43, 294–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshayes, S.; Ly, K.H.; Rieu, V.; Maigné, G.; Martin Silva, N.; Manrique, A.; Monteil, J.; de Boysson, H.; Aouba, A.; French Study Group for Large Vessel Vasculitis (GEFA). Steroid-sparing effect of anakinra in giant-cell arteritis: A case series with clinical, biological and iconographic long-term assessments. Rheumatology 2021, 61, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Rotar, Ž.; Tomšič, M.; Hočevar, A. Secukinumab for the maintenance of glucocorticoid-free remission in a patient with giant cell arteritis and psoriatic arthritis. Rheumatology 2018, 57, 934–936. [Google Scholar] [CrossRef] [Green Version]
- Venhoff, N.; Schmidt, W.A.; Lamprecht, P.; Tony, H.P.; App, C.; Sieder, C.; Legeler, C.; Jentzsch, C.; Thiel, J. Efficacy and safety of secukinumab in patients with giant cell arteritis: Study protocol for a randomized, parallel group, double-blind, placebo-controlled phase II trial. Trials 2021, 22, 543. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Gadina, M.; Schreiber, R.D. Cytokine signaling in 2002: New surprises in the Jak/Stat pathway. Cell 2002, 109 (Suppl. S1), S121–S131. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: Current and future prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Zhang, H.; Watanabe, R.; Berry, G.J.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Inhibition of JAK-STAT Signaling Suppresses Pathogenic Immune Responses in Medium and Large Vessel Vasculitis. Circulation 2018, 137, 1934–1948. [Google Scholar] [CrossRef]
- Régent, A.; Terrier, B.; Legendre, P.; Wartski, M.; Cohen, P.; Mouthon, L.; Le Jeunne, C. Efficacy of baricitinib for refractory large-vessel vasculitis. Rheumatology 2021, 60, e389–e391. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Gandhi, R.; Corbera-Bellalta, M.; Muralidharan, S.; Paolini, J.F. THU0008 GM-CSF pathway signature identified in temporal artery biopsies of patients with giant cell arteritis [abstract]. Ann. Rheum. Dis. 2019, 78 (Suppl. S2), 271–272. [Google Scholar]
- Terrier, B.; Geri, G.; Chaara, W.; Allenbach, Y.; Rosenzwajg, M.; Costedoat-Chalumeau, N.; Fouret, P.; Musset, L.; Benveniste, O.; Six, A.; et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. 2012, 64, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Dougan, M.; Dranoff, G.; Dougan, S.K. GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity 2019, 50, 796–811. [Google Scholar] [CrossRef]
- Watanabe, R.; Zhang, H.; Maeda, T.; Akiyama, M.; Gandhi, R.; Paolini, J.; Berry, G.; Weyand, C. GM-CSF Is a Pro-Inflammatory Cytokine in Experimental Vasculitis of Medium and Large Arteries [abstract]. Arthritis Rheumatol. 2019, 2019, 07030-5774. Available online: https://acrabstracts.org/abstract/gm-csf-is-a-pro-inflammatory-cytokine-in-experimental-vasculitis-of-medium-and-large-arteries/ (accessed on 9 March 2022).
- Corbera-Bellalta, M.; Alba-Rovira, R.; Muralidharan, S.; Espígol-Frigolé, G.; Ríos-Garcés, R.; Marco-Hernández, J.; Denuc, A.; Kamberovic, F.; Pérez-Galán, P.; Joseph, A.; et al. Blocking GM-CSF receptor α with mavrilimumab reduces infiltrating cells, pro-inflammatory markers and neoangiogenesis in ex vivo cultured arteries from patients with giant cell arteritis. Ann. Rheum. Dis. 2022, 81. [Google Scholar] [CrossRef]
- Cid, M.; Unizony, S.; Pupim, L.; Fang, F.; Pirrello, J.; Ren, A.; Samant, M.; Zhou, T.; Paolini, J.F. Mavrilimumab (anti GM-CSF receptor α monoclonal antibody) reduces time to flare and increases sustained remission in a phase 2 trial of patients with giant cell Arteritis [abstract]. Ann. Rheum. Dis. 2020, 80 (Suppl. S1), L06. [Google Scholar]
- Masaki, T. Historical review: Endothelin. Trends Pharmacol. Sci. 2004, 25, 219–224. [Google Scholar] [CrossRef]
- Lozano, E.; Segarra, M.; Corbera-Bellalta, M.; García-Martínez, A.; Espígol-Frigolé, G.; Plà-Campo, A.; Hernández-Rodríguez, J.; Cid, M.C. Increased expression of the endothelin system in arterial lesions from patients with giant-cell arteritis: Association between elevated plasma endothelin levels and the development of ischaemic events. Ann. Rheum. Dis. 2010, 69, 434–442. [Google Scholar] [CrossRef]
- Danesh-Meyer, H.; Savino, P.J.; Gamble, G.G. Poor prognosis of visual outcome after visual loss from giant cell arteritis. Ophthalmology 2005, 112, 1098–1103. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Castañeda, S.; Llorca, J. Giant Cell Arteritis: Visual Loss Is Our Major Concern. J. Rheumatol. 2016, 43, 1458–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
| Author, Year [Ref.] | Type of Study | n | Sex F (%) | Population/Median Follow-Up (FU) | Therapeutic Protocol | Prior DMARD n (%) | Main Efficacy Outcomes | Serious Adverse Events |
|---|---|---|---|---|---|---|---|---|
| Observational Studies | ||||||||
| Loricera et al., 2015 [60] | Retrospective multicenter open-label study | 22 | 17 (72.3) | Refractory GCA and/or with unacceptable GC-related AE. FU: 9 months | i.v. TCZ 8mg/kg/m (monotherapy n = 10, combined with MTX n = 12) | 19 (86.4) | Clinical remission (17/22) or improvement (2/22). Significant reduction of ESR, CRP and GC dose. Prior visual loss not reverted by TCZ in 2 patients | TCZ discontinuation (n = 3) due to SAE (neutropenia, recurrent pneumonia and CMV). 1 Death (stroke and infective endocarditis) |
| Régent et al., 2016 [61] | Retrospective multicenter open-label study | 34 | 27 (79.4) | GCA with unacceptable GC-related AEs (n = 31), severe disease (n = 2) or as a GC-sparing agent (n = 1). FU: 13 months | i.v. TCZ 8mg/kg/m (monotherapy n = 16, combined with MTX n = 18) | 20 (58.8) | Clinical improvement (28/34). Significant reduction of CRP and GC dose. Prior visual loss not reverted by TCZ (n = 1). Planned stop of TCZ (n = 20), with 6 relapses after a mean of 3.5 m | TCZ discontinuation (n = 3) due to SAE (liver cytolysis, neutropenia and TBC pericarditis). 1 Death (septic shock) |
| Samson et al., 2018 [39] | Prospective multicenter open-label study | 20 | 15 (75) | New-onset (95%) or recurrent GCA. FU: 52 weeks | 4 Infusions of i.v. TCZ 8 mg/kg/m from w0 to w12 | Prior DMARD within 6 m before inclusion were not allowed | Remission in 100% at w12 75% in remission with ≤0.1 mg/kg/d of GC at w26. 45% relapse-free survival at w52 Relapse (n = 10) more frequent if baseline aortitis (p = 0.048), CRP > 70 mg/L (p = 0.036) or Hb ≤ 10 g/dL (p = 0.015) | 3 SAE: 1 sudden death for unknown reasons and 2 hospitalizations (atrial fibrillation and hip replacement) |
| Adler et al., 2019 [62] | Open-label extension of iv TCZ trial | 17 | 13 (76.5) | Relapse-free patients from the TCZ arm. FU: 28.1 months | If relapse, TCZ or GC could be added at physician discretion | No GC or other DMARD were given after TCZ discontinua-tion | Lasting remission DMARD-free in > 50% of patients. 8 Patients relapsed: 6 within first 5 m and 2 at 13 m and 14 m, respectively. Relapsing patients were younger and had more signs of mural enhancement in MRA | NR |
| Nannini et al., 2019 [63] | Single-center prospective open-label study | 15 | NR | New-onset (n = 11) or recurrent GCA with thoracic LVV. FU: 18 months | i.v. TCZ 8mg/kg/m (n = 9) or s.c. TCZ 162 mg qw (n = 6) + GC taper of 2 m TCZ tapering from m6 to m10 TCZ interruption at m12 | NR | 100% Sustained remission during TCZ therapy. 66.7% Patients maintained remission after TCZ withdrawal. 5 Patients relapsed 2–4 m after TCZ interruption, responding to TCZ reintroduction at the last dose, allowing a second withdrawal of TCZ in 2 of them. Non-response correlated with some PET/CT parameters | No SAE |
| Calderón-Goercke et al., 2019 [64] | Multicenter retrospective series | 134 | 101 (75.4) | Refractory GCA (100%) | i.v. TCZ 8mg/kg/m (n = 106) or s.c.TCZ 162 mg qw (n = 28) + GC | csDMARD 98 (73.1) bDMARD 3 (2.2) | Clinical remission was achieved in 55.5%, 70.4%, 69.2% and 90% of patients at 6, 12, 18, 24 months, respectively | SAE: 32 (21.1 per 100 patients-year). Serious infections: 16 (10.6 per 100 patients-year). TCZ withdrawals: 17 (12.7 per 100 patients-year) |
| Amsler et al., 2021 [65] | Observational monocentric study | 186 | 116 (62) | MR angiography of aorta performed/ positive in: 170 (91%)/123 (72%). FU: NR | i.v. TCZ was added to GC in doses of 8 mg/kg body weight at 4-week intervals or at a dosage of 162 mg s.c. at weekly or bi-weekly intervals | NR | The occurrence of vision loss in a large GCA cohort treated with TCZ | Only visual AE described: Two patients (1.1%) developed vision loss, both at the initiation of TCZ treatment |
| Randomized clinical trials | ||||||||
| Villiger et al., 2016 [12] | Single center, phase 2, DB, PBO-controlled | 30 (20 TCZ, 10 PBO) 2:1 ratio | TCZ 13 (65) PBO 8 (80) | New-onset (80% TCZ, 70% PBO) or relapsing GCA. FU: 52 weeks | i.v. TCZ 8mg/kg/m + GC vs. PBO + GC Same GC tapering schedule and concomitant drugs TCZ was used as mono-therapy | Prior bDMARD not allowed No mention about prior csDMARD | Complete remission at week 12: 85% TCZ vs. 40% PBO (RD 45%, 95% CI 11–79; p = 0.03). Complete remission at week 52: 85% TCZ vs. 20% PBO (RD 65%, 95% CI 36–94; p = 0.001). Significantly lower time to stop GC and cumulative GC dose in TCZ group vs. PBO group | 35% TCZ vs. 50% PBO, (p = 0.46). TCZ: 1 headache and tinnitus, 3 GI (prepyloric ulcer perforation, viral hepatopathy and bleeding), 1 eye infection, 1 psychosis and 1 Stevens-Johnson syndrome. PBO: 3 CV events (syncope, coronary artery disease, lethal MI), 1 sigmoid perforation, 2 GC-induced myopathy and hyperglycemia, 2 back pain and 2 lumbar fractures and vertebroplasty |
| Stone et al., 2017 [13] Tuckwell et al., 2017 [66] | Multicenter, phase 3, DB, PBO-controlled (GiACTA trial) | 251 (150 TCZ groups *, 101 PBO groups #) 2:1:1:1 ratio | TCZ * 78 (78) 35 (70) PBO # 38 (76) 37 (73) | New-onset (47% and 52% TCZ groups *, 46% and 45% PBO groups #) or relapsing GCA. FU: 52 weeks | s.c. TCZ 162 mg qw + GC 26 w (n = 100); s.c. TCZ 162 mg q2w + GC 26 w (n = 50); PBO + GC 26 w (n = 50); PBO + GC 52 w (n = 51). Concomitant MTX was the only DMARD allowed Ϯ | Baseline MTX in 4 (3) newly diagnosed GCA and 23 (17) relapsing GCA | Sustained GC-free remission at week 52 significantly favored TCZ over PBO (p < 0.001): 56% TCZ qw, 53% TCZ q2w, 14% PBO + GC 26 w and 18% PBO + GC 52 w. Significantly lower risk of flare and cumulative GC dose in TCZ groups vs. PBO groups | 15% TCZ qw, 14% TCZ q2w, 22% PBO + GC 26 w and 25% PBO + GC 52 w, NS. Infections were the most frequent SAE: 7%, 4%, 7% and 12%, respectively. 1 case of AION during a flare in TCZ q2w group |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castañeda, S.; Prieto-Peña, D.; Vicente-Rabaneda, E.F.; Triguero-Martínez, A.; Roy-Vallejo, E.; Atienza-Mateo, B.; Blanco, R.; González-Gay, M.A. Advances in the Treatment of Giant Cell Arteritis. J. Clin. Med. 2022, 11, 1588. https://doi.org/10.3390/jcm11061588
Castañeda S, Prieto-Peña D, Vicente-Rabaneda EF, Triguero-Martínez A, Roy-Vallejo E, Atienza-Mateo B, Blanco R, González-Gay MA. Advances in the Treatment of Giant Cell Arteritis. Journal of Clinical Medicine. 2022; 11(6):1588. https://doi.org/10.3390/jcm11061588
Chicago/Turabian StyleCastañeda, Santos, Diana Prieto-Peña, Esther F. Vicente-Rabaneda, Ana Triguero-Martínez, Emilia Roy-Vallejo, Belén Atienza-Mateo, Ricardo Blanco, and Miguel A. González-Gay. 2022. "Advances in the Treatment of Giant Cell Arteritis" Journal of Clinical Medicine 11, no. 6: 1588. https://doi.org/10.3390/jcm11061588
APA StyleCastañeda, S., Prieto-Peña, D., Vicente-Rabaneda, E. F., Triguero-Martínez, A., Roy-Vallejo, E., Atienza-Mateo, B., Blanco, R., & González-Gay, M. A. (2022). Advances in the Treatment of Giant Cell Arteritis. Journal of Clinical Medicine, 11(6), 1588. https://doi.org/10.3390/jcm11061588