Abstract
Mutations in SF3B1 are found in 20% of myelodysplastic syndromes and 5–10% of myeloproliferative neoplasms, where they are considered important for diagnosis and therapy decisions. Sanger sequencing and NGS are the currently available methods to identify SF3B1 mutations, but both are time-consuming and expensive techniques that are not practicable in most small-/medium-sized laboratories. To identify the most frequent SF3B1 mutation, p.Lys700Glu, we developed a novel fast and cheap assay based on PNA-PCR clamping. After setting the optimal PCR conditions, the limit of detection of PNA-PCR clamping was evaluated, and the method allowed up to 0.1% of mutated SF3B1 to be identified. Successively, PNA-PCR clamping and Sanger sequencing were used to blind test 90 DNA from patients affected by myelodysplastic syndromes and myeloproliferative neoplasms for the SF3B1 p.Lys700Glu mutation. PNA-PCR clamping and Sanger sequencing congruently identified 75 negative and 13 positive patients. Two patients identified as positive by PNA-PCR clamping were missed by Sanger analysis. The discordant samples were analyzed by NGS, which confirmed the PNA-PCR clamping result, indicating that these samples contained the SF3B1 p.Lys700Glu mutation. This approach could easily increase the characterization of myelodysplastic syndromes and myeloproliferative neoplasms in small-/medium-sized laboratories, and guide patients towards more appropriate therapy.
1. Introduction
Splicing represents the process by which introns are excised from the precursor messenger RNA (pre-mRNA) and contiguous exons are joined together []. In higher eukaryotes, genes undergo different splicing processes, generating different mRNA isoforms that lead to the translation of proteins with distinct functions []. Splicing is catalyzed by the spliceosome, a macromolecule composed of five small nuclear RNA (snRNA), each associated with proteins to form small nuclear ribonucleoproteins (snRNP).
The splicing factor 3b subunit 1 (SF3B1) gene is located on chromosome 2 in position 2q33.1. It encodes subunit 1 of the SF3B splice complex, a 146 kDa protein that is essential for the spliceosome function. Mutations in SF3B1 are common in different hematological malignancies, and they were mostly found in myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN). MDS are clonal disorders of hematopoietic stem cells, characterized by ineffective hematopoiesis and the risk of evolving into acute myeloid leukemia (AML) []. Mutations in the SF3B1 gene were found in 20% of MDS patients, particularly in 65% of MDS patients with ring sideroblasts (MDS-RS). In most cases, SF3B1 mutations are missense and occur in heterozygosity. The most common mutation, present in 55–65% of cases, is NM_012433.4(SF3B1): c.2098 A>G, which leads to the substitution of lysine in glutamic acid in position 700 (p.Lys700Glu). Other sites that can be less frequently mutated are p.Glu622, p.Arg625, p.His662, p.Lys666, and p.Ile704 []. The strong association between mutations in SF3B1 and the RS phenotype indicates a causal relationship, which makes this the first gene to be associated with a specific morphological feature in MDS []. These data allowed the WHO classification to be revised, where the RS threshold necessary for diagnosing MDS-RS in SF3B1-mutated patients was reduced from 15% to 5% []. Despite MDS-RS being characterized by a lower risk of AML evolution, patients often become resistant to erythropoietin-stimulating agents (ESA) [] and dependent on red cell transfusions. Nowadays, a new therapeutic option for MDS-RS patients is available. Luspatercept is a recombinant fusion protein that works by binding and inhibiting the ligands of the TGFβ superfamily, interrupting TGFβ signaling, which is dysregulated in MDS []. The efficacy of luspatercept was evaluated in a phase II multicenter study, PACE [], and in a phase III, double-blind, multinational randomized study, MEDALIST []. Both yielded positive results in terms of improved hemoglobin levels and transfusion independence. For these reasons, the SF3B1 mutational status in MDS patients has acquired an important role in both the diagnostic and clinical settings, as it guides an important therapeutic choice.
MPN is a group of blood cancers characterized by overproduction of erythrocytes, leukocytes, or platelets in the bone marrow. In MPN, SF3B1 was found to be mutated in approximately 5% of polycythemia vera (PV) and essential thrombocythemia (ET) patients, and 10% of myelofibrosis (MF) patients. SF3B1 mutations in MPN do not affect survival [], though they seem to correlate with increased thrombotic risk []. The initial results from a phase II study suggest clinically significant activity of luspatercept in patients with MF-associated anemia [].
The reference technologies for the mutational analysis of the SF3B1 gene are Sanger sequencing and next-generation sequencing (NGS). Both methods are expensive and time consuming, so evaluation of the mutational status of SF3B1 is often not performed by small-/medium-sized laboratories.
Peptide nucleic acids (PNA), synthetic oligonucleotides with a skeleton of repeated N-(2-aminoethyl)-glycine residues joined by peptide bonds [], can be used to evaluate the presence of specific mutations in a DNA sample by PNA-PCR clamping. The principles upon which PNA-PCR clamping is based, and some of its applications, have been amply summarized by Fouz and Appella []. PNA has such a high affinity for complementary DNA that even one different base prevents hybridization []. Furthermore, the PNA/DNA dimer cannot be amplified by DNA polymerase []. PNA-PCR clamping is based on the competition between a PNA probe and a primer to bind the same DNA sequence, and exploits the ability of PNA to hybridize to DNA and suppress amplification [,,,,].
With the aim of identifying the p.Lys700Glu mutation, we developed a novel fast and cheap assay using PNA-PCR clamping. This approach could easily increase the characterization of MDS patients in small-/medium-sized laboratories, and guide them to more appropriate therapy.
2. Materials and Methods
2.1. Patient Cohort
After signing the informed consent form, 31 bone marrow (BM) and 59 peripheral blood (PB) samples were collected from 90 patients (57 MDS and 33 MPN). DNA was extracted as described by Saguna et al. [] and quantified by spectrophotometry. Patients’ characteristics are listed in the Results section tables. In addition to the patients enrolled for screening, we tested, in duplicate (both BM and PB), the DNA of 3 mutated SF3B1 p.Lys700Glu patients and 3 wild-type patients.
2.2. Cloning PCR Controls with pGEM®—T Easy Vector
Plasmids used as PCR positive controls were generated by amplifying SF3B1 p.Lys700Glu and wild-type (WT) from an MDS patient with the following primers (Sigma-Aldrich, St. Louis, MO, USA): forward 5′-TGACAGGCTATGGTTC-3′ and reverse 5′-GAAACATATCCAGTTTACAT-3′. PCR products were purified by Monarch PCR & DNA Cleanup Kit (New England Biolabs, Ipswich, MA, USA) and cloned in pGEM-T Easy Vector (Promega, Milan, Italy). The sequences were verified by the capillary Sanger sequence method. All reactions were performed following the manufacturer’s instructions.
2.3. Sanger Sequencing for SF3B1 p.Lys700Glu Evaluation
To perform Sanger sequencing, SF3B1 was amplified from DNA (100 ng) of patients and analyzed by sequencing with BigDye terminator v3.1 (Applied Biosystem, Foster City, CA, USA) and capillary electrophoresis on an ABI PRISM 3500XL genetic analyzer (Applied Biosystem, Foster City, CA, USA), using the primers described before. All reactions were performed following the manufacturer’s instructions. The limit of detection (LoD) of the method was previously estimated, by serial dilution experiments, to be approximately 15–20% [].
2.4. PNA-PCR Clamping for SF3B1 p.Lys700Glu Evaluation
The method for the detection of the SF3B1 p.Lys700Glu mutation by PNA-PCR clamping forecasted two PCR steps. Primers (Sigma-Aldrich, St. Louis, MO, USA) and PNA probe (Panagene, Daejeon, Korea) for SF3B1 amplification were designed on the DNA sequence NG_032903.2.
Step 1 (pre-amplification): DNA from patients (200 ng) and control plasmids (0.01 ng) were used to amplify a small area of SF3B1 around the c.2098 position. The primers used for the first step were the same as described before. The final volume of each reaction was 20 uL and the final concentrations of the reagents were as follows: MgCl2 [2.5 mM] (Applied Biosystem, Foster City, CA, USA), GeneAmp™ 10X PCR Buffer II [1X] (Applied Biosystem, Foster City, CA, USA), dNTP [200 nM] (Thermo Fisher Scientific, Waltham, MA, USA), SF3B1 Fwd [50 nM], SF3B1 Rev [50 nM], and AmpliTaq™ DNA polymerase [1U] (Applied Biosystem, Foster City, CA, USA). The amplification protocol was as follows: 94 °C × 3′, (94 °C × 30″, 55 °C × 30″, 68 °C × 40″) for 10 cycles, using a T100 thermal cycler (Bio-Rad, Hercules, CA, USA).
Step 2: 1 uL of the PCR product from the first step was used for the second step. PCR amplifications were carried out in duplicate, in the presence (TEST) or absence (CONTROL) of the PNA probe.
Primers and PNA probe sequences were as follows: forward 5′-ATGAGCAGCAGGAAG-3′, reverse 5′-GAAACATATCCAGTTTACAT-3′, and PNA probe 5′-AGCAGAAAGTTC-3′.
The final volume of the reaction was 20 uL and the final concentrations of the reagents were as follows: MgCl2 (Applied Biosystem, Foster City, CA, USA) [2.5 mM], GeneAmp™ 10X PCR Bfr II [1X] (Applied Biosystem, Foster City, CA, USA), dNTP mix [200 nM] (Thermo Fisher Scientific, Waltham, MA, USA), SF3B1 p.Lys700Glu Fwd [100 nM], SF3B1 p.Lys700Glu Rev [100 nM], PNA [800 nM], and AmpliTaq™ DNA polymerase [1U] (Applied Biosystem, Foster City, CA, USA).
The amplification protocol was as follows: 94 °C × 3′, (94 °C × 30″, 56 °C × 20″, 68 °C × 30″) for 33 cycles, 68 °C × 2′, using a T100 thermal cycler (Bio-Rad, Hercules, CA, USA).
After PCR amplification, each amplicon (20 uL) was mixed with DNA gel loading dye 6X (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions, and loaded onto 2% AgaPure™ Agarose LE (Canvax Reagents, Cordoba, Spain)-1x UltraPure™ TBE (Invitrogen, Waltham, MA, USA) gel with 5 ug/mL ethidium bromide (Thermo Fisher Scientific, Waltham, MA, USA) and run at 150 V for 30 min. The electrophoretic runs were acquired with ChemiDoc XRS+ (Biorad, Hercules, CA, USA) and analyzed with Image Lab software 4.0.1 (Biorad, Hercules, CA, USA).
2.5. Next-Generation Sequencing
According to the manufacturer’s recommendations, NGS libraries were obtained using the Myeloid Solution kit (Sophia Genetic, Lausanne, Switzerland) starting with the genomic DNA of the two patients with non-congruent results. The libraries were further sequenced onto a MiSeq Sequencing System (Illumina, San Diego, CA, USA) using the MiSeq Reagent Kit v3 (Illumina, San Diego, CA, USA). Data processing and BAM files were obtained by Sophia Genetics using DDMTMv5 Software (Sophia Genetic, Lausanne, Switzerland). The alignment of sequenced reads with the SF3B1 gene in the reference genome (hg19) was visualized using the “Integrative Genomics Viewer” (IGV) (https://software.broadinstitute.org/software/igv/ (accessed on 19 July 2021); Broad Institute, University of California).
2.6. Statistical Analysis
Diagnostic test equivalency was checked with the McNemar statistical test. Baseline characteristics were investigated using Fisher’s exact test for categorical variables and unpaired t-test for continuous variables. WT1 expression at diagnosis was dichotomized in normal- or high-expression group, as indicated in the literature [] (BM: 90 WT1/104 ABL1 copies; PB: 10 WT1/104 ABL1 copies). Statistical analyses were performed using GraphPad Prism 7 statistical software. All the analyses with a p-value less than or equal to 0.05 were indicated as significant.
3. Results
3.1. Detection of SF3B1 p.Lys700Glu Mutation by PNA-PCR Clamping
The PNA-PCR clamping method is based on direct competition between the PNA probe and one of the primers to bind the complementary genomic DNA sequence (gDNA). If the SF3B1 gene is WT, the PNA probe, drawn on the WT sequence, pairs perfectly with the gDNA and inhibits its amplification. On the contrary, the presence of the p.Lys700Glu mutation favors the bond between the gDNA and the primer, which is complementary to the mutated sequence, making mutated gDNA amplification possible (Figure 1).
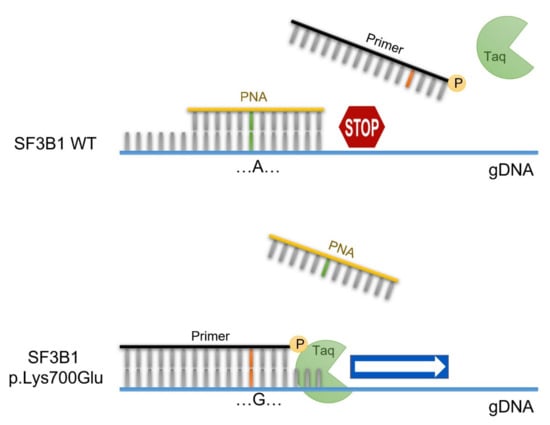
Figure 1.
PNA-PCR clamping experimental design. Amplification of SF3B1 is performed in presence of the PNA probe (yellow), designed on the WT sequence, and the primer competitor (black), designed on the mutated sequence. In these conditions, the PCR of the WT sequence is inhibited by the hybridization of PNA/DNA. In contrast, in the presence of the SF3B1 p.Lys700Glu genotype, the primer/DNA duplex allows the amplification of the target sequence.
This method involves the following two PCR steps: The first consists of pre-amplification for a short amount of time, to increase the specificity of the test, avoiding the non-specific amplification of other fragments by second-step primers. The second step consists of the mutational test, and is carried out in duplicate, in the presence (TEST) or absence (CONTROL) of the PNA probe. In CONTROL PCR, the DNA must always be amplified. Otherwise, the result will not be valid, and the assay will have to be repeated. If the CONTROL PCR is valid, TEST PCR returns the result about the mutational status of SF3B1; the patient will be SF3B1 WT if the DNA is not amplified, while the patient will be SF3B1 p.Lys700Glu if the DNA is amplified (Figure 2A).
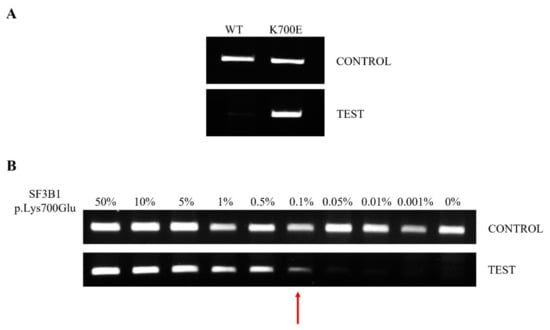
Figure 2.
Electrophoretic runs of PCR reactions: each amplicon was loaded on 2% agarose-TBE 1× gel with 5 μg/mL EtBr and run at 150 V for 30 min. PNA-PCR clamping for SF3B1 was carried out in the absence (CONTROL) and presence (TEST) of the PNA probe. (A) Example of PNA-PCR clamping in DNA from WT and p.Lys700Glu-mutated patients. CONTROL PCR represents an internal control and DNA must always be amplified. TEST PCR returns the result about the mutational status of SF3B1; if the patient is SF3B1 WT, the gDNA is not amplified, while if the patient is SF3B1 p.Lys700Glu, the gDNA is amplified. (B) PNA-PCR clamping LoD was assessed by mixing pGEMT-SF3B1 p.Lys700Glu and pGEMT-SF3B1 WT plasmids at different ratios in the same PCR reaction. Dilutions were as follows: 50, 10, 5, 1, 0.5, 0.1, 0.05, 0.05, 0,01, 0.001 and 0% pGEMT-SF3B1 p.Lys700Glu, all brought to 100% with the respective amount of pGEMT-SF3B1 WT template. The percentage of the mutated template is indicated above each amplicon. The red arrow indicates the LoD of the PNA-PCR clamping method.
The LoD of the method was evaluated by mixing, at different concentrations, the pGEMT-SF3B1 p.Lys700Glu and pGEMT-SF3B1 WT plasmids. The dilutions used were as follows: 50, 10, 5, 1, 0.5, 0.1, 0.01, 0.001 and 0% of pGEMT-SF3B1 p.Lys700Glu in pGEMT-SF3B1 WT. The method showed a very low LoD, allowing us to identify up to 0.1% of mutated SF3B1 (Figure 2B).
3.2. Comparison of Sanger Sequencing and PNA-PCR Clamping for Detection of SF3B1 p.Lys700Glu
Before patient screening was started, we tested, in duplicate (both BM and PB), the DNA of two mutated SF3B1 p.Lys700Glu and three wild-type patients with PNA-PCR clamping. No difference was observed between the results obtained for the BM and PB samples. Then, PNA-PCR clamping and Sanger sequencing were used to blind test 90 DNA from 57 MDS and 33 MPN patients for the SF3B1 p.Lys700Glu mutation. PNA-PCR clamping and Sanger sequencing congruently identified 75 negative and 13 positive samples, while two MDS patients identified as positive by PNA-PCR clamping were missed by Sanger analysis (Table 1).

Table 1.
PNA-PCR clamping and Sanger sequencing results.
The agreement between Sanger sequencing and PNA-PCR clamping was confirmed by McNemar’s test, which did not highlight differences in the proportion of disagreement data. To understand if the discordant samples were PNA-PCR clamping false-positives or Sanger sequencing false-negatives, we analyzed them with NGS. NGS confirmed the PNA-PCR clamping result, indicating that these samples possessed SF3B1 p.Lys700Glu with mutation percentages of 1.3% and 6.1%, respectively (Figure 3).
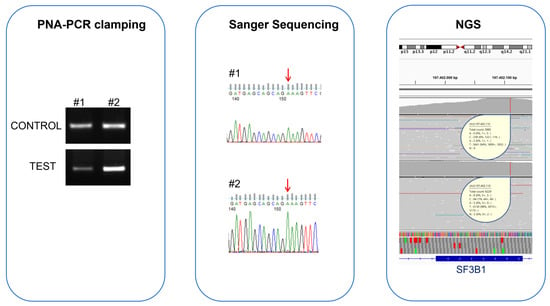
Figure 3.
Comparison of PNA-PCR clamping, Sanger sequencing, and NGS for the evaluation of SF3B1 status in two MDS patients with contrasting results. The electrophoretic runs of PNA-PCR clamping, carried out in the absence (CONTROL) and presence (TEST) of the PNA probe, showed amplification in both the samples, indicating the presence of the SF3B1 p.Lys700Glu mutation in these patients. In contrast, Sanger sequencing chromatograms showed the presence of only one allele (red arrows); the nucleotide “A” indicates that both patients were WT for SF3B1 p.Lys700Glu mutation. NGS confirmed the PNA-PCR clamping result, indicating that these samples contained SF3B1 p.Lys700Glu with mutation percentages of 1.3% and 6.1%, respectively. Both samples had mutation percentages lower than the Sanger sequencing LoD, but they were detected by PNA-PCR clamping methodology.
Interestingly, to identify enough mutated sequences to increase confidence in the results, it is necessary to perform deep sequencing with more than a thousand reads for each patient, even with NGS. In particular, for the first patient, we only identified 84 mutated reads out of the total 6229, while 238 out of 3885 total reads were mutated for the second patient.
3.3. Prevalence of SF3B1 p.Lys700Glu in MDS and MPN Samples
In our cohort of patients, we found the SF3B1 p.Lys700Glu mutation in 24.5% (14/57) of MDS patients and 3% (1/33) of MPN patients (Table 2).
Table 2.
MDS patients and MPN patients’ characteristics.
For the MDS samples, we compared the baseline characteristics of mutated and WT patients. We did not find any significant correlations between the SF3B1 p.Lys700Glu mutation and age, sex, or IPSS-R. Fisher’s exact test confirmed the significant strong association between the p.Lys700Glu mutation and the RS phenotype (p < 0.0001). Indeed, 85.7% of mutated patients were classified as MDS-RS, while only 14.3% were MDS-MLD. In the SF3B1 p.Lys700Glu WT group, only 9.3% of the patients showed RS. Furthermore, by dichotomizing the samples, based on WT1 expression at diagnosis in the normal- or high-expression group, as indicated in the literature [], we reported that the p.Lys700Glu mutation correlates with normal WT1 expression at diagnosis (p = 0.022). Because the expression of WT1 at normal levels is an indicator of a good prognosis, this result corroborates the role of SF3B1 status as a good prognosis marker.
Regarding the MPN cohort, we only found one mutated sample. The patient was a 69-year-old male, with an MF secondary to ET and intolerant to ruxolitinib treatment. The patient had a concomitant JAK2 V617F mutation and a deletion on chromosome 20q.
4. Discussion
Molecular characterization has become mandatory in the setting of myelodysplastic syndromes and myeloproliferative disorders. The 2016 WHO classification recognized MDS with ring sideroblasts as an entity defined by the presence of more than 15% of ring sideroblasts or more than 5% with the SF3B1 mutation.
MDS with the RS phenotype are associated with a shorter median duration of response to ESA, and with fibrosis [,]. For these reasons, the assessment of the mutational status of SF3B1 has become relevant not only for a correct diagnosis, but also for offering the possibility of an effective therapy. The most used methods for identifying SF3B1 mutations are Sanger sequencing and NGS. The main limit of Sanger sequencing is its low sensitivity, which means that it is unable to detect small clones. By contrast, NGS is much more sensitive, but is burdened by rather long times and fairly high costs. Although the search for mutations with prognostic significance should be sought in all patients for a correct diagnostic classification, in “real life”, the search for mutations with NGS is reserved for younger patients with therapeutic perspectives. Frequently, the patient chosen for supportive therapy only, including transfusions, is not molecularly typed in order to prevent overloading diagnostic laboratories. In this study, we designed and validated a new technology that is sensitive, fast, and cheap to identify the SF3B1 p.Lys700Glu mutation based on PNA-PCR clamping, and we compared this technique with Sanger sequencing. PNA-PCR clamping rapidly identified the SF3B1 p.Lys700Glu mutation in DNA samples. The PNA-PCR clamping technique showed high efficiency, sensitivity, and specificity, and it was faster and cheaper than Sanger sequencing. PNA-PCR clamping showed an LoD of about 0.1% of a mutation that was extrapolated in vitro by serial dilutions of mutated plasmids, and subsequently confirmed in DNA samples. This limit of detection is markedly lower than the Sanger method, which is estimated, in the literature, to be 15–20% []. Indeed, in our analysis, Sanger sequencing showed two cases of false-negatives, while the mutation was detected by PNA-PCR clamping, in agreement with NGS analysis. Furthermore, even with NGS, it is necessary to obtain a high sequence depth to obtain confident results, a depth that is not always guaranteed by routine diagnostic conditions. Malcovati et al. reported that the median variant allele frequency (VAF) for the SF3B1 mutations is 39.5% in MDS patients, with a high variable range from 5.4 to 70.3% []. The low LoD that our assay showed can easily cover the entire range of VAF described in the literature, minimizing the false-negative results of Sanger (LoD of about 15–20%) and/or high-resolution melting (HRM) analysis (LoD of about 10%) []. Few data are currently available about the role of small mutated SF3B1 clones, but it is known that SF3B1 mutations can propagate from hematopoietic stem cells to their progeny []. Furthermore, xenograft mice, in which SF3B1 subclones were <5% of the total grafted cells, also developed an MDS phenotype []. Although a clear prognostic role of small SF3B1-mutated clones is not known to date, having a diagnostic assay capable of identifying them may still be useful for a “watch and wait” approach, and for an early therapeutic decision.
A possible limitation of our test is the evaluation of only one of the SF3B1 mutations, the most frequent one. Although the p.Lys700Glu mutation covers only 55–65% of all SF3B1 mutations, the remaining 35–45% is characterized by a multitude of different mutations, including p.Glu622Asp, p.Tyr623Cys, p.Arg625His/Gly/Cys/Leu, p.Asn626Asp, p.His662Gln/Asp, p.Lys666Glu/Asn/Arg/Thr, and p.Ile704Val/Asn, each present with a very low frequency (<5%). From a prognostic point of view, the available data are very conflicting. Malcovalti et al. highlight the positive independent prognostic value of SF3B1 mutations in sideroblastic categories only, while, in MDS with excess blasts, the mutation did not have a significant effect on survival and risk of disease progression [,]. By contrast, Dalton et al. report that the p.Lys666Asn hotspot is associated with MDS with excess blasts and increased progression to acute myeloid leukemia []. More recently, Kanagal-Shamanna et al. found that only the p.Lys700Glu mutation independently predicts better overall survival in MDS, suggesting that the identification of this SF3B1 mutation type is important for risk stratification []. In this paper, we demonstrated the feasibility and utility of the PNA-PCR clamping approach in the identification of the most frequent SF3B1 mutations. For a more complete analysis, it will be necessary to improve our system by including the mutations in the p.Lys666 position to cover the majority of SF3B1 mutations.
Compared with other currently available methods, such as Sanger sequencing and HRM analysis, PNA-PCR clamping has been shown to have a better LoD and a lower cost, both in terms of reagents and instrumentation. Although the other two techniques cited allow other SF3B1 mutations to also be evaluated, PNA-PCR clamping can be easily implemented to evaluate other hotspots, without significantly affecting the cost and timing of execution, which remain lower than Sanger sequencing or HRM analysis.
In addition, we confirmed the role of mutated SF3B1 as a molecular biomarker of the MDS-RS subgroup [,], and we showed a correlation between the mutational status of SF3B1 and WT1 expression. Indeed, MDS patients with a mutated SF3B1 genotype showed lower WT1 expression values than the WT counterpart. Because the expression of WT1 at normal levels is an indicator of a good prognosis, this result corroborates the role of SF3B1 status as a good prognosis marker [,,,].
5. Conclusions
Considering the accuracy and sensitivity of PNA-PCR clamping, its rapidity, and its cost, we propose this method for basal molecular typing of MDS. Although it cannot replace NGS, PNA-PCR clamping will allow a percentage of patients to have a molecular assessment, which, although incomplete, offers the possibility of identifying an effective therapy.
Author Contributions
Conceptualization, J.P. and D.C.; data curation, F.I., C.F., M.A. and S.M.; formal analysis, J.P. and M.L.I.; funding acquisition, G.S.; methodology, J.P., E.S., E.G. and M.L.I.; project administration, J.P. and D.C.; resources, G.S. and D.C.; supervision, G.S. and D.C.; validation, J.P. and A.F.; writing—original draft, J.P., F.I., M.L.I. and D.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research and APC were funded by AIRC, Grant Number 23344.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of San Luigi Gonzaga Hospital, Orbassano, Turin (Number of approval 263/INT).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gilbert, W. Why genes in pieces? Nature 1978, 271, 501. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Adès, L.; Itzykson, R.; Fenaux, P. Myelodysplastic syndromes. Lancet 2014, 383, 2239–2252. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Hodnefield, J.M.; Knudson, R.A.; Ketterling, R.P.; Garcia-Manero, G.; Steensma, D.P.; Pardanani, A.; Hanson, C.A.; Tefferi, A. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 2012, 119, 569–572. [Google Scholar] [CrossRef]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 2011, 118, 6239–6246. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Mies, A.; Hermine, O.; Platzbecker, U. Activin Receptor II Ligand Traps and Their Therapeutic Potential in Myelodysplastic Syndromes with Ring Sideroblasts. Curr. Hematol. Malig. Rep. 2016, 11, 416–424. [Google Scholar] [CrossRef]
- Attie, K.M.; Allison, M.J.; McClure, T.; Boyd, I.E.; Wilson, D.M.; Pearsall, A.E.; Sherman, M.L. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am. J. Hematol. 2014, 89, 766–770. [Google Scholar] [CrossRef]
- Platzbecker, U.; Germing, U.; Götze, K.S.; Kiewe, P.; Mayer, K.; Chromik, J.; Radsak, M.; Wolff, T.; Zhang, X.; Laadem, A.; et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017, 18, 1338–1347. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Jimma, T.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SF3B1 mutations in primary myelofibrosis: Clinical, histopathology and genetic correlates among 155 patients. Leukemia 2012, 26, 1135–1137. [Google Scholar] [CrossRef]
- Visconte, V.; Makishima, H.; Jankowska, A.; Szpurka, H.; Traina, F.; Jerez, A.; O’Keefe, C.; Rogers, H.J.; Sekeres, M.A.; Maciejewski, J.P.; et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 2012, 26, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Gerds, A.T.; Vannucchi, A.M.; Passamonti, F.; Kremyanskaya, M.; Gotlib, J.R.; Palmer, J.M.; McCaul, K.; Ribrag, V.; Mead, A.J.; Harrison, C.N.; et al. A Phase 2 Study of Luspatercept in Patients with Myelofibrosis-Associated Anemia. Blood 2019, 134, 557. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Fouz, M.F.; Appella, D.H. PNA Clamping in Nucleic Acid Amplification Protocols to Detect Single Nucleotide Mutations Related to Cancer. Molecules 2020, 25, 786. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef]
- Kyger, E.M.; Krevolin, M.D.; Powell, M.J. Detection of the Hereditary Hemochromatosis Gene Mutation by Real-Time Fluorescence Polymerase Chain Reaction and Peptide Nucleic Acid Clamping. Anal. Biochem. 1998, 260, 142–148. [Google Scholar] [CrossRef][Green Version]
- Petiti, J.; Rosso, V.; Croce, E.; Franceschi, V.; Andreani, G.; Dragani, M.; De Gobbi, M.; Lunghi, M.; Saglio, G.; Fava, C.; et al. Highly Sensitive Detection of IDH2 Mutations in Acute Myeloid Leukemia. J. Clin. Med. 2020, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Petiti, J.; Rosso, V.; Lo Iacono, M.; Calabrese, C.; Signorino, E.; Gaidano, V.; Berger, M.; Saglio, G.; Cilloni, D. Prognostic significance of The Wilms’ Tumor-1 (WT1) rs16754 polymorphism in acute myeloid leukemia. Leuk. Res. 2018, 67, 6–11. [Google Scholar] [CrossRef]
- Rosso, V.; Petiti, J.; Bracco, E.; Pedrola, R.; Carnuccio, F.; Signorino, E.; Carturan, S.; Calabrese, C.; Bot-Sartor, G.; Ronconi, M.; et al. A novel assay to detect calreticulin mutations in myeloproliferative neoplasms. Oncotarget 2017, 8, 6399–6405. [Google Scholar] [CrossRef] [PubMed]
- Rosso, V.; Bracco, E.; Pedrola, R.; Carturan, S.; Signorino, E.; Petiti, J.; Calabrese, C.; Nicoli, P.; De Gobbi, M.; Gaidano, V.; et al. Detection of BCR-ABL T315I mutation by peptide nucleic acid directed PCR clamping and by peptide nucleic acid FISH. Biomark. Res. 2015, 3, 15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mancini, M.; Hasan, S.K.; Ottone, T.; Lavorgna, S.; Ciardi, C.; Angelini, D.F.; Agostini, F.; Venditti, A.; Lo-Coco, F. Two Novel Methods for Rapid Detection and Quantification of DNMT3A R882 Mutations in Acute Myeloid Leukemia. J. Mol. Diagn. 2015, 17, 179–184. [Google Scholar] [CrossRef]
- Suguna, S.; Nandal, D.H.; Kamble, S.; Bharatha, A.; Kunkulol, R. Genomic DNA isolation from human whole blood samples by non enzymatic salting out method. Int. J. Pharm. Pharm. Sci. 2014, 6, 198–199. [Google Scholar]
- Oliner, K.; Juan, T.; Suggs, S.; Wolf, M.; Sarosi, I.; Freeman, D.J.; Gyuris, T.; Baron, W.; Bakker, A.; Parker, A.; et al. A comparability study of 5 commercial KRAS tests. Diagn. Pathol. 2010, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Gottardi, E.; Messa, F.; Fava, M.; Scaravaglio, P.; Bertini, M.; Girotto, M.; Marinone, C.; Ferrero, D.; Gallamini, A.; et al. Significant Correlation Between the Degree of WT1 Expression and the International Prognostic Scoring System Score in Patients with Myelodysplastic Syndromes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 1988–1995. [Google Scholar] [CrossRef]
- Visconte, V.; Makishima, H.; Maciejewski, J.P.; Tiu, R.V. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia 2012, 26, 2447–2454. [Google Scholar] [CrossRef]
- Mizuta, S.; Yamane, N.; Komai, T.; Koba, Y.; Ukyo, N.; Tamekane, A.; Watanabe, M. Evaluation of SF3B1 Mutation Screening by High-Resolution Melting Analysis and its Clinical Utility for Myelodysplastic Syndrome with Ring Sideroblasts at the Point of Diagnosis. Lab. Med. 2019, 50, 254–262. [Google Scholar] [CrossRef]
- Mian, S.A.; Rouault-Pierre, K.; Smith, A.E.; Seidl, T.; Pizzitola, I.; Kizilors, A.; Kulasekararaj, A.G.; Bonnet, D.; Mufti, G.J. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat. Commun. 2015, 6, 10004. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef]
- Dalton, W.B.; Helmenstine, E.; Pieterse, L.; Li, B.; Gocke, C.D.; Donaldson, J.; Xiao, Z.; Gondek, L.P.; Ghiaur, G.; Gojo, I.; et al. The K666N mutation in SF3B1 is associated with increased progression of MDS and distinct RNA splicing. Blood Adv. 2020, 4, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R.; Montalban-Bravo, G.; Sasaki, K.; Darbaniyan, F.; Jabbour, E.; Bueso-Ramos, C.; Wei, Y.; Chien, K.; Kadia, T.; Ravandi, F.; et al. Only SF3B1 mutation involving K700E independently predicts overall survival in myelodysplastic syndromes. Cancer 2021, 127, 3552–3565. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Invernizzi, R. Ring sideroblasts and sideroblastic anemias. Haematologica 2011, 96, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Rossi, M.; Malcovati, L.; on behalf of the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood 2013, 121, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Hosono, N. Genetic abnormalities and pathophysiology of MDS. Int. J. Clin. Oncol. 2019, 24, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Venable, E.R.; Chen, D.; Chen, C.P.; Bessonen, K.R.; Nguyen, P.L.; Oliveira, J.L.; Reichard, K.K.; Hoyer, J.D.; Althoff, S.D.; Roh, D.J.; et al. Pathologic Spectrum and Molecular Landscape of Myeloid Disorders Harboring SF3B1 Mutations. Am. J. Clin. Pathol. 2021, 156, 679–690. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).