
Antibodies to Porphyromonas gingivalis Are Increased in Patients with Severe Periodontitis, and Associate with Presence of Specific Autoantibodies and Myocardial Infarction

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
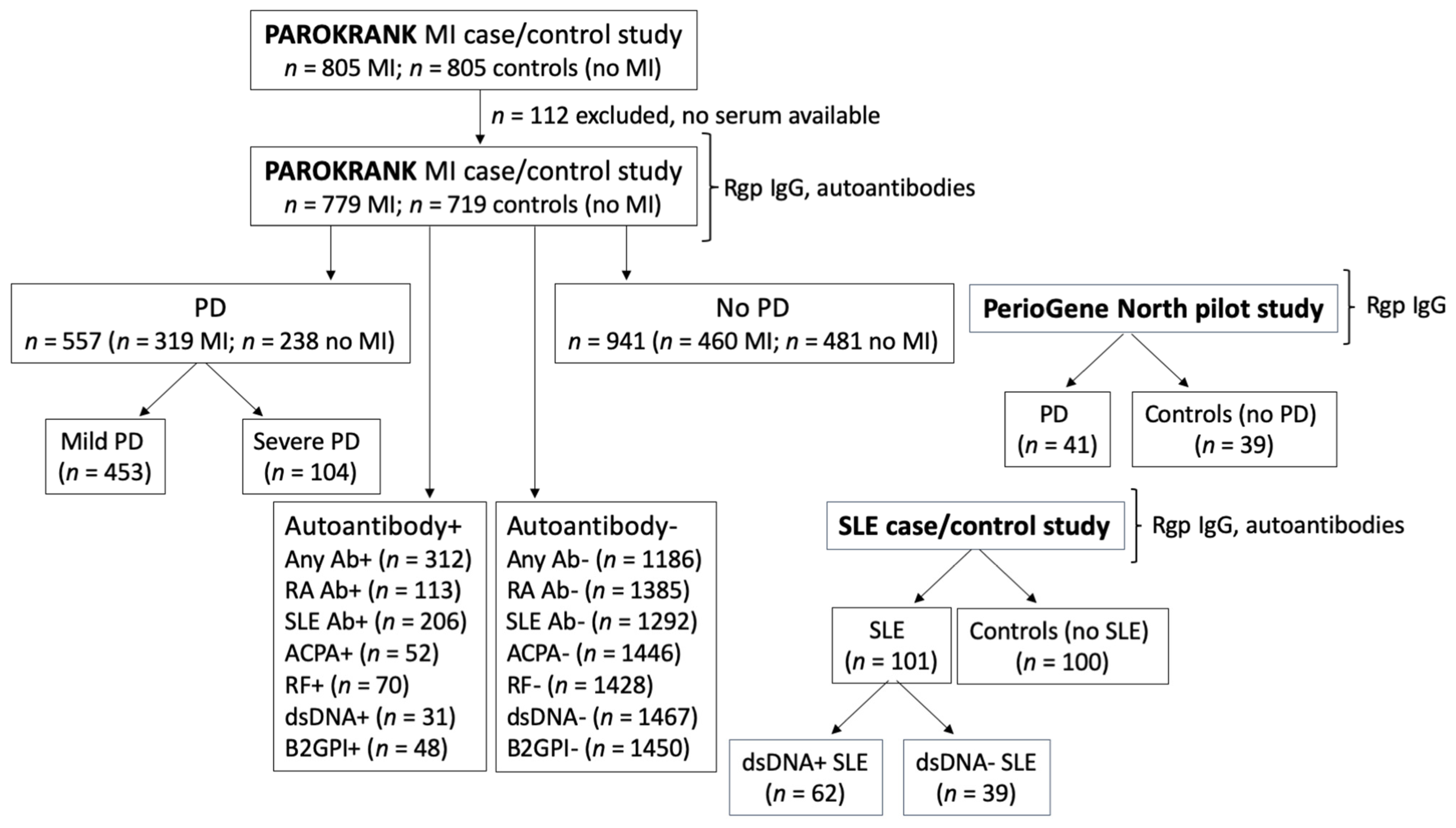
2.1. Study Design
2.2. Study Populations
2.3. Anti-Rgp IgG ELISA
2.4. Detection of Autoantibodies
2.5. Statistical Methods
3. Results
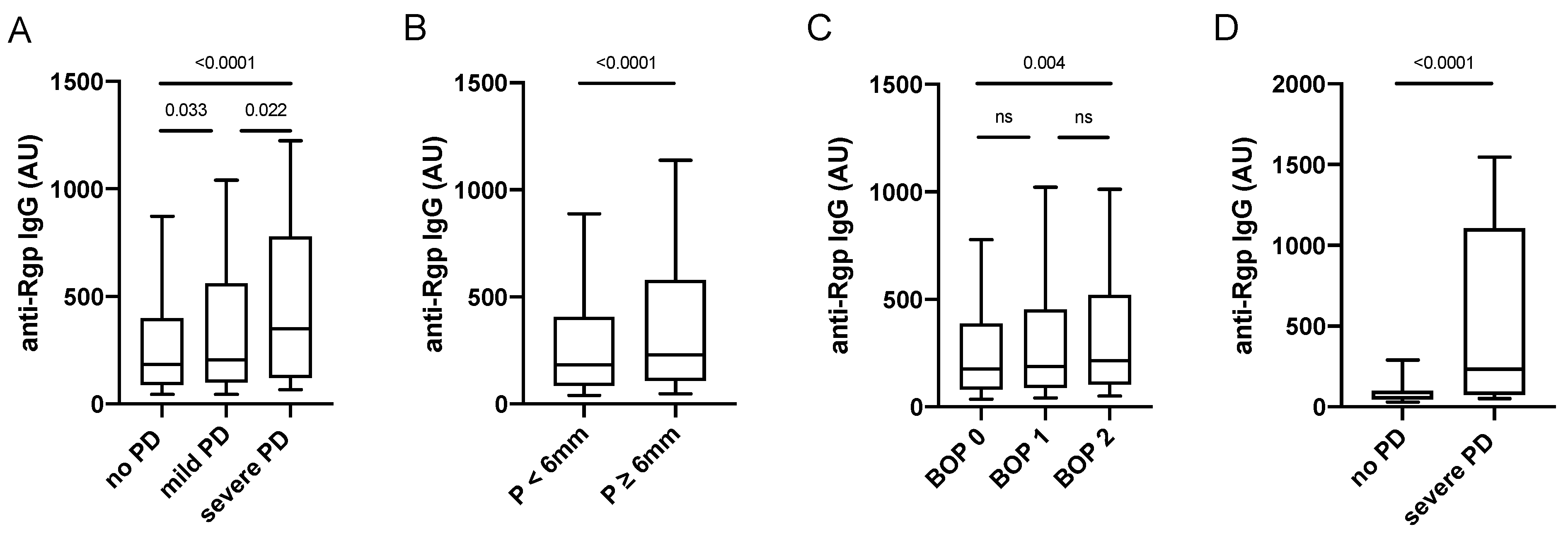
3.1. Increased Anti-Rgp IgG Levels Associate with Periodontitis Severity
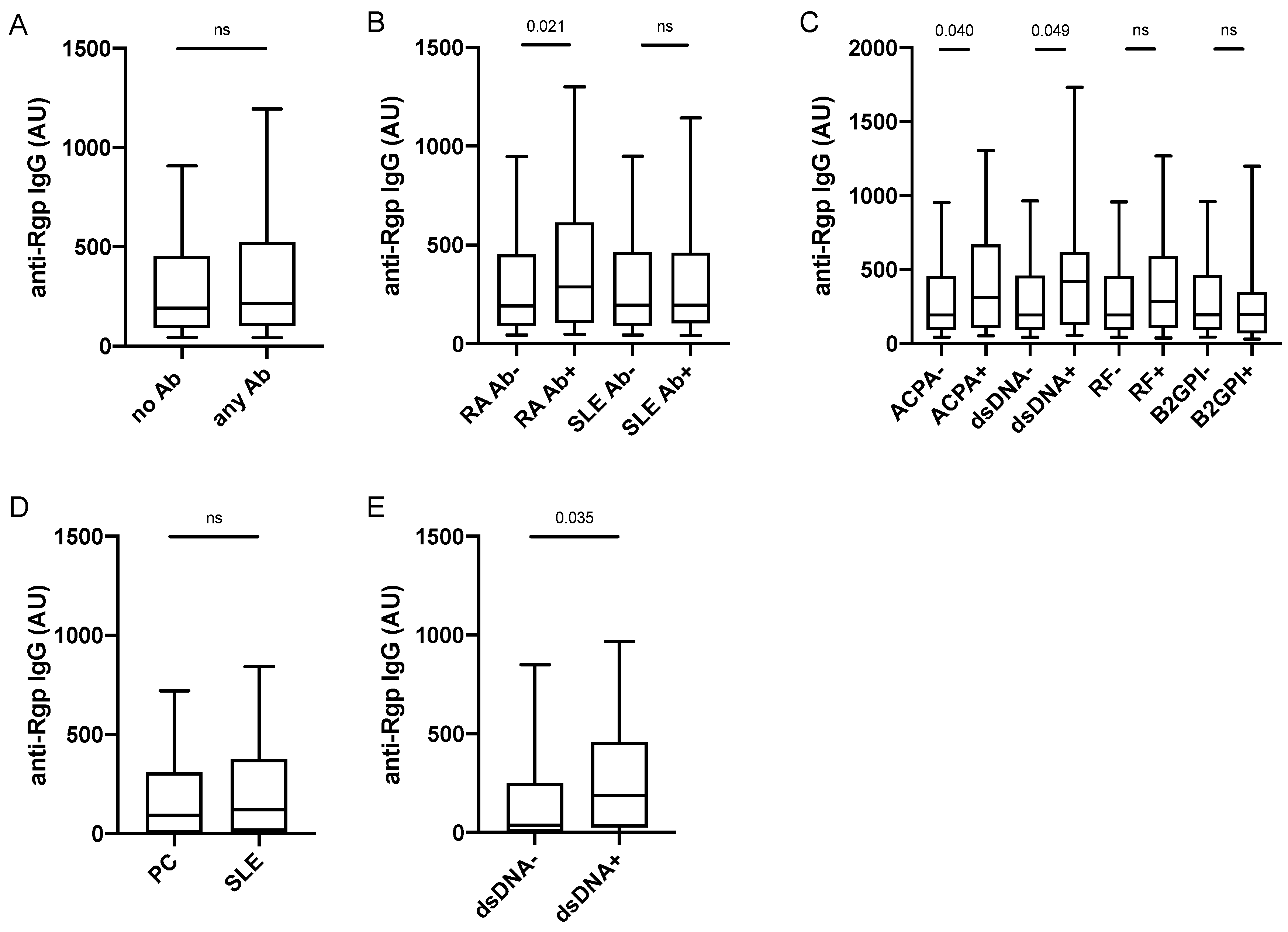
3.2. Increased Anti-Rgp IgG Levels Associate with Presence of ACPA and Anti-dsDNA Antibodies
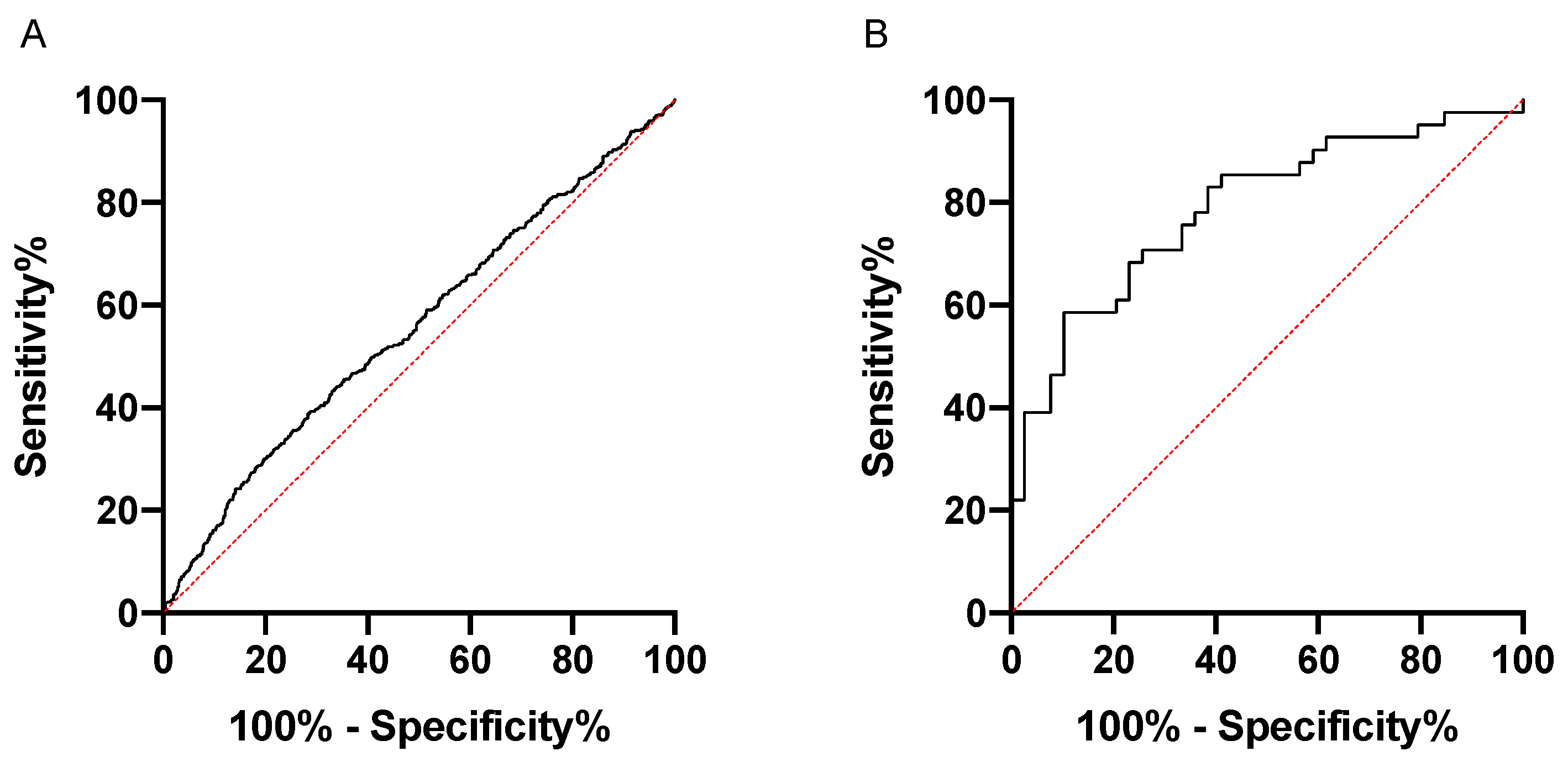
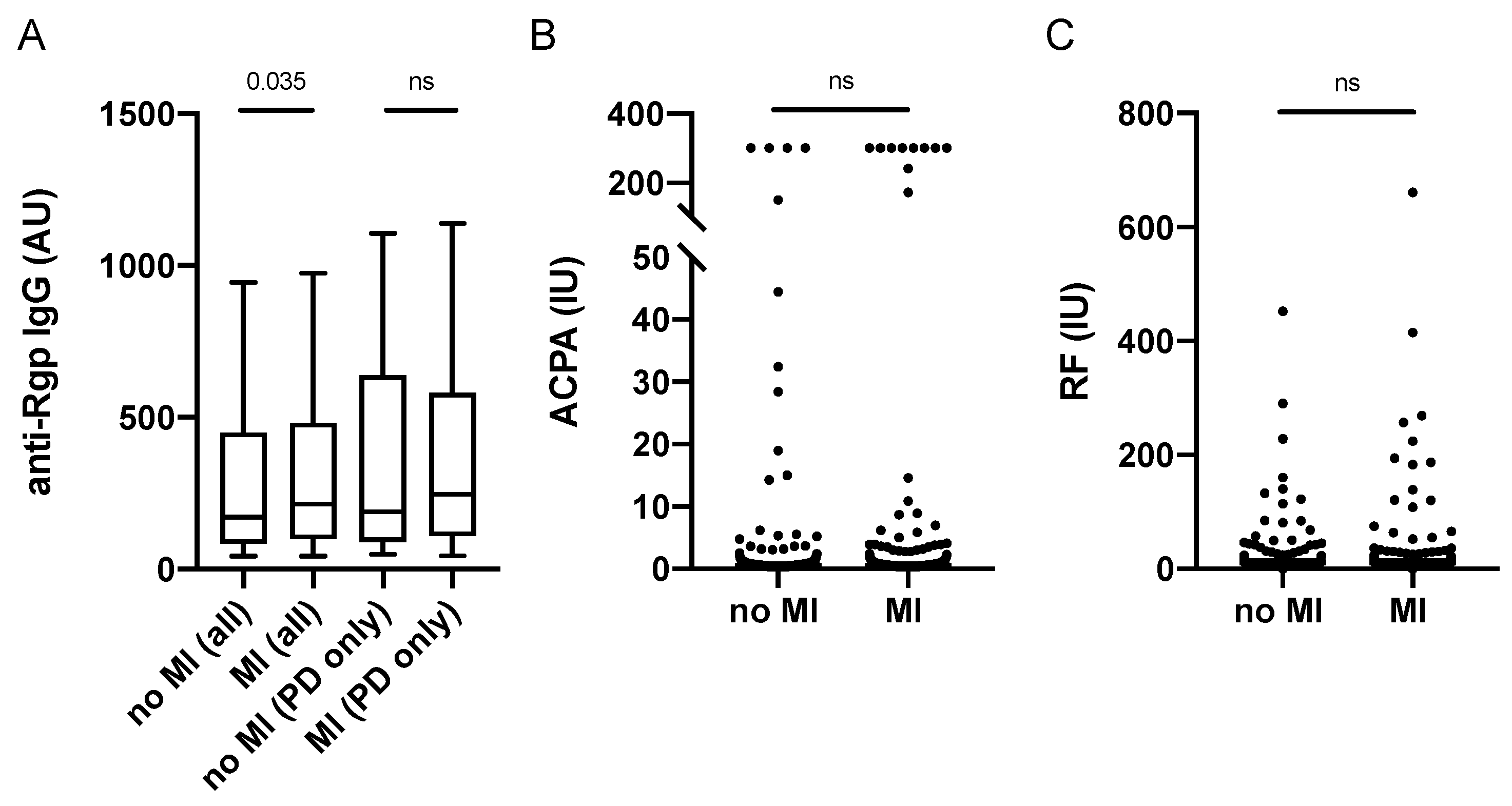
3.3. Increased Anti-Rgp IgG Levels Associate with Myocardial Infarction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hajishengallis, G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindstedt, E.; Johansson, L.; Palmqvist, P.; Koskinen Holm, C.; Kokkonen, H.; Johansson, I.; Rantapää Dahlqvist, S.; Lundberg, P. Association Between Marginal Jawbone Loss and Onset of Rheumatoid Arthritis and Relationship to Plasma Levels of RANKL. Arthritis Rheumatol. 2018, 70, 508–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuggle, N.R.; Smith, T.O.; Kaul, A.; Sofat, N. Hand to Mouth: A Systematic Review and Meta-Analysis of the Association between Rheumatoid Arthritis and Periodontitis. Front. Immunol. 2016, 7, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zardawi, F.; Gul, S.; Abdulkareem, A.; Sha, A.; Yates, J. Association Between Periodontal Disease and Atherosclerotic Cardiovascular Diseases: Revisited. Front. Cardiovasc. Med. 2021, 7, 625579. [Google Scholar] [CrossRef] [PubMed]
- Rutter-Locher, Z.; Smith, T.O.; Giles, I.; Sofat, N. Association between Systemic Lupus Erythematosus and Periodontitis: A Systematic Review and Meta-analysis. Front. Immunol. 2017, 8, 1295. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef]
- Paraskevas, S.; Huizinga, J.D.; Loos, B.G. A systematic review and meta-analyses on C-reactive protein in relation to periodontitis. J. Clin. Periodontol. 2008, 35, 277–290. [Google Scholar] [CrossRef]
- D’Aiuto, F.; Gkranias, N.; Bhowruth, D.; Khan, T.; Orlandi, M.; Suvan, J.; Masi, S.; Tsakos, G.; Hurel, S.; Hingorani, A.D.; et al. Systemic effects of periodontitis treatment in patients with type 2 diabetes: A 12 month, single-centre, investigator-masked, randomised trial. Lancet Diabetes Endocrinol. 2018, 6, 954–965. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Zheng, S.; Yu, S.; Fan, X.; Zhang, Y.; Sun, Y.; Lin, L.; Wang, H.; Pan, Y.; Li, C. Porphyromonas gingivalis survival skills: Immune evasion. J. Periodontal Res. 2021, 56, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Nguyen, K.-A.; Potempa, J. Dichotomy of gingipains action as virulence factors: From cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol 2000 2010, 54, 15–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hočevar, K.; Potempa, J.; Turk, B. Host cell-surface proteins as substrates of gingipains, the main proteases of Porphyromonas gingivalis. Biol. Chem. 2018, 399, 1353–1361. [Google Scholar] [CrossRef]
- Hirai, K.; Yamaguchi-Tomikawa, T.; Eguchi, T.; Maeda, H.; Takashiba, S. Identification and Modification of Porphyromonas gingivalis Cysteine Protease, Gingipain, Ideal for Screening Periodontitis. Front. Immunol. 2020, 11, 1017. [Google Scholar] [CrossRef]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.M.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to Porphyromonas gingivalis Indicate Interaction Between Oral Infection, Smoking, and Risk Genes in Rheumatoid Arthritis Etiology. Arthritis Rheumatol. 2016, 68, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Johansson, L.; Sherina, N.; Kharlamova, N.; Potempa, B.; Larsson, B.; Israelsson, L.; Potempa, J.; Rantapää-Dahlqvist, S.; Lundberg, K. Concentration of antibodies against Porphyromonas gingivalis is increased before the onset of symptoms of rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 201. [Google Scholar] [CrossRef] [Green Version]
- McGraw, W.T.; Potempa, J.; Farley, D.; Travis, J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect. Immun. 1999, 67, 3248–3256. [Google Scholar] [CrossRef] [Green Version]
- Rosenstein, E.D.; Greenwald, R.A.; Kushner, L.J.; Weissmann, G. Hypothesis: The humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation 2004, 28, 311–318. [Google Scholar] [CrossRef]
- Potempa, J.; Mydel, P.; Koziel, J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 606–620. [Google Scholar] [CrossRef]
- Bender, P.; Bürgin, W.B.; Sculean, A.; Eick, S. Serum antibody levels against Porphyromonas gingivalis in patients with and without rheumatoid arthritis–A systematic review and meta-analysis. Clin. Oral Investig. 2017, 21, 33–42. [Google Scholar] [CrossRef] [PubMed]
- De Pablo, P.; Dietrich, T.; Chapple, I.L.; Milward, M.; Chowdhury, M.; Charles, P.J.; Buckley, C.D.; Venables, P.J. The autoantibody repertoire in periodontitis: A role in the induction of autoimmunity to citrullinated proteins in rheumatoid arthritis? Ann. Rheum. Dis. 2014, 73, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Lappin, D.F.; Apatzidou, D.; Quirke, A.M.; Oliver-Bell, J.; Butcher, J.P.; Kinane, D.F.; Riggio, M.P.; Venables, P.; McInnes, I.B.; Culshaw, S. Influence of periodontal disease, Porphyromonas gingivalis and cigarette smoking on systemic anti-citrullinated peptide antibody titres. J. Clin. Periodontol. 2013, 40, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.; Fei, G.; Lundmark, A.; Benchimol, D.; Lee, L.; Hu, Y.O.O.; Kats, A.; Saevarsdottir, S.; Catrina, A.I.; Klinge, B.; et al. Periodontal Health and Oral Microbiota in Patients with Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 630. [Google Scholar] [CrossRef] [Green Version]
- Loutan, L.; Alpizar-Rodriguez, D.; Courvoisier, D.S.; Finckh, A.; Mombelli, A.; Giannopoulou, C. Periodontal status correlates with anti-citrullinated protein antibodies in first-degree relatives of individuals with rheumatoid arthritis. J. Clin. Periodontol. 2019, 46, 690–698. [Google Scholar] [CrossRef]
- Mankia, K.; Cheng, Z.; Do, T.; Hunt, L.; Meade, J.; Kang, J.; Clerehugh, V.; Speirs, A.; Tugnait, A.; Hensor, E.M.A.; et al. Prevalence of Periodontal Disease and Periodontopathic Bacteria in Anti-Cyclic Citrullinated Protein Antibody-Positive At-Risk Adults without Arthritis. JAMA Netw. Open 2019, 2, e195394. [Google Scholar] [CrossRef]
- Rodríguez-Lozano, B.; González-Febles, J.; Garnier-Rodríguez, J.L.; Dadlani, S.; Bustabad-Reyes, S.; Sanz, M.; Sánchez-Alonso, F.; Sánchez-Piedra, C.; González-Dávila, E.; Díaz-González, F. Association between severity of periodontitis and clinical activity in rheumatoid arthritis patients: A case-control study. Arthritis Res. Ther. 2019, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Harvey, G.P.; Fitzsimmons, T.R.; Dhamarpatni, A.A.; Marchant, C.; Haynes, D.R.; Bartold, P.M. Expression of peptidylarginine deiminase-2 and -4, citrullinated proteins and anti-citrullinated protein antibodies in human gingiva. J. Periodontal Res. 2013, 48, 252–261. [Google Scholar] [CrossRef]
- Corrêa, J.D.; Calderaro, D.C.; Ferreira, G.A.; Mendonça, S.M.; Fernandes, G.R.; Xiao, E.; Teixeira, A.L.; Leys, E.J.; Graves, D.T.; Silva, T.A. Subgingival microbiota dysbiosis in systemic lupus erythematosus: Association with periodontal status. Microbiome 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Bagavant, H.; Dunkleberger, M.L.; Wolska, N.; Sroka, M.; Rasmussen, A.; Adrianto, I.; Montgomery, C.; Sivils, K.; Guthridge, J.M.; James, J.A.; et al. Antibodies to periodontogenic bacteria are associated with higher disease activity in lupus patients. Clin. Exp. Rheumatol. 2019, 37, 106–111. [Google Scholar]
- Rydén, L.; Buhlin, K.; Ekstrand, E.; Faire, U.d.; Gustafsson, A.; Holmer, J.; Kjellström, B.; Lindahl, B.; Norhammar, A.; Nygren, Å.; et al. Periodontitis Increases the Risk of a First Myocardial Infarction. Circulation 2016, 133, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Boström, E.A.; Kindstedt, E.; Sulniute, R.; Palmqvist, P.; Majster, M.; Holm, C.K.; Zwicker, S.; Clark, R.; Önell, S.; Johansson, I.; et al. Increased Eotaxin and MCP-1 Levels in Serum from Individuals with Periodontitis and in Human Gingival Fibroblasts Exposed to Pro-Inflammatory Cytokines. PLoS ONE 2015, 10, e0134608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, J.T.; Herlitz Lindberg, M.; Gunnarsson, I.; Pettersson, S.; Elvin, K.; Öhrvik, J.; Larsson, A.; Jensen-Urstad, K.; Svenungsson, E. Excess atherosclerosis in systemic lupus erythematosus,—A matter of renal involvement: Case control study of 281 SLE patients and 281 individually matched population controls. PLoS ONE 2017, 12, e0174572. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.M.; Cohen, A.S.; Fries, J.F.; Masi, A.T.; McShane, D.J.; Rothfield, N.F.; Schaller, J.G.; Talal, N.; Winchester, R.J. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982, 25, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Quirke, A.M.; Lugli, E.B.; Wegner, N.; Hamilton, B.C.; Charles, P.; Chowdhury, M.; Ytterberg, A.J.; Zubarev, R.A.; Potempa, J.; Culshaw, S.; et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: A potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veillard, F.; Potempa, B.; Guo, Y.; Ksiazek, M.; Sztukowska, M.N.; Houston, J.A.; Koneru, L.; Nguyen, K.A.; Potempa, J. Purification and characterisation of recombinant His-tagged RgpB gingipain from Porphymonas gingivalis. Biol. Chem. 2015, 396, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, D.; Imai, K.; King, G.; Stuart, E.A. MatchIt: Nonparametric Preprocessing for Parametric Causal Inference. J. Stat. Softw. 2011, 42, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Buhlin, K.; Gustafsson, A.; Ahnve, S.; Janszky, I.; Tabrizi, F.; Klinge, B. Oral health in women with coronary heart disease. J. Periodontol. 2005, 76, 544–550. [Google Scholar] [CrossRef]
- Grosso, G.; Sippl, N.; Kjellström, B.; Amara, K.; de Faire, U.; Elvin, K.; Lindahl, B.; Näsman, P.; Rydén, L.; Norhammar, A.; et al. Antiphospholipid Antibodies in Patients with Myocardial Infarction. Ann. Intern. Med. 2018, 170, 277–280. [Google Scholar] [CrossRef]
- Eke, P.I.; Dye, B.A.; Wei, L.; Slade, G.D.; Thornton-Evans, G.O.; Borgnakke, W.S.; Taylor, G.W.; Page, R.C.; Beck, J.D.; Genco, R.J. Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. J. Periodontol. 2015, 86, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Tonetti, M.S.; Greenwell, H.; Kornman, K.S. Staging and grading of periodontitis: Framework and proposal of a new classification and case definition. J. Periodontol. 2018, 89, S159–S172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapple, I.L.C.; Mealey, B.L.; Van Dyke, T.E.; Bartold, P.M.; Dommisch, H.; Eickholz, P.; Geisinger, M.L.; Genco, R.J.; Glogauer, M.; Goldstein, M.; et al. Periodontal health and gingival diseases and conditions on an intact and a reduced periodontium: Consensus report of workgroup 1 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Clin. Periodontol. 2018, 45 (Suppl. 20), S68–S77. [Google Scholar] [CrossRef] [PubMed]
- Caton, J.G.; Armitage, G.; Berglundh, T.; Chapple, I.L.C.; Jepsen, S.; Kornman, K.S.; Mealey, B.L.; Papapanou, P.N.; Sanz, M.; Tonetti, M.S. A new classification scheme for periodontal and peri-implant diseases and conditions—Introduction and key changes from the 1999 classification. J. Clin. Periodontol. 2018, 45 (Suppl. 20), S1–S8. [Google Scholar] [CrossRef] [PubMed]
- Dahlen, G.; Basic, A.; Bylund, J. Importance of Virulence Factors for the Persistence of Oral Bacteria in the Inflamed Gingival Crevice and in the Pathogenesis of Periodontal Disease. J. Clin. Med. 2019, 8, 1339. [Google Scholar] [CrossRef] [Green Version]
- Kudo, C.; Naruishi, K.; Maeda, H.; Abiko, Y.; Hino, T.; Iwata, M.; Mitsuhashi, C.; Murakami, S.; Nagasawa, T.; Nagata, T.; et al. Assessment of the plasma/serum IgG test to screen for periodontitis. J. Dent. Res. 2012, 91, 1190–1195. [Google Scholar] [CrossRef]
- Diaz-Gallo, L.M.; Oke, V.; Lundström, E.; Elvin, K.; Ling Wu, Y.; Eketjäll, S.; Zickert, A.; Gustafsson, J.T.; Jönsen, A.; Leonard, D.; et al. Four Systemic Lupus Erythematosus Subgroups, Defined by Autoantibodies Status, Differ Regarding HLA-DRB1 Genotype Associations and Immunological and Clinical Manifestations. ACR Open Rheumatol. 2022, 4, 27–39. [Google Scholar] [CrossRef]
- Bryzek, D.; Ciaston, I.; Dobosz, E.; Gasiorek, A.; Makarska, A.; Sarna, M.; Eick, S.; Puklo, M.; Lech, M.; Potempa, B.; et al. Triggering NETosis via protease-activated receptor (PAR)-2 signaling as a mechanism of hijacking neutrophils function for pathogen benefits. PLoS Pathog. 2019, 15, e1007773. [Google Scholar] [CrossRef]
- Magán-Fernández, A.; Rasheed Al-Bakri, S.M.; O’Valle, F.; Benavides-Reyes, C.; Abadía-Molina, F.; Mesa, F. Neutrophil Extracellular Traps in Periodontitis. Cells 2020, 9, 1494. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef]
- Jenning, M.; Marklein, B.; Ytterberg, J.; Zubarev, R.A.; Joshua, V.; van Schaardenburg, D.; van de Stadt, L.; Catrina, A.I.; Nonhoff, U.; Häupl, T.; et al. Bacterial citrullinated epitopes generated by Porphyromonas gingivalis infection-a missing link for ACPA production. Ann. Rheum. Dis. 2020, 79, 1194–1202. [Google Scholar] [CrossRef]
- Nesse, W.; Westra, J.; van der Wal, J.E.; Abbas, F.; Nicholas, A.P.; Vissink, A.; Brouwer, E. The periodontium of periodontitis patients contains citrullinated proteins which may play a role in ACPA (anti-citrullinated protein antibody) formation. J. Clin. Periodontol. 2012, 39, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebbag, M.; Simon, M.; Vincent, C.; Masson-Bessière, C.; Girbal, E.; Durieux, J.J.; Serre, G. The antiperinuclear factor and the so-called antikeratin antibodies are the same rheumatoid arthritis-specific autoantibodies. J. Clin. Investig. 1995, 95, 2672–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liccardo, D.; Cannavo, A.; Spagnuolo, G.; Ferrara, N.; Cittadini, A.; Rengo, C.; Rengo, G. Periodontal Disease: A Risk Factor for Diabetes and Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mougeot, J.L.C.; Stevens, C.B.; Paster, B.J.; Brennan, M.T.; Lockhart, P.B.; Mougeot, F.K.B. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J. Oral Microbiol. 2017, 9, 1281562. [Google Scholar] [CrossRef] [Green Version]
- England, B.R.; Thiele, G.M.; Anderson, D.R.; Mikuls, T.R. Increased cardiovascular risk in rheumatoid arthritis: Mechanisms and implications. BMJ Clin. Res. Ed. 2018, 361, k1036. [Google Scholar] [CrossRef]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef]
- Westerlind, H.; Rönnelid, J.; Hansson, M.; Alfredsson, L.; Mathsson-Alm, L.; Serre, G.; Cornillet, M.; Holmdahl, R.; Jakobsson, P.J.; Skriner, K.; et al. Anti-Citrullinated Protein Antibody Specificities, Rheumatoid Factor Isotypes, and Incident Cardiovascular Events in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 1658–1667. [Google Scholar] [CrossRef]
- Rantapää-Dahlqvist, S.; de Jong, B.A.; Berglin, E.; Hallmans, G.; Wadell, G.; Stenlund, H.; Sundin, U.; van Venrooij, W.J. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2741–2749. [Google Scholar] [CrossRef]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Periodontitis (n = 557) | No Periodontitis (n = 941) | p Value 1 | |
|---|---|---|---|
| MI patient, n (%) | 319 (57%) | 460 (49%) | 0.002 |
| Male sex, n (%) | 435 (78%) | 783 (83%) | 0.017 |
| Smoking, ever, n (%) 2 | 408 (74%) | 473 (51%) | <0.001 |
| Age, median years (range) | 66 (40–75) | 62 (28–75) | <0.001 |
| HbA1C, median mmol/mol (range) | 39 (26–117) | 38 (20–94) | <0.001 |
| BOP index, median % (range) 3 | 25 (0.9–100) | 21 (0.9–100.) | <0.002 |
| Boneloss, median % (range) | 24.5 (20.0–100) | 15.4 (6.4–19.9) | <0.001 |
| Presence of pockets ≥ 6 mm, n (%) | 251 (55.5%) | 309 (26.7%) | <0.001 |
| PerioGene North | Periodontitis (n = 41) | No Periodontitis (n = 39) | p Value 1 |
|---|---|---|---|
| Male sex, n (%) | 21 (51.2) | 15 (38.5) | ns |
| Smoking, ever, n (%) | 29 (70.7) | 9 (23.1) | <0.001 |
| Age, median years (range) | 55 (28–76) | 44 (35–63) | <0.001 |
| BOP index, median % (range) | 35 (8–100) | 6 (0–27) | <0.001 |
| Number of teeth with bone loss, median (range) | 18 (7–29) | 0 | <0.001 |
| Number of teeth with pocket ≥ 4 mm, median (range) | 21 (3–28) | 0 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Vries, C.; Ruacho, G.; Kindstedt, E.; Potempa, B.A.; Potempa, J.; Klinge, B.; Lundberg, P.; Svenungsson, E.; Lundberg, K. Antibodies to Porphyromonas gingivalis Are Increased in Patients with Severe Periodontitis, and Associate with Presence of Specific Autoantibodies and Myocardial Infarction. J. Clin. Med. 2022, 11, 1008. https://doi.org/10.3390/jcm11041008
de Vries C, Ruacho G, Kindstedt E, Potempa BA, Potempa J, Klinge B, Lundberg P, Svenungsson E, Lundberg K. Antibodies to Porphyromonas gingivalis Are Increased in Patients with Severe Periodontitis, and Associate with Presence of Specific Autoantibodies and Myocardial Infarction. Journal of Clinical Medicine. 2022; 11(4):1008. https://doi.org/10.3390/jcm11041008
Chicago/Turabian Stylede Vries, Charlotte, Guillermo Ruacho, Elin Kindstedt, Barbara Aleksandra Potempa, Jan Potempa, Björn Klinge, Pernilla Lundberg, Elisabet Svenungsson, and Karin Lundberg. 2022. "Antibodies to Porphyromonas gingivalis Are Increased in Patients with Severe Periodontitis, and Associate with Presence of Specific Autoantibodies and Myocardial Infarction" Journal of Clinical Medicine 11, no. 4: 1008. https://doi.org/10.3390/jcm11041008
APA Stylede Vries, C., Ruacho, G., Kindstedt, E., Potempa, B. A., Potempa, J., Klinge, B., Lundberg, P., Svenungsson, E., & Lundberg, K. (2022). Antibodies to Porphyromonas gingivalis Are Increased in Patients with Severe Periodontitis, and Associate with Presence of Specific Autoantibodies and Myocardial Infarction. Journal of Clinical Medicine, 11(4), 1008. https://doi.org/10.3390/jcm11041008