
The Pathophysiology of Portal Vein Thrombosis in Cirrhosis: Getting Deeper into Virchow’s Triad

 , , and
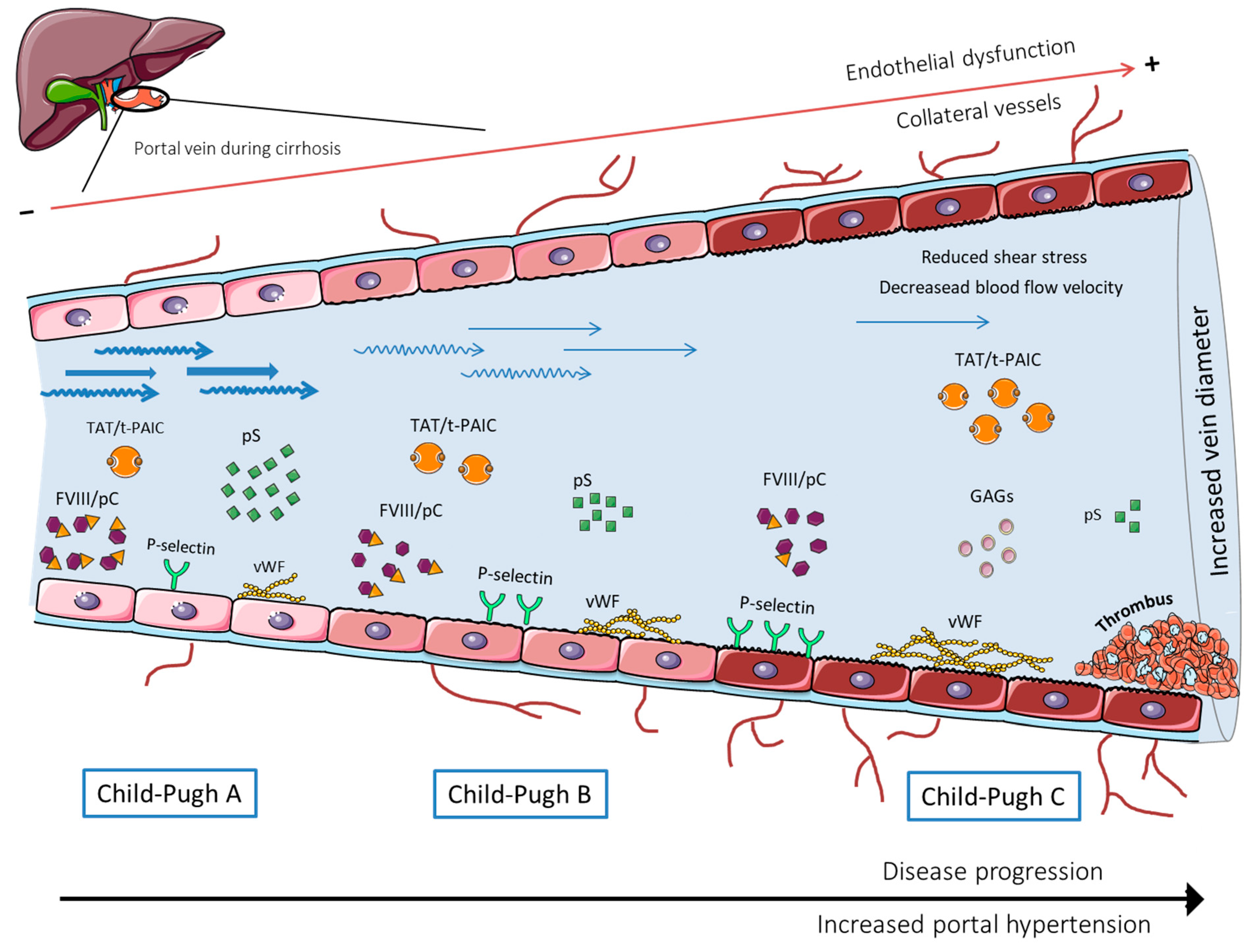
, , and 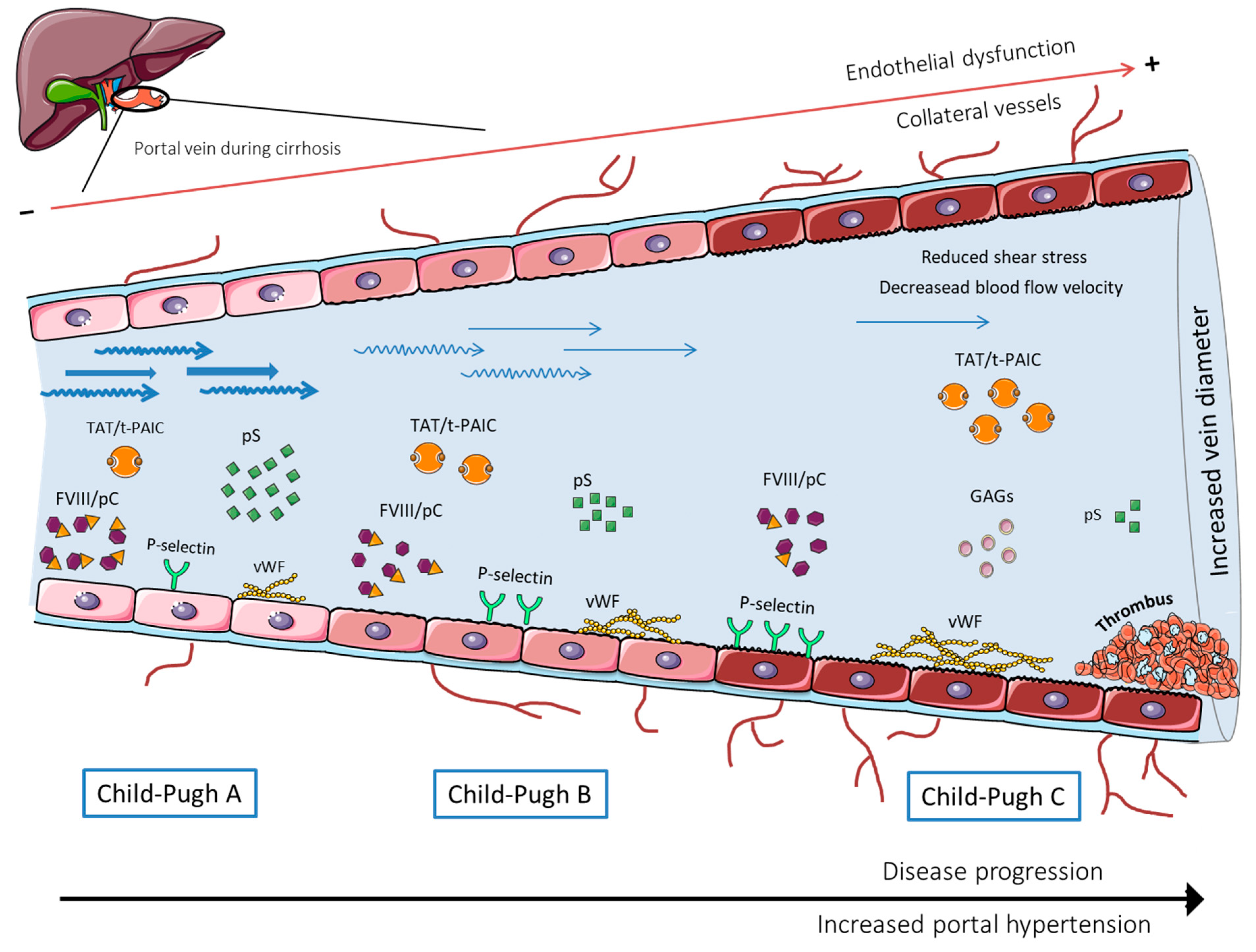{kind=link}
Abstract
:1. Introduction
2. Blood Hypercoagulability
3. Portal Vein Hemodynamics and Blood Flow
4. Intravascular Vessel Wall (Endothelial) Damage
5. Thrombus Composition
6. Main Challenges and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, H.; Turon, F.; Hernández-Gea, V.; Fuster, J.; García-Criado, A.; Barrufet, M.; Darnell, A.; Fondevila, C.; Garcia-Valdecasas, J.C.; Garcia-Pagan, J.C. Nontumoral portal vein thrombosis in patients awaiting liver transplantation. Liver Transplant. 2016, 22, 352–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caiano, L.M.; Riva, N.; Carrier, M.; Gatt, A.; Ageno, W. Treatment of portal vein thrombosis: An updated narrative review. Minerva Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yan, Z.; Luo, J.; Liu, Q.; Wang, J.; Qiu, S. Rational Classification of Portal Vein Thrombosis and Its Clinical Significance. PLoS ONE 2014, 9, e112501. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, C.; Brito, J.; Bilreiro, C.; Barros, M.; Bahia, C.; Santiago, I.; Caseiro-Alves, F. All about portal vein: A pictorial display to anatomy, variants and physiopathology. Insights Imaging 2019, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Gea, V.; De Gottardi, A.; Leebeek, F.W.G.; Rautou, P.E.; Salem, R.; Garcia-Pagan, J.C. Current knowledge in pathophysiology and management of Budd-Chiari syndrome and non-cirrhotic non-tumoral splanchnic vein thrombosis. J. Hepatol. 2019, 71, 175–199. [Google Scholar] [CrossRef] [Green Version]
- Zocco, M.A.; Di Stasio, E.; De Cristofaro, R.; Novi, M.; Ainora, M.E.; Ponziani, F.R.; Riccardi, L.; Lancellotti, S.; Santoliquido, A.; Flore, R.A.; et al. Thrombotic risk factors in patients with liver cirrhosis: Correlation with MELD scoring system and portal vein thrombosis development. J. Hepatol. 2009, 51, 682–689. [Google Scholar] [CrossRef]
- Intagliata, N.; Caldwell, S.H.; Tripodi, A. Diagnosis, Development, and Treatment of Portal Vein Thrombosis in Patients With and Without Cirrhosis. Gastroenterology 2019, 156, 1582–1599. [Google Scholar] [CrossRef]
- Ferreira, C.N.; Marinho, R.T.; Cortez-Pinto, H.; Ferreira, P.; Dias, M.S.; Vasconcelos, M.; Alexandrino, P.; Serejo, F.; Pedro, A.J.; Gonçalves, A.; et al. Incidence, predictive factors and clinical significance of development of portal vein thrombosis in cirrhosis: A prospective study. Liver Int. 2019, 39, 1459–1467. [Google Scholar] [CrossRef]
- Stine, J.; Shah, P.M.; Cornella, S.L.; Rudnick, S.R.; Ghabril, M.; Stukenborg, G.J.; Northup, P.G. Portal vein thrombosis, mortality and hepatic decompensation in patients with cirrhosis: A meta-analysis. World J. Hepatol. 2015, 7, 2774–2780. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, H.; Okugawa, H.; Takahashi, M.; Yokosuka, O. De novo Portal Vein Thrombosis in Virus-Related Cirrhosis: Predictive Factors and Long-Term Outcomes. Am. J. Gastroenterol. 2013, 108, 568–574. [Google Scholar] [CrossRef]
- Nery, F.; Chevret, S.; Condat, B.; de Raucourt, E.; Boudaoud, L.; Rautou, P.-E.; Plessier, A.; Roulot, D.; Chaffaut, C.; Bourcier, V.; et al. Causes and consequences of portal vein thrombosis in 1243 patients with cirrhosis: Results of a longitudinal study. Hepatology 2015, 61, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Sun, K.Y.; Fu, Z.R. Tailored classification of portal vein thrombosis for liver transplantation: Focus on strategies for portal vein inflow reconstruction. World J. Gastroenterol. 2020, 26, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Bhangui, P.; Fernandes, E.S.M.; Di Benedetto, F.; Joo, D.J.; Nadalin, S. Current management of portal vein thrombosis in liver transplantation. Int. J. Surg. 2020, 82, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Zanetto, A.; Rodriguez-Kastro, K.-I.; Germani, G.; Ferrarese, A.; Cillo, U.; Burra, P.; Senzolo, M. Mortality in liver transplant recipients with portal vein thrombosis—An updated meta-analysis. Transpl. Int. 2018, 31, 1318–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghabril, M.; Agarwal, S.; Lacerda, M.; Chalasani, N.; Kwo, P.; Tector, A.J. Portal Vein Thrombosis Is a Risk Factor for Poor Early Outcomes After Liver Transplantation. Transplantation 2016, 100, 126–133. [Google Scholar] [CrossRef]
- Xian, J.; Tang, Y.; Shao, H.; Wang, X.; Zhang, M.; Xing, T. Effect of portal vein thrombosis on the prognosis of patients with cirrhosis without a liver transplant: A systematic review and meta-analysis. Medicine 2021, 100, e25439. [Google Scholar] [CrossRef]
- Loffredo, L.; Pastori, D.; Farcomeni, A.; Violi, F. Effects of Anticoagulants in Patients With Cirrhosis and Portal Vein Thrombosis: A Systematic Review and Meta-analysis. Gastroenterology 2017, 153, 480–487. [Google Scholar] [CrossRef] [Green Version]
- Luca, A.; Caruso, S.; Milazzo, M.; Marrone, G.; Mamone, G.; Crinò, F.; Maruzzelli, L.; Miraglia, R.; Floridia, G.; Vizzini, G. Natural Course of Extrahepatic Nonmalignant Partial Portal Vein Thrombosis in Patients with Cirrhosis. Radiology 2012, 265, 124–132. [Google Scholar] [CrossRef]
- Martín-Llahí, M.; Albillos, A.; Bañares, R.; Berzigotti, A.; García-Criado, M.; Genescà, J.; Hernández-Gea, V.; Llop-Herrera, E.; Masnou-Ridaura, H.; Mateo, J.; et al. Vascular diseases of the liver. Clinical Guidelines from the Catalan Society of Digestology and the Spanish Association for the Study of the Liver. Gastroenterol. Hepatol. 2017, 40, 538–580. [Google Scholar] [CrossRef]
- De Franchis, R.; Faculty, B.V. Expanding consensus in portal hypertension: Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J. Hepatol. 2015, 63, 743–752. [Google Scholar] [CrossRef] [Green Version]
- EASL. Clinical Practice Guidelines: Vascular diseases of the liver. J. Hepatol. 2016, 64, 179–202. [Google Scholar] [CrossRef] [PubMed]
- Turon, F.; Hernández-Gea, V.; García-Pagán, J.C. Portal vein thrombosis. Curr. Opin. Organ Transplant. 2018, 23, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.G.; Seijo, S.; Yepes, I.; Achécar, L.; Catalina, M.V.; García–Criado, A.; Abraldes, J.G.; de la Peña, J.; Bañares, R.; Albillos, A.; et al. Efficacy and Safety of Anticoagulation on Patients With Cirrhosis and Portal Vein Thrombosis. Clin. Gastroenterol. Hepatol. 2012, 10, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.R.; Hanlin, E.R.; Glurich, I.; Mazza, J.J.; Yale, S.H. Virchow’s contribution to the understanding of thrombosis and cellular biology. Clin. Med. Res. 2010, 8, 168–172. [Google Scholar] [CrossRef] [Green Version]
- Wolberg, A.S.; Aleman, M.M.; Leiderman, K.; Machlus, K.R. Procoagulant activity in hemostasis and thrombosis: Virchow’s triad revisited. Anesth. Analg. 2012, 114, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Fortea, J.I.; Carrera, I.G.; Puente, A.; Cuadrado, A.; Huelin, P.; Tato, C.; Fernández, P.; Montes, M.D.R.P.; Céspedes, J.N.; López, A.B.; et al. Portal Thrombosis in Cirrhosis: Role of Thrombophilic Disorders. J. Clin. Med. 2020, 9, 2822. [Google Scholar] [CrossRef]
- Nicoară-Farcău, O.; Soy, G.; Magaz, M.; Baiges, A.; Turon, F.; Garcia-Criado, A.; Barrufet, M.; Burrel, M.; Hernández-Gea, V.; García-Pagán, J.C. New Insights into the Pathogenesis, Risk Factors, and Treatment of Portal Vein Thrombosis in Patients with Cirrhosis. Semin. Thromb. Hemost. 2020, 46, 673–681. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, X.-P.; Zhong, B.-Y.; Lau, W.Y.; Madoff, D.; Davidson, J.C.; Qi, X.; Cheng, S.-Q.; Teng, G.-J. Management of patients with hepatocellular carcinoma and portal vein tumour thrombosis: Comparing east and west. Lancet Gastroenterol. Hepatol. 2019, 4, 721–730. [Google Scholar] [CrossRef]
- Lisman, T.; Porte, R.J. Rebalanced hemostasis in patients with liver disease: Evidence and clinical consequences. Blood 2010, 116, 878–885. [Google Scholar] [CrossRef] [Green Version]
- Tripodi, A.; Primignani, M.; Chantarangkul, V.; Dell’Era, A.; Clerici, M.; de Franchis, R.; Colombo, M.; Mannucci, P.M. An Imbalance of Pro- vs Anti-Coagulation Factors in Plasma From Patients With Cirrhosis. Gastroenterology 2009, 137, 2105–2111. [Google Scholar] [CrossRef]
- Turon, F.; Driever, E.G.; Baiges, A.; Cerda, E.; García-Criado, Á.; Gilabert, R.; Bru, C.; Berzigotti, A.; Nuñez, I.; Orts, L.; et al. Predicting portal thrombosis in cirrhosis: A prospective study of clinical, ultrasonographic and hemostatic factors. J. Hepatol. 2021, 75, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Kalambokis, G.N.; Oikonomou, A.; Christou, L.; Baltayiannis, G. High von Willebrand factor antigen levels and procoagulant imbalance may be involved in both increasing severity of cirrhosis and portal vein thrombosis. Hepatology 2016, 64, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Zhang, J.; Chen, Y.; Wen, M.; Su, Y.; Zhao, Y.; Lu, S.; Wu, J. Evaluation of Coagulation, Fibrinolysis and Endothelial Biomarkers in Cirrhotic Patients With or Without Portal Venous Thrombosis. Clin. Appl. Thromb. 2020, 26, 1076029620982666. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, B.; Balcar, L.; Nussbaumer, R.J.; Weinzierl, J.; Paternostro, R.; Simbrunner, B.; Hartl, L.; Jachs, M.; Bauer, D.; Stättermayer, A.F.; et al. Factor VIII/protein C ratio independently predicts liver-related events but does not indicate a hypercoagulable state in ACLD. J. Hepatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qi, X.; He, C.; Yin, Z.; Fan, D.; Han, G. Coagulation imbalance may not contribute to the development of portal vein thrombosis in patients with cirrhosis. Thromb. Res. 2013, 131, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wang, Y.; Zhao, X.; Wang, X.; Zhang, T.; Ou, X.; Shou, W.; You, H.; Jia, J. Procoagulant imbalance aggravated with falling liver function reserve, but not associated with the presence of portal vein thrombosis in cirrhosis. Eur. J. Gastroenterol. Hepatol. 2015, 27, 672–678. [Google Scholar] [CrossRef]
- La Mura, V.; Tripodi, A.; Tosetti, G.; Cavallaro, F.; Chantarangkul, V.; Colombo, M.; Primignani, M. Resistance to thrombomodulin is associated with de novo portal vein thrombosis and low survival in patients with cirrhosis. Liver Int. 2016, 36, 1322–1330. [Google Scholar] [CrossRef]
- Fimognari, F.L.; De Santis, A.; Piccheri, C.; Moscatelli, R.; Gigliotti, F.; Vestri, A.; Attili, A.; Violi, F. Evaluation of D-dimer and factor VIII in cirrhotic patients with asymptomatic portal venous thrombosis. J. Lab. Clin. Med. 2005, 146, 238–243. [Google Scholar] [CrossRef]
- Rossetto, V.; Spiezia, L.; Senzolo, M.; Rodriguez-Castro, K.I.; Maggiolo, S.; Simioni, P. Whole blood rotation thromboelastometry (ROTEM®) profiles in subjects with non-neoplastic portal vein thrombosis. Thromb. Res. 2013, 132, 9–12. [Google Scholar] [CrossRef]
- Mangia, A.; Villani, M.R.; Cappucci, G.; Santoro, R.; Ricciardi, R.; Facciorusso, D.; Leandro, G.; Caruso, N.; Andriulli, A. Causes of portal venous thrombosis in cirrhotic patients: The role of genetic and acquired factors. Eur. J. Gastroenterol. Hepatol. 2005, 17, 745–751. [Google Scholar] [CrossRef]
- Tremblay, D.; Naymagon, L.; Troy, K.; Cromwell, C.; Edwards, C.; Schiano, T.; Kremyanskaya, M.; Mascarenhas, J. The utility of thrombophilia testing in patients with newly diagnosed portal vein thrombosis. Blood Coagul. Fibrinolysis 2020, 31, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Delahousse, B.; Labat-Debelleix, V.; Decalonne, L.; D’Alteroche, L.; Perarnau, J.M.; Gruel, Y. Comparative study of coagulation and thrombin generation in the portal and jugular plasma of patients with cirrhosis. Thromb. Haemost. 2010, 104, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praktiknjo, M.; Trebicka, J.; Carnevale, R.; Pastori, D.; Queck, A.; Ettorre, E.; Violi, F. Von Willebrand and Factor VIII Portosystemic Circulation Gradient in Cirrhosis: Implications for Portal Vein Thrombosis. Clin. Transl. Gastroenterol. 2020, 11, e00123. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Fan, X.; Zhang, R.; Jiang, S.; Yang, K.; Chen, S. Systemic inflammation and portal vein thrombosis in cirrhotic patients with gastroesophageal varices. Eur. J. Gastroenterol. Hepatol. 2020, 32, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Fernandez-Carrillo, C.; Dai, D.; Dana, J.; Clemente-Sanchez, A.; Dharmayan, S.; Kaltenmeier, C.; Liu, H.; Behari, J.; Rachakonda, V.; et al. Portal vein thrombosis and renal dysfunction: A national comparative study of liver transplant recipients for NAFLD versus alcoholic cirrhosis. Transpl. Int. 2021, 34, 1105–1122. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M. Increased prevalence of portal vein thrombosis in patients with nonalcoholic steatohepatitis-cirrhosis due to increased proinflammatory cytokines releasing from abdominal adipose tissue. Eur. J. Gastroenterol. Hepatol. 2020, 32, 458. [Google Scholar] [CrossRef]
- Zanetto, A.; Senzolo, M.; Campello, E.; Bulato, C.; Gavasso, S.; Saggiorato, G.; Feltracco, P.; Farinati, F.; Russo, F.P.; Burra, P.; et al. Determinants of increased thrombotic tendency in NASH cirrhosis: Not there yet! Transpl. Int. 2021, 34, 1325–1327. [Google Scholar] [CrossRef]
- Bos, S.; Boom, B.V.D.; Kamphuisen, P.; Adelmeijer, J.; Blokzijl, H.; Schreuder, T.; Lisman, T. Haemostatic Profiles are Similar across All Aetiologies of Cirrhosis. Thromb. Haemost. 2019, 119, 246–253. [Google Scholar] [CrossRef]
- García-Pagán, J.C.; Gracia-Sancho, J.; Bosch, J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J. Hepatol. 2012, 57, 458–461. [Google Scholar] [CrossRef]
- Haag, K.; Rössle, M.; Ochs, A.; Huber, M.; Siegerstetter, V.; Olschewski, M.; Berger, E.; Lu, S.; E Blum, H. Correlation of duplex sonography findings and portal pressure in 375 patients with portal hypertension. Am. J. Roentgenol. 1999, 172, 631–635. [Google Scholar] [CrossRef]
- Bosch, J.; Abraldes, J.G.; Fernández, M.; García-Pagán, J.C. Hepatic endothelial dysfunction and abnormal angiogenesis: New targets in the treatment of portal hypertension. J. Hepatol. 2010, 53, 558–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Razik, A.; Mousa, N.; Elhelaly, R.; Tawfik, A. De-novo portal vein thrombosis in liver cirrhosis: Risk factors and correlation with the Model for End-stage Liver Disease scoring system. Eur. J. Gastroenterol. Hepatol. 2015, 27, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Huang, X.-Q.; Zhu, Y.-L.; Ding, H.; Li, F.; Chen, S.-Y. Increased portal vein diameter is predictive of portal vein thrombosis development in patients with liver cirrhosis. Ann. Transl. Med. 2021, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.G.; Wang, J.; Shah, P.M.; Argo, C.K.; Intagliata, N.; Uflacker, A.; Caldwell, S.H.; Northup, P.G. Decreased portal vein velocity is predictive of the development of portal vein thrombosis: A matched case-control study. Liver Int. 2017, 38, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Guo, X.; De Stefano, V.; Silva-Junior, G.; Goyal, H.; Bai, Z.; Zhao, Q.; Qi, X. Nonselective beta-blockers and development of portal vein thrombosis in liver cirrhosis: A systematic review and meta-analysis. Hepatol. Int. 2019, 13, 468–481. [Google Scholar] [CrossRef]
- Wang, M.; Hao, H.; Leeper, N.J.; Zhu, L. Thrombotic regulation from the endothelial cell perspectives. Arterioscler. Thromb. Vasc. Biol. 2018, 38, E90–E95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 564–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Poredos, P.; Jezovnik, M.K. Endothelial Dysfunction and Venous Thrombosis. Angiology 2018, 69, 564–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferroni, P.; Mammarella, A.; Martini, F.; Paoletti, V.; Cardarello, C.M.; Labbadia, G.; Donnarumma, L.; De Matteis, A.; Gazzaniga, P.P.; Musca, A.; et al. Increased Soluble P-Selectin Levels in Hepatitis C Virus-Related Chronic Hepatitis. J. Investig. Med. 2001, 49, 407–412. [Google Scholar] [CrossRef]
- Raparelli, V.; Basili, S.; Carnevale, R.; Napoleone, L.; Del Ben, M.; Nocella, C.; Bartimoccia, S.; Lucidi, C.; Talerico, G.; Riggio, O.; et al. Low-grade endotoxemia and platelet activation in cirrhosis. Hepatology 2016, 65, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Wannhoff, A.; Müller, O.J.; Friedrich, K.; Rupp, C.; Klöters-Plachky, P.; Leopold, Y.; Brune, M.; Senner, M.; Weiss, K.-H.; Stremmel, W.; et al. Effects of Increased Von Willebrand Factor Levels on Primary Hemostasis in Thrombocytopenic Patients with Liver Cirrhosis. PLoS ONE 2014, 9, e112583. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T.; Bongers, T.N.; Adelmeijer, J.; Janssen, H.L.; De Maat, M.P.; De Groot, P.G.; Leebeek, F.W. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology 2006, 44, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Gîrleanu, I.; Trifan, A.; Stanciu, C.; Sfarti, C. Portal vein thrombosis in cirrhotic patients—It is always the small pieces that make the big picture. World J. Gastroenterol. 2018, 24, 4419. [Google Scholar] [CrossRef]
- Ferlitsch, M.; Reiberger, T.; Hoke, M.; Salzl, P.; Schwengerer, B.; Ulbrich, G.; Payer, B.A.; Trauner, M.; Peck-Radosavljevic, M.; Ferlitsch, A. Von Willebrand factor as new noninvasive predictor of portal hypertension, decompensation and mortality in patients with liver cirrhosis. Hepatology 2012, 56, 1439–1447. [Google Scholar] [CrossRef]
- Carnevale, R.; Raparelli, V.; Nocella, C.; Bartimoccia, S.; Novo, M.; Severino, A.; De Falco, E.; Cammisotto, V.; Pasquale, C.; Crescioli, C.; et al. Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implications for hypercoagulability in cirrhosis. J. Hepatol. 2017, 67, 950–956. [Google Scholar] [CrossRef]
- Violi, F.; Ferro, D.; Basili, S.; Lionetti, R.; Rossi, E.; Merli, M.; Riggio, O.; Bezzi, M.; Capocaccia, L. Ongoing Prothrombotic State in the Portal Circulation of Cirrhotic Patients. Thromb. Haemost. 1997, 77, 44–47. [Google Scholar] [CrossRef]
- Shalaby, S.; Simioni, P.; Campello, E.; Spiezia, L.; Gavasso, S.; Bizzaro, D.; Cardin, R.; D’Amico, F.; Gringeri, E.; Cillo, U.; et al. Endothelial Damage of the Portal Vein is Associated with Heparin-Like Effect in Advanced Stages of Cirrhosis. Thromb. Haemost. 2020, 120, 1173–1181. [Google Scholar] [CrossRef]
- Mooberry, M.J.; Key, N.S. Microparticle Analysis in Disorders of Hemostasis and Thrombosis. Cytom. Part A 2016, 89, 111. [Google Scholar] [CrossRef] [Green Version]
- Lindberg-Larsen, V.; Ostrowski, S.R.; Lindberg-Larsen, M.; Rovsing, M.L.; Johansson, P.I.; Kehlet, H. The effect of pre-operative methylprednisolone on early endothelial damage after total knee arthroplasty: A randomised, double-blind, placebo-controlled trial. Anaesthesia 2017, 72, 1217–1224. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Jiang, Y.; Luo, L.; Huang, R.; Su, L.; Hou, M.; Wang, X.; Deng, J.; Hu, Y. Evaluation the combined diagnostic value of TAT, PIC, tPAIC, and sTM in disseminated intravascular coagulation: A multi-center prospective observational study. Thromb. Res. 2018, 173, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Driever, E.G.; von Meijenfeldt, F.A.; Adelmeijer, J.; de Haas, R.J.; Heuvel, M.C.V.D.; Nagasami, C.; Weisel, J.W.; Fondevila, C.; Porte, R.J.; Blasi, A.; et al. Nonmalignant portal vein thrombi in patients with cirrhosis consist of intimal fibrosis with or without a fibrin-rich thrombus. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Favaloro, E.J. Venous and Arterial Thromboses: Two Sides of the Same Coin? Semin. Thromb. Hemost. 2018, 44, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.A.; Saha, P.; Cooley, B.; Palmer, O.R.; Grover, S.; Mackman, N.; Wakefield, T.W.; Henke, P.K.; Smith, A.; Lal, B.K. Choosing a mouse model of venous thrombosis: A consensus assessment of utility and application. J. Thromb. Haemost. 2019, 17, 699–707. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anton, A.; Campreciós, G.; Pérez-Campuzano, V.; Orts, L.; García-Pagán, J.C.; Hernández-Gea, V. The Pathophysiology of Portal Vein Thrombosis in Cirrhosis: Getting Deeper into Virchow’s Triad. J. Clin. Med. 2022, 11, 800. https://doi.org/10.3390/jcm11030800
Anton A, Campreciós G, Pérez-Campuzano V, Orts L, García-Pagán JC, Hernández-Gea V. The Pathophysiology of Portal Vein Thrombosis in Cirrhosis: Getting Deeper into Virchow’s Triad. Journal of Clinical Medicine. 2022; 11(3):800. https://doi.org/10.3390/jcm11030800
Chicago/Turabian StyleAnton, Aina, Genís Campreciós, Valeria Pérez-Campuzano, Lara Orts, Joan Carles García-Pagán, and Virginia Hernández-Gea. 2022. "The Pathophysiology of Portal Vein Thrombosis in Cirrhosis: Getting Deeper into Virchow’s Triad" Journal of Clinical Medicine 11, no. 3: 800. https://doi.org/10.3390/jcm11030800
APA StyleAnton, A., Campreciós, G., Pérez-Campuzano, V., Orts, L., García-Pagán, J. C., & Hernández-Gea, V. (2022). The Pathophysiology of Portal Vein Thrombosis in Cirrhosis: Getting Deeper into Virchow’s Triad. Journal of Clinical Medicine, 11(3), 800. https://doi.org/10.3390/jcm11030800