

T Cell-Mediated Tumor Killing-Related Classification of the Immune Microenvironment and Prognosis Prediction of Lung Adenocarcinoma

 ,
, 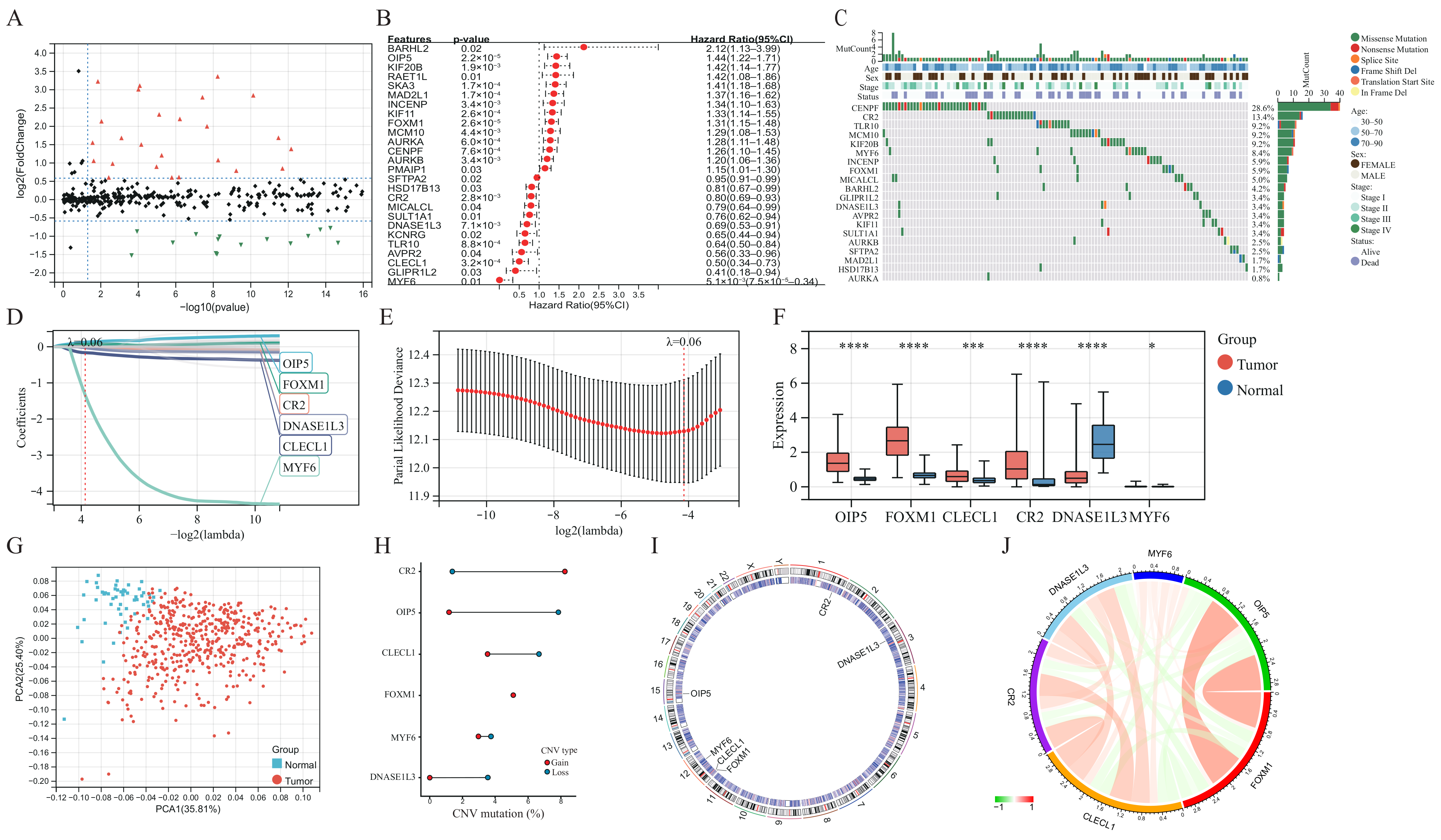{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Exploration of Key Genes
2.3. Unsupervised Clustering
2.4. Differential Analysis Based on Whole-Genome Data
2.5. Functional Enrichment Analysis
2.6. Gene Set Enrichment Analysis (GSEA)
2.7. Single Samples Gene Set Enrichment Analysis (ssGSEA)
2.8. Immune Landscape Comparison between Two GSTTK Subgroups
2.9. Establishment of a Risk Model
2.10. Survival Analysis
2.11. Construction and Evaluation of the Integrated Nomogram
2.12. Statistical Analyses
3. Results
3.1. Determination and Characterization of GSTTK Involved in LUAD
3.2. Identification of Two TTK Modes in LUAD
3.3. Somatic Mutations Related to the Two TTK Modes
3.4. Functional Enrichment Analysis
3.5. Different TIME in the Two TTK Modes
3.6. Different TIME Status in the Two Molecular Modes
3.7. Construction of The GSTTK Risk Signature in the Training Set
3.8. Verification of the GSTTK Risk Signature in the Validation Set
3.9. Construction and Calibration of an Integrated Nomogram
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Yao, H.; Li, C.; Fang, J.-Y.; Xu, J. Regulation of PD-L1: Emerging Routes for Targeting Tumor Immune Evasion. Front. Pharmacol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Torlakovic, E.; Lim, H.J.; Adam, J.; Barnes, P.; Bigras, G.; Chan, A.W.H.; Cheung, C.C.; Chung, J.-H.; Couture, C.; Fiset, P.O.; et al. “Interchangeability” of PD-L1 immunohistochemistry assays: A meta-analysis of diagnostic accuracy. Mod. Pathol. 2019, 33, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Elfving, H.; Mattsson, J.S.M.; Lindskog, C.; Backman, M.; Menzel, U.; Micke, P. Programmed Cell Death Ligand 1 Immunohisto-chemistry: A Concordance Study Between Surgical Specimen, Biopsy, and Tissue Microarray. Clin. Lung Cancer 2019, 20, 258–262. [Google Scholar] [CrossRef]
- Brunnström, H.; Johansson, A.; Fremer, S.W.; Backman, M.; Djureinovic, D.; Patthey, A.; Isaksson-Mettävainio, M.; Gulyas, M.; Micke, P. PD-L1 immunohistochemistry in clinical diagnostics of lung cancer: Inter-pathologist variability is higher than assay variability. Mod. Pathol. 2017, 30, 1411–1421. [Google Scholar] [CrossRef]
- Tsao, M.S.; Kerr, K.M.; Kockx, M.; Beasley, M.-B.; Borczuk, A.C.; Botling, J.; Bubendorf, L.; Chirieac, L.; Chen, G.; Chou, T.-Y.; et al. PD-L1 Immunohistochemistry Comparability Study in Real-Life Clinical Samples: Results of Blueprint Phase 2 Project. J. Thorac. Oncol. 2018, 13, 1302–1311. [Google Scholar] [CrossRef]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 Immunohistochemistry Assays for Lung Cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef]
- Rimm, D.L.; Han, G.; Taube, J.M.; Yi, E.S.; Bridge, J.A.; Flieder, D.B.; Homer, R.; West, W.W.; Wu, H.; Roden, A.C.; et al. A Prospective, Multi-institutional, Pathologist-Based Assessment of 4 Immunohistochemistry Assays for PD-L1 Expression in Non–Small Cell Lung Cancer. JAMA Oncol. 2017, 3, 1051. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Wei, W.; Gupta, S.; Zugazagoitia, J.; Robbins, C.; Adamson, B.; Rimm, D.L. PD-L1 tumor proportion score and overall survival from first line pembrolizumab in patients with nonsquamous versus squamous non-small cell lung cancer. J. Thorac. Oncol. 2021, 16, 2139–2143. [Google Scholar] [CrossRef]
- Camidge, D.R.; Doebele, R.C.; Kerr, K.M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 2019, 16, 341–355. [Google Scholar] [CrossRef]
- Genova, C.; Dellepiane, C.; Carrega, P.; Sommariva, S.; Ferlazzo, G.; Pronzato, P.; Gangemi, R.; Filaci, G.; Coco, S.; Croce, M. Therapeutic Implications of Tumor Micro-environment in Lung Cancer: Focus on Immune Checkpoint Blockade. Front. Immunol. 2021, 12, 799455. [Google Scholar] [CrossRef]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines re-sistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor–immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Taminau, J.; Meganck, S.; Lazar, C.; Steenhoff, D.; Coletta, A.; Molter, C.; Duque, R.; Schaetzen, V.D.; Weiss Solís, D.Y.; Bersini, H.; et al. Unlocking the potential of publicly available mi-croarray data using inSilicoDb and inSilicoMerging R/Bioconductor packages. BMC Bioinform. 2012, 13, 335. [Google Scholar] [CrossRef]
- Johnson, W.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tis-sue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Ye, Z.; Shen, R.; Yu, G.; Wu, J.; Xiong, Y.; Zhou, R.; Qiu, W.; Huang, N.; Sun, L.; et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 2021, 12, 687975. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Shen, W.; Song, Z.; Zhong, X.; Huang, M.; Shen, D.; Gao, P.; Qian, X.; Wang, M.; He, X.; Wang, T.; et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta 2022, 1, e36. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Busund, L.T.; Kilvær, T.L.; Andersen, S.; Richardsen, E.; Paulsen, E.E.; Hald, S.; Khanehkenari, M.R.; Cooper, W.A.; Kao, S.C.; et al. The Role of Tumor-Infiltrating Lymphocytes in Development, Progression, and Prognosis of Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 789–800. [Google Scholar] [CrossRef]
- Xu, X.; Li, J.; Zou, J.; Feng, X.; Zhang, C.; Zheng, R.; Duanmu, W.; Saha-Mandal, A.; Ming, Z.; Wang, E. Association of Germline Variants in Natural Killer Cells with Tumor Immune Microenvironment Subtypes, Tumor-Infiltrating Lymphocytes, Immunotherapy Response, Clinical Outcomes, and Cancer Risk. JAMA Netw. Open 2019, 2, e199292. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2020, 18, 842–859. [Google Scholar] [CrossRef]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef]
- Wang, S.-S.; Liu, W.; Ly, D.; Xu, H.; Qu, L.; Zhang, L. Tumor-infiltrating B cells: Their role and application in anti-tumor immunity in lung cancer. Cell. Mol. Immunol. 2018, 16, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Solimando, A.; Pezzella, F. The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer. Cancers 2021, 13, 3433. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Sistigu, A.; Manic, G.; Rudqvist, N.-P.; Trajanoski, Z.; Galluzzi, L. Mutational and Antigenic Landscape in Tumor Progression and Cancer Immunotherapy. Trends Cell Biol. 2019, 29, 396–416. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. P53 and prognosis: New insights and further complexity. Cell 2005, 120, 7–10. [Google Scholar]
- Mendiratta, G.; Ke, E.; Aziz, M.; Liarakos, D.; Tong, M.; Stites, E.C. Cancer gene mutation frequencies for the U.S. population. Nat. Commun. 2021, 12, 5961. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, P.; Gao, C.; Chen, J.; Li, J.; Chen, Z.; Xu, M.; Shao, J.; Zhang, Y.; Xie, J. Down-regulation of LRP1B in colon cancer promoted the growth and mi-gration of cancer cells. Exp. Cell Res. 2017, 357, 1–8. [Google Scholar] [CrossRef]
- Koinuma, J.; Akiyama, H.; Fujita, M.; Hosokawa, M.; Tsuchiya, E.; Kondo, S.; Nakamura, Y.; Daigo, Y. Characterization of an Opa interacting protein 5 involved in lung and esophageal carcinogenesis. Cancer Sci. 2012, 103, 577–586. [Google Scholar] [CrossRef]
- Kalathil, D.; John, S.; Nair, A.S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front. Oncol. 2021, 10, 626836. [Google Scholar] [CrossRef]
- Arceci, A.; Bonacci, T.; Wang, X.; Stewart, K.; Damrauer, J.S.; Hoadley, K.; Emanuele, M.J. FOXM1 Deubiquitination by USP21 Regulates Cell Cycle Progression and Paclitaxel Sensitivity in Basal-like Breast Cancer. Cell Rep. 2019, 26, 3076–3086.e6. [Google Scholar] [CrossRef]
- Tassi, R.A.; Todeschini, P.; Siegel, E.R.; Calza, S.; Cappella, P.; Ardighieri, L.; Cadei, M.; Bugatti, M.; Romani, C.; Bandiera, E.; et al. FOXM1 expression is significantly associated with chemotherapy resistance and adverse prognosis in non-serous epithelial ovarian cancer patients. J. Exp. Clin. Cancer Res. 2017, 36, 63. [Google Scholar] [CrossRef]
- Xia, L.; Huang, W.; Tian, D.; Zhu, H.; Zhang, Y.; Hu, H.; Fan, D.; Nie, Y.; Wu, K. Upregulated FoxM1 expression induced by hepatitis B virus X protein promotes tumor metastasis and indicates poor prognosis in hepatitis B virus-related hepatocellular carcinoma. J. Hepatol. 2012, 57, 600–612. [Google Scholar] [CrossRef]
- Liang, S.K.; Hsu, C.C.; Song, H.L.; Huang, Y.C.; Kuo, C.W.; Yao, X.; Li, C.C.; Yang, H.C.; Hung, Y.L.; Chao, S.Y.; et al. FOXM1 is required for small cell lung cancer tumorigenesis and associated with poor clinical prognosis. Oncogene 2021, 40, 4847–4858. [Google Scholar] [CrossRef]
- Drouin, M.; Saenz, J.; Chiffoleau, E. C-Type Lectin-Like Receptors: Head or Tail in Cell Death Immunity. Front. Immunol. 2020, 11, 251. [Google Scholar] [CrossRef]
- Ryan, E.J.; Magaletti, D.; Draves, K.E.; Clark, E.A. Ligation of dendritic cell–associated lectin–1 induces partial maturation of human monocyte derived dendritic cells. Hum. Immunol. 2009, 70, 1–5. [Google Scholar] [CrossRef][Green Version]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Vaes, R.D.; Reynders, K.; Sprooten, J.; Nevola, K.T.; Rouschop, K.M.; Vooijs, M.; Garg, A.D.; Lambrecht, M.; Hendriks, L.E.; Rucevic, M.; et al. Identification of Potential Prognostic and Predictive Immunological Biomarkers in Patients with Stage I and Stage III Non-Small Cell Lung Cancer (NSCLC): A Prospective Exploratory Study. Cancers 2021, 13, 6259. [Google Scholar] [CrossRef]
- Errami, Y.; Naura, A.S.; Kim, H.; Ju, J.; Suzuki, Y.; El-Bahrawy, A.H.; Ghonim, M.A.; Hemeida, R.A.; Mansy, M.S.; Zhang, J.; et al. Apoptotic DNA Fragmentation May Be a Cooperative Activity between Caspase-activated Deoxyribonuclease and the Poly(ADP-ribose) Polymerase-regulated DNAS1L3, an Endoplasmic Reticulum-localized Endonuclease That Translocates to the Nucleus during Apoptosis. J. Biol. Chem. 2013, 288, 3460–3468. [Google Scholar] [CrossRef]
- Han, D.S.; Ni, M.; Chan, R.W.; Chan, V.W.; Lui, K.O.; Chiu, R.W.; Lo, Y.D. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020, 106, 202–214. [Google Scholar] [CrossRef]
- Chen, J.; Ding, J.; Huang, W.; Sun, L.; Chen, J.; Liu, Y.; Zhan, Q.; Gao, G.; He, X.; Qiu, G.; et al. DNASE1L3 as a Novel Diagnostic and Prognostic Biomarker for Lung Adenocarcinoma Based on Data Mining. Front. Genet. 2021, 12, 699242. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, H.; Ma, K.; Sun, J.; Feng, X.; Geng, J.; Gu, J.; Wang, W.; Zhang, H.; He, Y.; et al. Abnormal methylation of seven genes and their associations with clinical characteristics in early stage non-small cell lung cancer. Oncol. Lett. 2013, 5, 1211–1218. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, P.; Liu, L.; Bin, Y.; Huang, Y.; Chen, L.; Wen, L.; Zhang, R.; Tong, F.; Dong, X. T Cell-Mediated Tumor Killing-Related Classification of the Immune Microenvironment and Prognosis Prediction of Lung Adenocarcinoma. J. Clin. Med. 2022, 11, 7223. https://doi.org/10.3390/jcm11237223
Ding P, Liu L, Bin Y, Huang Y, Chen L, Wen L, Zhang R, Tong F, Dong X. T Cell-Mediated Tumor Killing-Related Classification of the Immune Microenvironment and Prognosis Prediction of Lung Adenocarcinoma. Journal of Clinical Medicine. 2022; 11(23):7223. https://doi.org/10.3390/jcm11237223
Chicago/Turabian StyleDing, Peng, Lichao Liu, Yawen Bin, Yu Huang, Lingjuan Chen, Lu Wen, Ruiguang Zhang, Fan Tong, and Xiaorong Dong. 2022. "T Cell-Mediated Tumor Killing-Related Classification of the Immune Microenvironment and Prognosis Prediction of Lung Adenocarcinoma" Journal of Clinical Medicine 11, no. 23: 7223. https://doi.org/10.3390/jcm11237223
APA StyleDing, P., Liu, L., Bin, Y., Huang, Y., Chen, L., Wen, L., Zhang, R., Tong, F., & Dong, X. (2022). T Cell-Mediated Tumor Killing-Related Classification of the Immune Microenvironment and Prognosis Prediction of Lung Adenocarcinoma. Journal of Clinical Medicine, 11(23), 7223. https://doi.org/10.3390/jcm11237223