
Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment





{kind=link}
{kind=link}
Abstract
1. Introduction
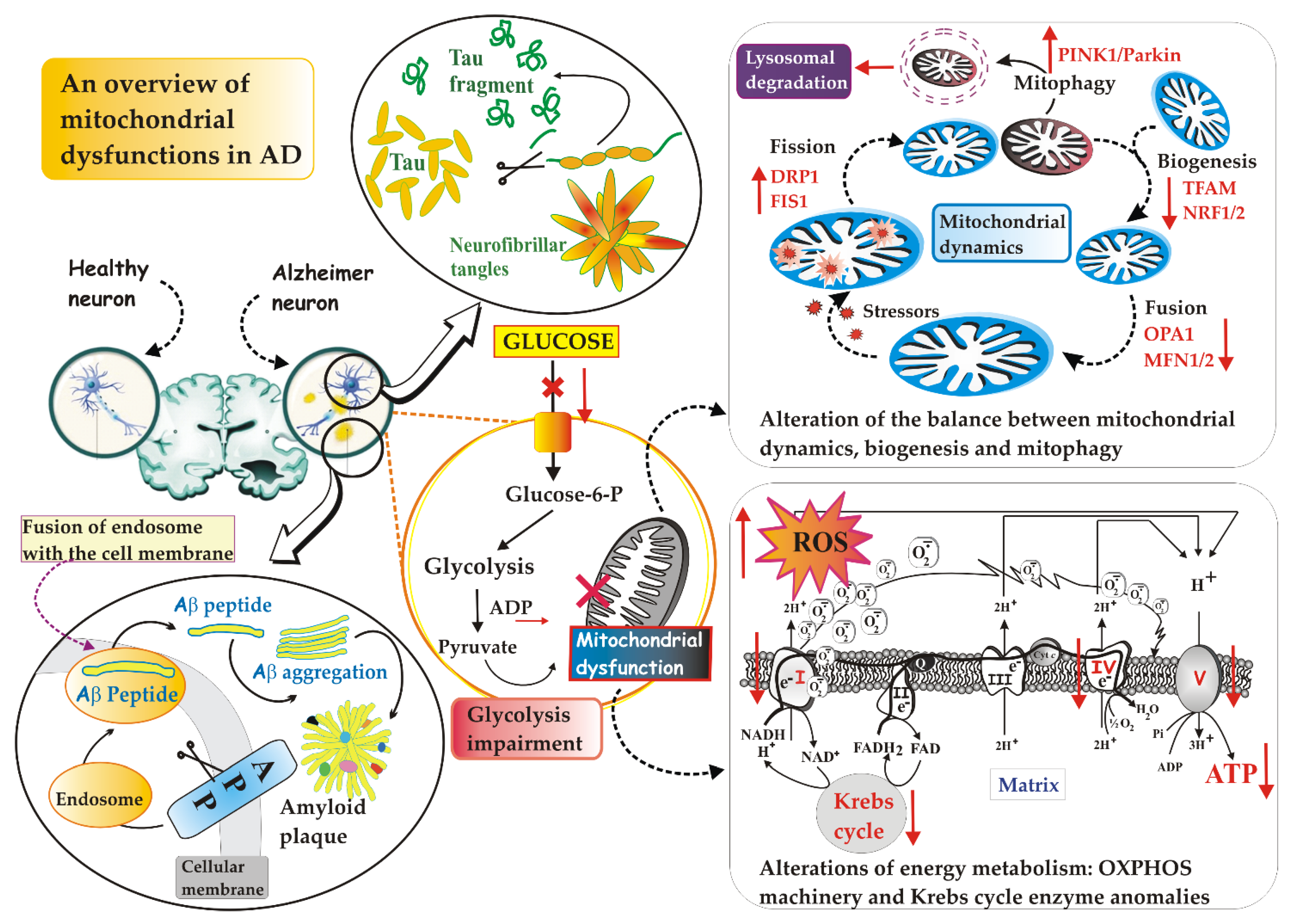
2. The Mitochondrion: Its Dysfunctions in AD
2.1. Defective Mitochondrial Dynamics in AD
2.2. Defective Mitophagy in AD
2.3. Defective Mitochondrial Biogenesis in AD
2.4. Defective Mitochondrial Energy Metabolism in AD
3. Therapeutic Strategies Targeting Mitochondria for the Treatment of AD
3.1. Therapeutic Strategies Targeting Mitochondrial Bioenergetics
3.2. Therapeutic Strategies Targeting Mitochondrial Biogenesis
3.3. Therapeutic Strategies Targeting Mitochondrial Dynamics
3.4. Therapeutic Strategies Targeting Mitophagy
3.5. Therapeutic Strategies Targeting Mitochondria-Dependent Oxidative Damage
3.6. The Use of Nanoparticles Targeting Mitochondria in the Treatment of AD
- Most drugs do not cross the BBB [208,209]: this is why various efforts have been made to overcome the BBB. Apart from parenteral administration, according to which the drug would be injected directly into the spinal fluid [208], other methods involve chemical modification of the drug, the use of viruses/exosomes as vectors for the administration of the drug to the central nervous system [210,211], intranasal administration, in order to circumvent the obstacle of the BBB [208], the destruction of the BBB by ultrasound or radiotherapy [212] and finally, strategies based on nanotechnology.
- Selectively targeting drugs to mitochondria in vivo [208] needs to take into account the existence of a proton gradient (negative inner Δψm), which produces a strong negative potential on the IMM of approximately −160–180 mV [208] and influences the entry of the drug into the mitochondria, the presence of phospholipid cardiolipin and, last but not least, the impermeability of the mitochondria. If we add to all this the fact that four parts are distinguishable in the mitochondria, namely the MOM, the MIM, the intermembrane space (IMS) and the mitochondrial matrix, the task becomes very difficult. Both high and small molecular weight lipophilic particles (< and >500 mw) can freely penetrate through the MOM, but the Δψm makes the MIM resistant to anionic molecules [215]. Therefore, these barriers prevent the potential drug from being delivered to the mitochondrial space.
3.7. Intercellular Mitochondrial Transfer as Potential Therapeutic Approach for AD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, X.; Li, G.; Zhang, Y.; Wu, Y.; Song, W. Modifications and Trafficking of APP in the Pathogenesis of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: Impact of genetic risk factors. Mol. Neurodegener. 2019, 14, 20. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal Alzheimer Aβ42 Accumulates in Multivesicular Bodies and Is Associated with Synaptic Pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Zoltowska, K.M.; Maesako, M.; Berezovska, O. Interrelationship between Changes in the Amyloid β 42/40 Ratio and Presenilin 1 Conformation. Mol. Med. 2016, 22, 329–337. [Google Scholar] [CrossRef]
- Kuperstein, I.; Broersen, K.; Benilova, I.; Rozenski, J.; Jonckheere, W.; Debulpaep, M.; Vandersteen, A.; Segers-Nolten, I.; Van Der Werf, K.; Subramaniam, V.; et al. Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 2010, 29, 3408–3420. [Google Scholar] [CrossRef]
- Jan, A.; Gokce, O.; Luthi-Carter, R.; Lashuel, H.A. The ratio of monomeric to aggregated forms of Abeta40 and Abeta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J. Biol. Chem. 2008, 283, 28176–28189. [Google Scholar] [CrossRef]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203. [Google Scholar] [CrossRef]
- Bilousova, T.; Miller, C.A.; Poon, W.W.; Vinters, H.V.; Corrada, M.; Kawas, C.; Hayden, E.Y.; Teplow, D.B.; Glabe, C.; Albay, R., 3rd; et al. Synaptic Amyloid-beta Oligomers Precede p-Tau and Differentiate High Pathology Control Cases. Am. J. Pathol. 2016, 186, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Hodson, R. Alzheimer’s disease. Nature 2018, 559, S1. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.; Hyman, B. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honige, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis. 2018, 64 (Suppl. S1), S611–S631. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropath. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64 (Suppl. S1), S567–S610. [Google Scholar] [CrossRef]
- Koike, H.; Iguchi, Y.; Sahashi, K.; Katsuno, M. Significance of Oligomeric and Fibrillar Species in Amyloidosis: Insights into Pathophysiology and Treatment. Molecules 2021, 26, 5091. [Google Scholar] [CrossRef]
- Lleó, A. Current Therapeutic Options for Alzheimer’s Disease. Curr. Genom. 2007, 8, 550–558. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Amat-Ur-Rasool, H.; Ahmed, M.; Hasnain, S.; Carter, W.J. Anti-Cholinesterase Combination Drug Therapy as a Potential Treatment for Alzheimer’s Disease. Brain Sci. 2021, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Eldufani, J.; Blaise, G. The role of acetylcholinesterase inhibitors such as neostigmine and rivastigmine on chronic pain and cognitive function in aging: A review of recent clinical applications. Alzheimer’s Dement. 2019, 5, 175–183. [Google Scholar] [CrossRef]
- Pyun, J.-M.; Ryoo, N.; Park, Y.H.; Kim, S.Y. Change in cognitive function according to cholinesterase inhibitor use and amyloid PET positivity in patients with mild cognitive impairment. Alzheimer’s Res. Ther. 2021, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, I.; Sorrentino, L.; Paoletti, A.; Marra, R.; Arbitrio, M. The State of The Art on Acetylcholinesterase Inhibitors in the Treatment of Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211029113. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gómez Moreta, M.; Burgos-Alonso, N.; Torrecilla, M.; Marco-Contelles, J.; Bruzos-Cidón, C. Efficacy of Acetylcholinesterase Inhibitors on Cognitive Function in Alzheimer’s Disease. Review of Reviews. Biomedicines 2021, 9, 1689. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, A.; Tripathi, T.; Kumar, A. Muscarinic and nicotinic acetylcholine receptor agonists: Current scenario in Alzheimer’s disease therapy. J. Pharm. Pharmacol. 2018, 70, 985–993. [Google Scholar] [CrossRef]
- Johnson, C.R.; Kangas, B.D.; Jutkiewicz, E.M.; Bergman, J.; Coop, A. Drug Design Targeting the Muscarinic Receptors and the Implications in Central Nervous System Disorders. Biomedicines 2022, 10, 398. [Google Scholar] [CrossRef]
- Zeng, H.; Zhang, Y.; Peng, L.; Shao, H.; Menon, N.K.; Yang, J.; Salomon, A.R.; Freidland, R.P.; Zagorski, M.G. Nicotine and amyloid formation. Biol. Psychiatry 2001, 49, 248–257. [Google Scholar] [CrossRef]
- Fu, H.J.; Liu, B.; Frost, J.L.; Lemere, C.A. Amyloid-beta immunotherapy for Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 197–206. [Google Scholar] [CrossRef]
- Vassilakopoulou, V.; Karachaliou, C.-E.; Evangelou, A.; Zikos, C.; Livaniou, E. Peptide-Based Vaccines for Neurodegenerative Diseases: Recent Endeavors and Future Perspectives. Vaccines 2021, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, X.; Lang, Y.; Li, Q.; Liu, X.; Cai, C.; Hao, J.; Li, G.; Yu, G. Low anticoagulant heparin oligosaccharides as inhibitors of BACE-1, the Alzheimer’s β-secretase. Carbohydr. Polym. 2016, 151, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.P.; Lefterov, I.M.; Staufenbiel, M.; Wolfe, D.; Huang, S.; Glorioso, J.C.; Walter, M.; Roth, M.G.; Lazo, J.S. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J. Biol. Chem. 2005, 280, 4079–4088. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372. [Google Scholar] [CrossRef]
- Beshir, S.A.; Aadithsoorya, A.M.; Parveen, A.; Goh, S.S.L.; Hussain, N.; Menon, V.B. Aducanumab Therapy to Treat Alzheimer’s Disease: A Narrative Review. Int. J. Alzheimers Dis. 2022, 2022, 9343514. [Google Scholar] [CrossRef]
- Walsh, S.; Merrick, R.; Milne, R.; Brayne, C. Aducanumab for Alzheimer’s disease? BMJ 2021, 374, 1682. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Haddad, H.W.; Malone, G.W.; Comardelle, N.J.; Degueure, A.E.; Kaye, A.M.; Kaye, A.D. Aducanumab, a Novel Anti-Amyloid Monoclonal Antibody, for the Treatment of Alzheimer’s Disease: A Comprehensive Review. Health Psychol. Res. 2022, 10, 31925. [Google Scholar] [CrossRef]
- Giacobini, E.; Gold, G. Alzheimer disease therapy—Moving from amyloid-β to tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef]
- Latina, V.; Giacovazzo, G.; Calissano, P.; Atlante, A.; La Regina, F.; Malerba, F.; Dell’Aquila, M.; Stigliano, E.; Balzamino, B.O.; Micera, A.; et al. Tau Cleavage Contributes to Cognitive Dysfunction in Strepto-Zotocin-Induced Sporadic Alzheimer’s Disease (sAD) Mouse Model. Int. J. Mol. Sci. 2021, 22, 12158. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, V.; Borreca, A.; Latina, V.; Giacovazzo, G.; Pignataro, A.; Krashia, P.; Natale, F.; Cocco, S.; Rinaudo, M.; Malerba, F.; et al. Passive immunotherapy for N-truncated tau ameliorates the cognitive deficits in two mouse Alzheimer’s disease models. Brain Commun. 2020, 2, fcaa039. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Neuropathology of the Alzheimer’s continuum: An update. Free Neuropathol. 2020, 1, 32. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells. Int. J. Mol. Sci. 2022, 23, 7722. [Google Scholar] [CrossRef]
- Buccellato, F.R.; D’Anca, M.; Fenoglio, C.; Scarpini, E.; Galimberti, D. Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef]
- Peng, P.; Gao, P.; Shi, L.; Chen, L.; Liu, J.; Long, J. Central and Peripheral Metabolic Defects Contribute to the Pathogenesis of Alzheimer’s Disease: Targeting Mitochondria for Diagnosis and Prevention. Antioxid. Redox. Signal. 2020, 32, 1188–1236. [Google Scholar] [CrossRef]
- Abe, T.; Tohgi, H.; Isobe, C.; Murata, T.; Sato, C. Remarkable increase in the concentration of 8-hydroxyguanosine in cerebrospinal fluid from patients with Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 447–450. [Google Scholar] [CrossRef]
- Cioffi, F.; Adam, R.H.I.; Bansal, R.; Broersen, K. A Review of Oxidative Stress Products and Related Genes in Early Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 83, 977–1001. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-W.; Han, R.; He, H.J.; Wang, Z.; Luan, X.Q.; Li, J.; Feng, L.; Chen, S.-Y.; Aman, Y.; Xie, C.-L. Delineating the Role of Mitophagy Inducers for Alzheimer Disease Patients. Aging Dis. 2021, 12, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Zhuang, X.-X.; Niu, Z.; Ai, R.; Lautrup, S.; Zheng, S.; Jiang, Y.; Han, R.; Gupta, T.S.; Cao, S.; et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat. Biomed. Eng. 2022, 6, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–1300. [Google Scholar] [CrossRef]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Drose, S.; Brandt, U.; Müller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santos, S.; Swerdlow, R.H.; Oliveira, C.R. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001, 15, 1439–1441. [Google Scholar] [CrossRef]
- Reiss, A.B.; Ahmed, S.; Dayaramani, C.; Glass, A.D.; Gomolin, I.H.; Pinkhasov, A.; Stecker, M.M.; Wisniewski, T.; De Leon, J. The role of mitochondrial dysfunction in Alzheimer’s disease: A potential pathway to treatment. Exp. Gerontol. 2022, 164, 111828. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Wu, H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J.; Barnham, K.J.; Duce, J.A.; Blake, R.E.; Masters, C.L.; Trounce, I.A. Copper-dependent inhibition of cytochrome c oxidase by Abeta (1–42) requires reduced methionine at residue 35 of the Abeta peptide. J. Neurochem. 2006, 99, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Mitochondrially-Targeted Therapeutic Strategies for Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 753–771. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.K.; Jara, C.; Park-Kang, H.S.; Polanco, C.M.; Tapia, D.; Alarcón, F.; de la Peña, A.; Llanquinao, J.; Vargas-Mardones, G.; Indo, J.A.; et al. Synaptic Mitochondria: An Early Target of Amyloid-β and Tau in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 84, 1391–1414. [Google Scholar] [CrossRef]
- Epremyan, K.K.; Goleva, T.N.; Zvyagilskaya, R.A. Effect of Tau Protein on Mitochondrial Functions. Biochemistry 2022, 87, 689–701. [Google Scholar] [CrossRef]
- Lee, S.-E.; Kwon, D.; Shin, D.; Kong, D.; Kim, N.G.; Kim, H.-Y.; Kim, M.-J.; Choi, S.W.; Kang, K.-S. Accumulation of APP-CTF induces mitophagy dysfunction in the iNSCs model of Alzheimer’s disease. Cell Death Discov. 2022, 8, 1. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Pulina, M.V.; Hopkins, M.; Haroutunian, V.; Greengard, P.; Bustos, V. C99 selectively accumulates in vulnerable neurons in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 273–282. [Google Scholar] [CrossRef]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Oikawa, N.; Walter, J. Presenilins and γ-Secretase in Membrane Proteostasis. Cells 2019, 8, 209. [Google Scholar] [CrossRef]
- Atlante, A.; Gagliardi, S.; Marra, E.; Calissano, P. Neuronal apoptosis in rats is accompanied by rapid impairment of cellular respiration and is prevented by scavengers of reactive oxygen species. Neurosci. Lett. 1998, 245, 127–130. [Google Scholar] [CrossRef]
- Bobba, A.; Petragallo, V.A.; Marra, E.; Atlante, A. Alzheimer’s proteins, oxidative stress, and mitochondrial dysfunction interplay in a neuronal model of Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2010, 2010, 621870. [Google Scholar] [CrossRef][Green Version]
- Bobba, A.; Amadoro, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Dissecting the molecular mechanism by which NH2htau and Aβ1-42 peptides impair mitochondrial ANT-1 in Alzheimer disease. Biochim. Biophys. Acta 2013, 1827, 848–860. [Google Scholar] [CrossRef]
- Atlante, A.; de Bari, L.; Bobba, A.; Amadoro, G. A disease with a sweet tooth: Exploring the Warburg effect in Alzheimer’s disease. Biogerontology 2017, 18, 301–319. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Majekova, M.; Medina, M.; Valoti, M. Key Targets for Multi-Target Ligands Designed to Combat Neurodegeneration. Front. Neurosci. 2016, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, J.M.P.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Pedriali, G.; Sbano, L.; Iannitti, T.; Giorgi, C.; Pinton, P. Intersection of mitochondrial fission and fusion machinery with apoptotic pathways: Role of Mcl-1. Biol. Cell 2016, 108, 279–293. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Stetler, R.A.; Leak, R.; Gao, Y.; Chen, J. The dynamics of the mitochondrial organelle as a potential therapeutic organelle. J. Cereb. Blood Flow Metab. 2013, 33, 22–32. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Nicolin, V.; Nori, S.L.; Calissano, P. Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: The pink-parkin pathway. Front. Aging Neurosci. 2014, 6, 18. [Google Scholar] [CrossRef]
- Rey, F.; Ottolenghi, S.; Zuccotti, G.V.; Samaja, M.; Carelli, S. Mitochondrial dysfunctions in neurodegenerative diseases: Role in disease pathogenesis, strategies for analysis and therapeutic prospects. Neural Regen. Res. 2022, 17, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Babylon, L.; Schmitt, F.; Franke, Y.; Hubert, T.; Eckert, G.P. Effects of Combining Biofactors on Bioenergetic Parameters, Aβ Levels and Survival in Alzheimer Model Organisms. Int. J. Mol. Sci. 2022, 23, 8670. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Drerup, C.M. Axonal Transport and Mitochondrial Function in Neurons. Front. Cell Neurosci. 2019, 13, 373. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Hu, D.; Liu, Z.; Qi, X. Mitochondrial Quality Control Strategies: Potential Therapeutic Targets for Neurodegenerative Diseases? Front. Neurosci. 2021, 15, 746873. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Oliver, D.; Reddy, P.H. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019, 8, 961. [Google Scholar] [CrossRef]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. Mitochondrial Fission Protein 1: Emerging Roles in Organellar Form and Function in Health and Disease. Front. Endocrinol. 2021, 12, 660095. [Google Scholar] [CrossRef]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Munshi, S.; Frank, D.E.; Gibson, G.E. Abnormal Glucose Metabolism in Alzheimer’s Disease: Relation to Autophagy/Mitophagy and Therapeutic Approaches. Neurochem. Res. 2015, 40, 2557–2569. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Palacín, M.; Zorzano, A. Mitochondrial dynamics: Coupling mitochondrial fitness with healthy aging. Trends Mol. Med. 2017, 23, 201–215. [Google Scholar] [CrossRef]
- Simula, L.; Nazio, F.; Campello, S. The Mitochondrial dynamics in cancer and immune-surveillance. Semin. Cancer Biol. 2017, 47, 29–42. [Google Scholar] [CrossRef]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Fry, D.; Suneetha, Y.; Sesaki, H.; Reddy, P.H. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 4881–4897. [Google Scholar] [CrossRef]
- Guha, S.; Johnson, G.V.W.; Nehrke, K. The Crosstalk between Pathological Tau Phosphorylation and Mitochondrial Dysfunction as a Key to Understanding and Treating Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 5103–5120. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef] [PubMed]
- de la Cueva, M.; Antequera, D.; Ordoñez-Gutierrez, L.; Wandosell, F.; Camins, C.; Carro, E.; Bartolome, F. Amyloid-β impairs mitochondrial dynamics and autophagy in Alzheimer’s disease experimental models. Sci. Rep. 2022, 12, 10092. [Google Scholar] [CrossRef]
- Xie, N.; Wang, C.; Lian, Y.; Wu, C.; Zhang, H.; Zhang, Q. Inhibition of mitochondrial fission attenuates a beta-induced microglia apoptosis. Neuroscience 2014, 256, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Vincow, E.S.; Merrihew, G.; Thomas, R.E.; Shulman, N.J.; Beyer, R.P.; MacCoss, M.J.; Pallanck, L.J. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 6400–6405. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Zimmermann, M.; Reichert, A.S. How to get rid of mitochondria: Crosstalk and regulation of multiple mitophagy pathways. Biol. Chem. 2017, 399, 29–45. [Google Scholar] [CrossRef]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol. Psychiatry 2022, 1–15. [Google Scholar] [CrossRef]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14, 612757. [Google Scholar] [CrossRef]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, T.; Ge, X.; Chen, J.; Zhao, Y.; Fu, J. Parkin overexpression attenuates Aβ-induced mitochondrial dysfunction in HEK293 cells by restoring impaired mitophagy. Life Sci. 2020, 244, 117322. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Md Hasan-Olive, M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Signore, A.P.; Iwai, M.; Cao, G.; Gao, Y.; Chen, J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 2008, 39, 3057–3063. [Google Scholar] [CrossRef] [PubMed]
- Cantò, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Han, S.; Zhang, M.; Jeong, Y.Y.; Margolis, D.J.; Cai, Q. The role of mitophagy in the regulation of mitochondrial energetic status in neurons. Autophagy 2021, 17, 4182–4201. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef]
- Yamada, E.S.; Respondek, G.; Müssner, S.; de Andrade, A.; Höllerhage, M.; Depienne, C.; Rastetter, A.; Tarze, A.; Friguet, B.; Salama, M.; et al. Annonacin, a natural lipophilic mitochondrial complex I inhibitor, increases phosphorylation of tau in the brain of FTDP-17 transgenic mice. Exp. Neurol. 2014, 253, 113–125. [Google Scholar] [CrossRef]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W.E. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Qi, G.; Diaz Brinton, R.; Yin, F. Mitochondria-Targeted Therapeutics for Alzheimer’s Disease: The Good, the Bad, the Potential. Antioxid. Redox. Signal. 2021, 34, 611–630. [Google Scholar] [CrossRef]
- Kanamaru, T.; Kamimura, N.; Yokota, T.; Iuchi, K.; Nishimaki, K.; Takami, S.; Akashiba, H.; Shitaka, Y.; Katsura, K.-I.; Kimura, K. Oxidative stress accelerates amyloid deposition and memory impairment in a double-transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2015, 587, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Liang, J.; Zhou, B. Glucose Metabolic Dysfunction in Neurodegenerative Diseases-New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G. Modulation of mitochondrial bioenergetics as a therapeutic strategy in Alzheimer’s disease. Neural Regen. Res. 2018, 13, 19–25. [Google Scholar] [CrossRef]
- Pantiya, P.; Thonusin, C.; Chattipakorn, N.; Chattipakorn, S.C. Mitochondrial abnormalities in neurodegenerative models and possible interventions: Focus on Alzheimer’s disease, Parkinson’s disease, Huntington’s disease. Mitochondrion 2020, 55, 14–47. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; La Piana, G.; Calissano, P.; Atlante, A. Glycolytic enzyme upregulation and numbness of mitochondrial activity characterize the early phase of apoptosis in cerebellar granule cells. Apoptosis 2015, 20, 10–28. [Google Scholar] [CrossRef]
- Newington, J.T.; Pitts, A.; Chien, A.; Arseneault, R.; Schubert, D.; Cumming, R.C. Amyloid beta resistance in nerve cell lines is mediated by the Warburg effect. PLoS ONE 2011, 6, e19191. [Google Scholar] [CrossRef]
- Santangelo, R.; Giuffrida, M.L.; Satriano, C.; Tomasello, M.F.; Zimbone, S.; Copani, A. β-amyloid monomers drive up neuronal aerobic glycolysis in response to energy stressors. Aging 2021, 13, 18033–18050. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Gordon, B.A.; Goyal, M.S.; Su, Y.; Blazey, T.M.; Durbin, T.J.; Couture, L.E.; Christensen, J.J.; Jafri, H.; Morris, J.C.; et al. Aerobic glycolysis and tau deposition in preclinical Alzheimer’s disease. Neurobiol. Aging 2018, 67, 95–98. [Google Scholar] [CrossRef]
- Kato, T.; Inui, Y.; Nakamura, A.; Ito, K. Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Res. Rev. 2016, 30, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.T.; Leon, G.V.O.; Provance, D.W., Jr.; de Mello, F.G.; Sorenson, M.M.; Salerno, V.P. Exogenous β-amyloid peptide interferes with GLUT4 localization in neurons. Brain Res. 2015, 1615, 42–50. [Google Scholar] [CrossRef]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox. Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Rodríguez, L.R.; Lapeña-Luzón, T.; Benetó, N.; Beltran-Beltran, V.; Pallardó, F.V.; Gonzalez-Cabo, P.; Navarro, J.A. Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases? Antioxidants 2022, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, G.K.; Gupta, A.; Pahwa, P.; Khullar, N.; Singh, S.; Navik, U.; Kumar, S.; Mastana, S.S.; Reddy, A.P.; Reddy, P.H.; et al. Targeting mitochondrial bioenergetics as a promising therapeutic strategy in metabolic and neurodegenerative diseases. Biomed. J. 2022, 45, 733–748. [Google Scholar] [CrossRef]
- Austad, S.N.; Ballinger, S.; Buford, T.W.; Carter, C.S.; Smith, D.L., Jr.; Darley-Usmar, V.; Zhang, J. Targeting whole body metabolism and mitochondrial bioenergetics in the drug development for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 511–531. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- McDade, E.; Llibre-Guerra, J.J.; Holtzman, D.M.; Morris, J.C.; Bateman, R.J. The informed road map to prevention of Alzheimer Disease: A call to arms. Mol. Neurodegener. 2021, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Mckean, N.E.; Handley, R.R.; Snell, R.G. A Review of the Current Mammalian Models of Alzheimer’s Disease and Challenges That Need to Be Overcome. Int. J. Mol. Sci. 2021, 22, 13168. [Google Scholar] [CrossRef] [PubMed]
- Belenguer, P.; Duarte, J.M.N.; Schuck, P.F.; Ferreira, G.C. Mitochondria and the brain: Bioenergetics and beyond. Neurotox. Res. 2019, 36, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial complex I as a therapeutic target for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 483–495. [Google Scholar] [CrossRef]
- Ke, J.; Tian, Q.; Xu, Q.; Fu, Z.; Fu, Q. Mitochondrial dysfunction: A potential target for Alzheimer’s disease intervention and treatment. Drug Discov. Today 2021, 26, 1991–2002. [Google Scholar] [CrossRef]
- Goldberg, J.; Currais, A.; Prior, M.; Fischer, W.; Chiruta, C.; Ratliff, E.; Daugherty, D.; Dargusch, R.; Finley, K.; Esparza-Moltó, P.B.; et al. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell 2018, 17, e12715. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Sui, X.; Zhang, Y.; Sun, Q.; Qu, Z.; Luo, X.; Chang, R.C.C.; Ni, J.; Liu, J.; Yang, X. Identification of novel key molecules involved in spatial memory impairment in triple transgenic mice of Alzheimer’s disease. Mol. Neurobiol. 2017, 54, 3843–3858. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Muraleva, N.A.; Skulachev, V.P.; Kolosova, N.G. Alzheimer’s disease-like pathology in senescence accelerated OXYS rats can be partially retarded with mitochondria-targeted antioxidant SkQ1. J. Alzheimer’s Dis. 2014, 38, 681–694. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Kandimalla, R. Mitochondria targeted small molecule SS31: A potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet 2017, 26, 1483–1496. [Google Scholar] [CrossRef]
- Yao, J.; Zhao, L.; Mao, Z.; Chen, S.; Wong, K.C.; To, J.; Brinton, R.D. Potentiation of brain mitochondrial function by S-equol and R/S-equol estrogen receptor β-selective phytoSERM treatments. Brain Res. 2013, 1514, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Buchke, S.; Sharma, M.; Bora, A.; Relekar, M.; Bhanu, P.; Kumar, J. Mitochondria-Targeted, Nanoparticle-Based Drug-Delivery Systems: Therapeutics for Mitochondrial Disorders. Life 2022, 12, 657. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-b peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef]
- Pettegrew, J.W.; Levine, J.; McClure, R.J. Acetyl-L-carnitine physical-chemical, metabolic, and therapeutic properties: Relevance for its mode of action in Alzheimer’s disease and geriatric depression. Mol. Psychiatry 2000, 5, 616–632. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Mitochondrial links between brain aging and Alzheimer’s disease. Transl. Neurodegener. 2021, 10, 33. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.W.; Shang, Y.; Jiang, L.; Shi, T.; Wang, L. The peroxisome proliferators activated receptor-gamma agonists as therapeutics for the treatment of Alzheimer’s disease and mild-to-moderate Alzheimer’s disease: A metaanalysis. Int. J. Neurosci. 2016, 126, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 97–101. [Google Scholar] [CrossRef]
- Hamano, T.; Shirafuji, N.; Makino, C.; Yen, S.H.; Kanaan, N.M.; Ueno, A.; Suzuki, J.; Ikawa, M.; Matsunaga, A.; Yamamura, O.; et al. Pioglitazone prevents tau oligomerization. Biochem. Biophys. Res. Commun. 2016, 478, 1035–1042. [Google Scholar] [CrossRef]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-c agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone. A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, Z.; Shen, R.; Zhong, W.; Zheng, H.; Chen, Z.; Zhu, J. Resveratrol improves mitochondrial biogenesis function and activates PGC-1α pathway in a preclinical model of early brain injury following subarachnoid hemorrhage. Front. Mol. Biosci. 2021, 8, 223. [Google Scholar] [CrossRef]
- Onyango, I.G.; Lu, J.; Rodova, M.; Lezi, E.; Crafter, A.B.; Swerdlow, R.H. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim. Biophys. Acta 2010, 1802, 228–234. [Google Scholar] [CrossRef]
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Regulation of neuronal bioenergetics as a therapeutic strategy in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1467–1482. [Google Scholar] [CrossRef]
- Ganguly, B.B.; Kadam, N.N. Triplication of HSA21 on alterations in structure and function of mitochondria. Mitochondrion 2022, 65, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.T.; Shaw, R.M.; Zhu, J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef]
- Kuruva, C.S.; Manczak, M.; Yin, X.; Ogunmokun, G.; Reddy, A.P.; Reddy, P.H. Aqua-soluble DDQ reduces the levels of Drp1 and Abeta and inhibits abnormal interactions between Abeta and Drp1 and protects Alzheimer’s disease neurons from Abeta- and Drp1-induced mitochondrial and synaptic toxicities. Hum. Mol. Genet. 2017, 26, 3375–3395. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2017, 9, 6128–6143. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, J.T.; Miao, D.; Wu, Z.C.; Tan, M.S.; Tan, L. Targeting the mTOR signaling network for Alzheimer’s disease therapy. Mol. Neurobiol 2014, 49, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Eckert, S.H.; Gaca, J.; Kolesova, N.; Friedland, K.; Eckert, G.P.; Muller, W.E. Mitochondrial pharmacology of dimebon (latrepirdine) calls for a new look at its possible therapeutic potential in Alzheimer’s disease. Aging Dis. 2018, 9, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Hwang, E.S. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell 2009, 8, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, Q.; Wu, X.; Zhang, D.; Xing, D. Activation of PKA/SIRT1 signaling pathway by photobiomodulation therapy reduces A beta levels in Alzheimer’s disease models. Aging Cell 2020, 19, e13054. [Google Scholar] [CrossRef] [PubMed]
- Esselun, C.; Theyssen, E.; Eckert, G.P. Effects of Urolithin A on Mitochondrial Parameters in a Cellular Model of Early Alzheimer Disease. Int. J. Mol. Sci. 2021, 22, 8333. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Mitophagy enhancers against phosphorylated Tau-induced mitochondrial and synaptic toxicities in Alzheimer disease. Pharmacol. Res. 2021, 174, 105973. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Protective effects of mitophagy enhancers against amyloid beta-induced mitochondrial and synaptic toxicities in Alzheimer disease. Hum. Mol. Genet. 2022, 31, 423–439. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Alvir, R.V.; Pradeepkiran, J.A.; Hindle, A.; Vijayan, M.; Ramasubramaniam, B.; Kumar, S.; Reddy, A.P.; Reddy, P.H. A Combination Therapy of Urolithin A+EGCG Has Stronger Protective Effects than Single Drug Urolithin A in a Humanized Amyloid Beta Knockin Mice for Late-Onset Alzheimer’s Disease. Cells 2022, 11, 2660. [Google Scholar] [CrossRef]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Rege, S.D.; Geetha, T.; Griffin, G.D.; Broderick, T.L.; Babu, J.R. Neuroprotective effects of resveratrol in Alzheimer disease pathology. Front. Aging Neurosci. 2014, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, T.; Li, W.; Gao, N.; Zhang, T. Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol. Lett. 2018, 282, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; De Amicis, F.; Sirianni, R.; Sinicropi, M.S.; Puoci, F.; Casaburi, I.; Saturnino, C.; Pezzi, V. Progress to improve oral bioavailability and beneficial effects of resveratrol. Int. J. Mol. Sci. 2019, 20, 1381. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Benson, G.; Schwarz, C.; Köbe, T.; Grittner, U.; Schmitz, D.; Sigrist, S.J.; Bohlken, J.; Stekovic, S.; Madeo, F.; et al. The effect of spermidine on memory performance in older adults at risk for dementia: A randomized controlled trial. Cortex 2018, 109, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.E.; Saleh, T.M.; Kalisch, B.E. Naturally Occurring Antioxidant Therapy in Alzheimer’s Disease. Antioxidants 2022, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Wongrakpanich, A.; Geary, S.M.; Joiner, M.-L.A.; Anderson, M.E.; Salem, A.K. Mitochondria-targeting particles. Nanomedicine 2014, 9, 2531–2543. [Google Scholar] [CrossRef]
- Yatin, S.M.; Varadarajan, S.; Butterfield, D.A. Vitamin E prevents Alzheimer’s amyloid b-peptide (1–42)-induced neuronal protein oxidation and reactive oxygen species production. J. Alzheimer’s Dis. 2000, 2, 123–131. [Google Scholar] [CrossRef]
- Fukui, K.; Takatsu, H.; Shinkai, T.; Suzuki, S.; Abe, K.; Urano, S. Appearance of amyloid b-like substances and delayed-type apoptosis in rat hippocampus CA1 region through aging and oxidative stress. J. Alzheimer’s Dis. 2005, 8, 299–309. [Google Scholar] [CrossRef]
- Smith, G.; Gallo, G. To mdivi-1 or not to mdivi-1: Is that the question? Dev. Neurobiol. 2017, 77, 1260–1268. [Google Scholar] [CrossRef]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s Disease: Current Therapeutic Significance and Future Prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-J.; Li, Z.-H.; Liu, L.; Tang, W.-X.; Wang, Y.; Dong, M.-R.; Xiao, C. Curcumin Attenuates Beta-Amyloid-Induced Neuroinflammation via Activation of Peroxisome Proliferator-Activated Receptor-Gamma Function in a Rat Model of Alzheimer’s Disease. Front. Pharmacol. 2016, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NFKB/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, M.O. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3b pathway in the mouse hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Smith, A.; Lin, X.; Yuan, F.; Copes, N.; Delic, V.; Tan, J.; Cao, C.; Shytle, R.D.; Bradshaw, P.C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2011, 26, 507–521. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Bellver-Sanchis, A.; Izquierdo, V.; Corpas, R.; Roig-Soriano, J.; Chillón, M.; Andres-Lacueva, C.; Somogyvári, M.; Sőti, C.; Sanfeliu, C.; et al. The pleiotropic neuroprotective effects of resveratrol in cognitive decline and Alzheimer’s disease pathology: From antioxidant to epigenetic therapy. Ageing Res. Rev. 2021, 67, 101271. [Google Scholar] [CrossRef]
- Kumar, A.; Naidu, P.S.; Seghal, N.; Padi, S.S.V. Neuroprotective effects of resveratrol against intracerebroventricular colchicine-induced cognitive impairment and oxidative stress in rats. Pharmacology 2007, 79, 17–26. [Google Scholar] [CrossRef]
- Corpas, R.; Griñán-Ferré, C.; Rodríguez-Farré, E.; Pallàs, M.; Sanfeliu, C. Resveratrol induces brain resilience against alzheimer neurodegeneration through proteostasis enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef]
- Wahl, D.; Bernier, M.; Simpson, S.J.; De Cabo, R.; Le Couteur, D.G. Future directions of resveratrol research. Nutr. Health Aging 2018, 4, 287–290. [Google Scholar] [CrossRef]
- Ahmed, T.; Javed, S.; Javed, S.; Tariq, A.; Šamec, D.; Tejada, S.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Resveratrol and Alzheimer’s Disease: Mechanistic Insights. Mol. Neurobiol. 2017, 54, 2622–2635. [Google Scholar] [CrossRef]
- Rege, S.; Geetha, T.; Broderick, T.; Babu, J. Resveratrol Protects β Amyloid-Induced Oxidative Damage and Memory Associated Proteins in H19-7 Hippocampal Neuronal Cells. Curr. Alzheimer Res. 2015, 12, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.J.; Fischer, N.; Efferth, T. Phytochemicals as inhibitors of NF-κB for treatment of Alzheimer’s disease. Pharmacol. Res. 2018, 129, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Johri, A. Disentangling Mitochondria in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11520. [Google Scholar] [CrossRef] [PubMed]
- Ambekar, T.; Pawar, J.; Rathod, R.; Patel, M.; Fernandes, V.; Kumar, R.; Singh, S.B.; Khatri, D.K. Mitochondrial quality control: Epigenetic signatures and therapeutic strategies. Neurochem. Int. 2021, 148, 105095. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q10 Analogues: Benefits and Challenges for Therapeutics. Antioxidants 2021, 10, 236. [Google Scholar] [CrossRef]
- Tauskela, J.S. MitoQ—A mitochondria-targeted antioxidant. IDrugs 2007, 10, 399–412. [Google Scholar]
- Zhang, Y.; Yang, H.; Wei, D.; Zhang, X.; Wang, J.; Wu, X.; Chang, J. Mitochondria-targeted nanoparticles in treatment of neurodegenerative diseases. Exploration 2021, 1, 20210115. [Google Scholar] [CrossRef]
- Fang, J.; Pieper, A.A.; Nussinov, R.; Lee, G.; Bekris, L.; Leverenz, J.B.; Cummings, J.; Cheng, F. Harnessing endophenotypes and network medicine for Alzheimer’s drug repurposing. Med. Res. Rev. 2020, 40, 2386–2426. [Google Scholar] [CrossRef]
- Duskey, J.T.; Belletti, D.; Pederzoli, F.; Vandelli, M.A.; Forni, F.; Ruozi, B.; Tosi, G. Current Strategies for the Delivery of Therapeutic Proteins and Enzymes to Treat Brain Disorders. Int. Rev. Neurobiol. 2017, 137, 1–28. [Google Scholar] [CrossRef]
- Colao, I.L.; Corteling, R.; Bracewell, D.; Wall, I. Manufacturing Exosomes: A Promising Therapeutic Platform. Trends Mol. Med. 2018, 24, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Poovaiah, N.; Davoudi, Z.; Peng, H.; Schlichtmann, B.; Mallapragada, S.; Narasimhan, B.; Wang, Q. Treatment of neurodegenerative disorders through the blood-brain barrier using nanocarriers. Nanoscale 2018, 10, 16962. [Google Scholar] [CrossRef] [PubMed]
- Durazo, S.A.; Kompella, U.B. Functionalized nanosystems for targeted mitochondrial delivery. Mitochondrion 2012, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Mu, W.; Wei, D.; Zhang, Y.; Duan, Y.; Gao, J.-X.; Gong, X.Q.; Wang, H.J.; Wu, X.L.; Tao, H.; et al. A Novel Targeted and High-Efficiency Nanosystem for Combinational Therapy for Alzheimer’s Disease. Adv. Sci. 2020, 7, 1902906. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Liang, M.Z.; Chen, L. Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl. Neurodegener. 2019, 8, 17. [Google Scholar] [CrossRef]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef]
- Rolandi, E.; Zaccaria, D.; Vaccaro, R.; Abbondanza, S.; Pettinato, L.; Davin, A.; Guaita, A. Estimating the potential for dementia prevention through modifiable risk factors elimination in the real-world setting: A population-based study. Alzheimer’s Res. Ther. 2020, 12, 94. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atlante, A.; Amadoro, G.; Latina, V.; Valenti, D. Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment. J. Clin. Med. 2022, 11, 6742. https://doi.org/10.3390/jcm11226742
Atlante A, Amadoro G, Latina V, Valenti D. Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment. Journal of Clinical Medicine. 2022; 11(22):6742. https://doi.org/10.3390/jcm11226742
Chicago/Turabian StyleAtlante, Anna, Giuseppina Amadoro, Valentina Latina, and Daniela Valenti. 2022. "Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment" Journal of Clinical Medicine 11, no. 22: 6742. https://doi.org/10.3390/jcm11226742
APA StyleAtlante, A., Amadoro, G., Latina, V., & Valenti, D. (2022). Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment. Journal of Clinical Medicine, 11(22), 6742. https://doi.org/10.3390/jcm11226742