

The Pancreas and Known Factors of Acute Pancreatitis

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
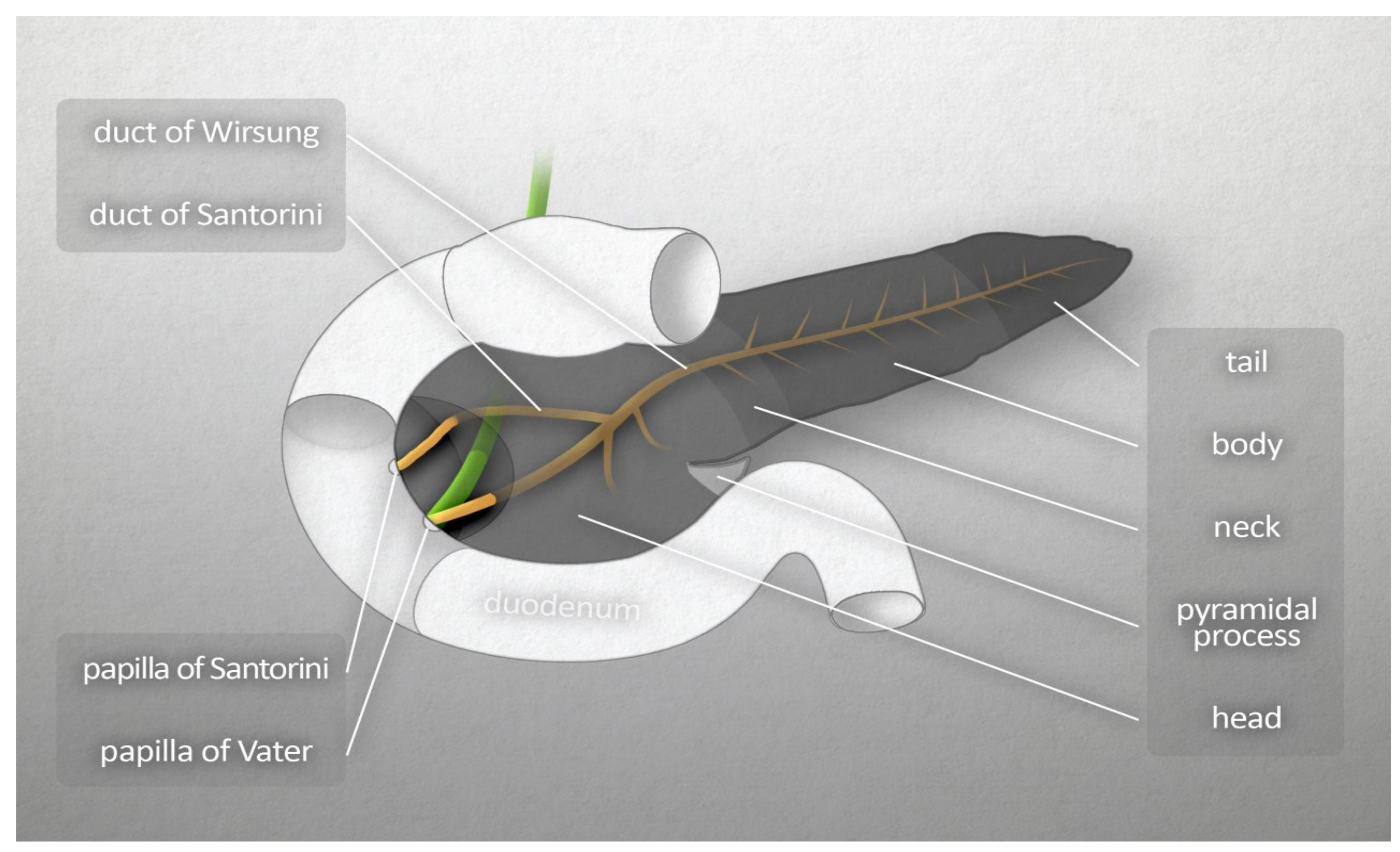
2. Morphology and Physiology of the Pancreas
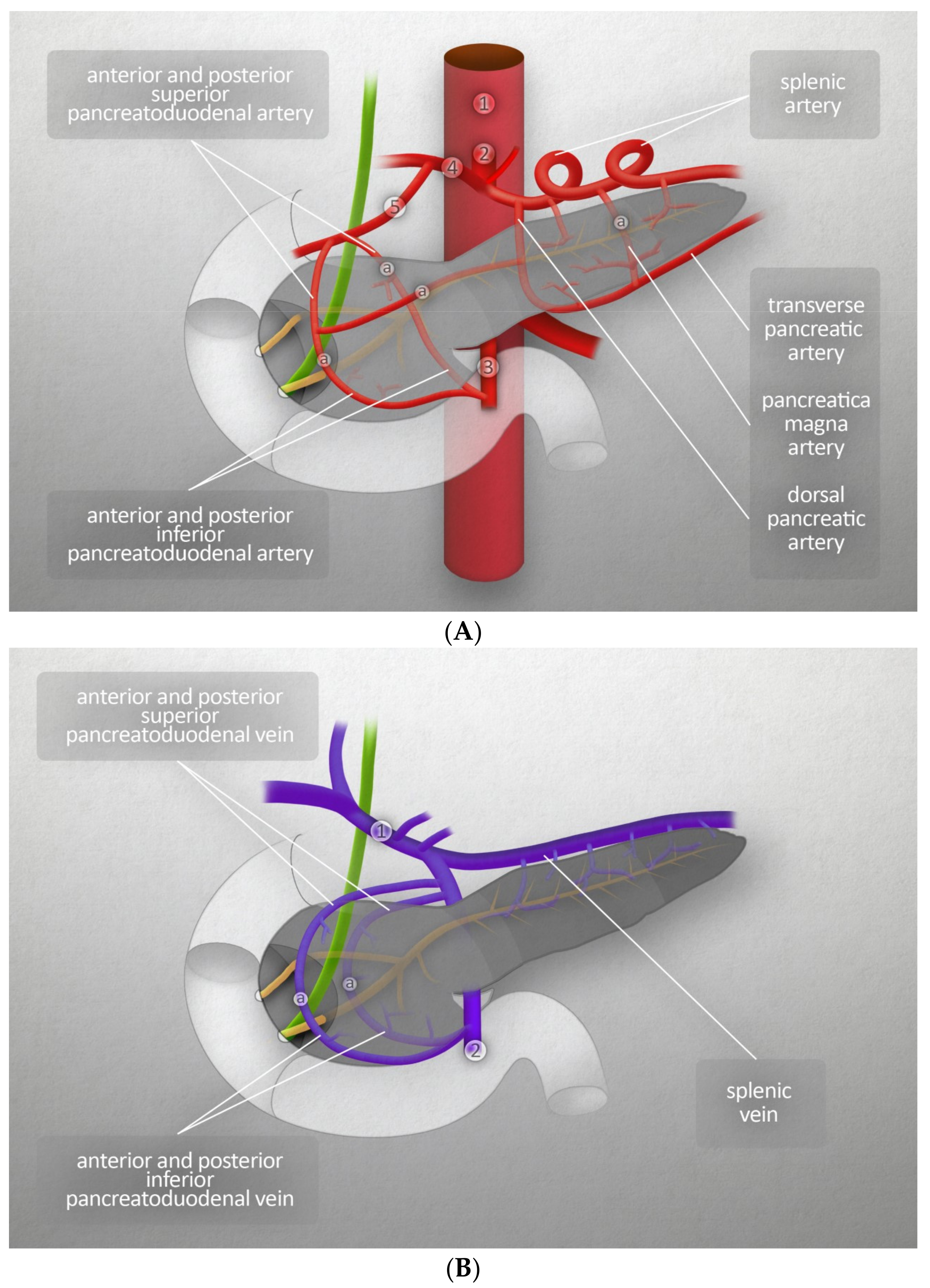
2.1. Vascularization
2.2. Innervation
2.3. Lymphatic System
2.4. Exocrine and Endocrine Functions
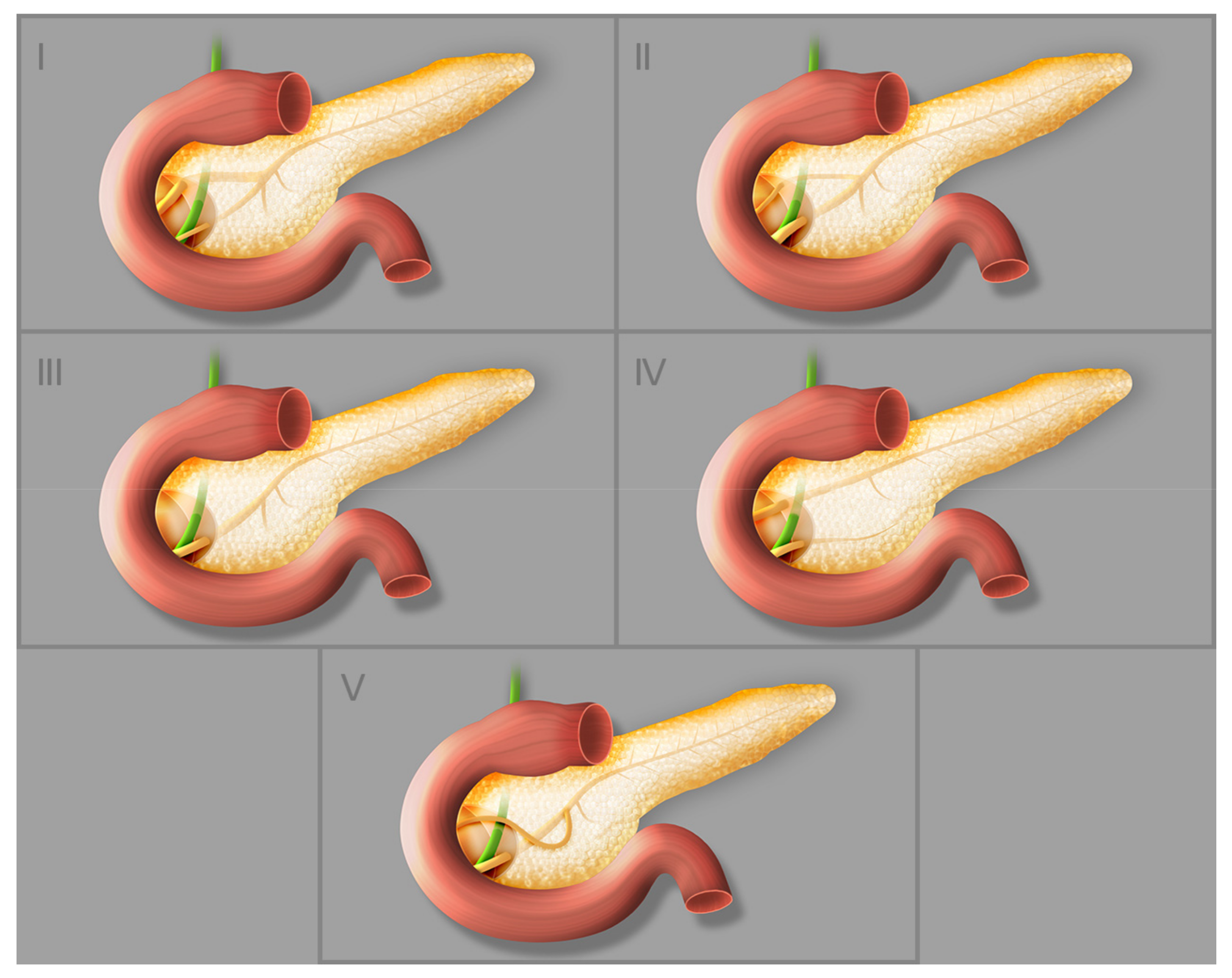
3. Anatomical Variations
4. Embryology and Histology of the Pancreas
4.1. Embryology
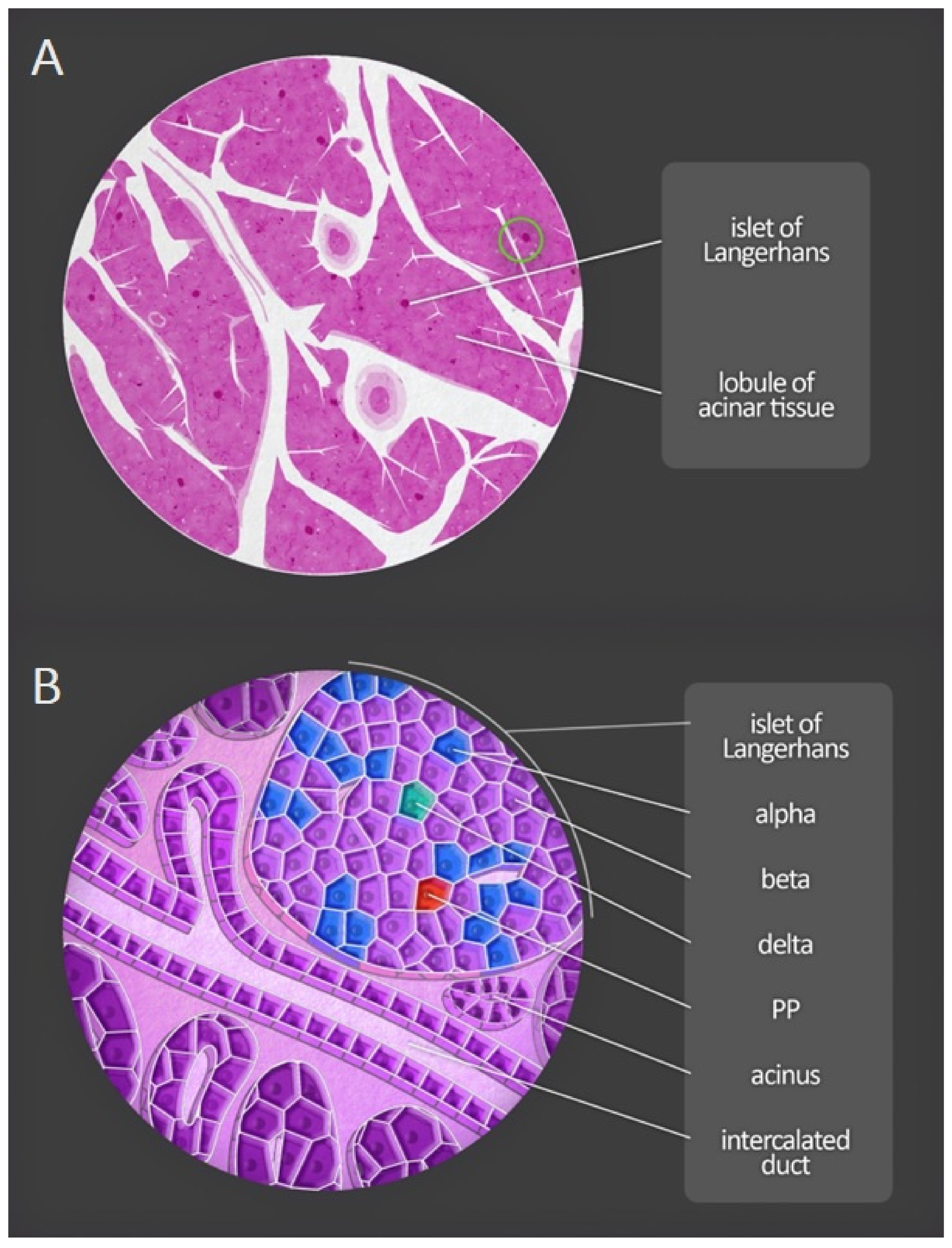
4.2. Histology
4.3. Histopathology of Acute Pancreatitis
5. The Course of Pancreatitis
5.1. Acute Pancreatitis
5.2. Recurrent Acute Pancreatitis
5.3. Chronic Pancreatitis
5.4. Clinical Aspects
6. Known Risk Factors and Pathomechanism of Acute Pancreatitis
6.1. Gallstones
6.2. Alcohol Abuse
6.3. Hypertriglyceridemia
6.4. Endoscopic Retrograde Cholangiopancreatography (ERCP)
6.5. Pancreas Divisium
6.6. Intraduct Papillary Mucinous Tumor
6.7. Autoimmune Pancreatitis
6.8. Genetic Risk
6.9. Rare Causes
6.9.1. Hypercalcemia
6.9.2. Drugs
6.9.3. Viruses
6.10. Clinical Aspects
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dimitriou, I.; Katsourakis, A.; Nikolaidou, E.; Noussios, G. The Main Anatomical Variations of the Pancreatic Duct System: Review of the Literature and Its Importance in Surgical Practice. J. Clin. Med. Res. 2018, 10, 370–375. [Google Scholar] [CrossRef]
- Suda, K.; Nobukawa, B.; Takase, M.; Hayashi, T. Pancreatic segmentation on an embryological and anatomical basis. J. Hepato-Biliary-Pancreat. Surg. 2006, 13, 146–148. [Google Scholar] [CrossRef]
- Dolenšek, J.; Rupnik, M.S.; Stožer, A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015, 7, e1024405. [Google Scholar] [CrossRef] [PubMed]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef]
- Arsenijevic, T.; Perret, J.; van Laethem, J.L.; Delporte, C. Aquaporins involvement in pancreas physiology and in pancreatic diseases. Int. J. Mol. Sci. 2019, 20, 5052. [Google Scholar] [CrossRef]
- Weiss, F.U.; Laemmerhirt, F.; Lerch, M.M. Etiology and risk factors of acute and chronic pancreatitis. Visc. Med. 2019, 35, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Bachet, J.-B.; Ayav, A.; Huguet, F.; Lambert, A.; Caramella, C.; Maréchal, R.; Van Laethem, J.-L.; Ducreux, M. Current standards and new innovative approaches for treatment of pancreatic cancer. Eur. J. Cancer 2016, 57, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Madácsy, T.; Pallagi, P.; Maleth, J. Cystic Fibrosis of the Pancreas: The Role of CFTR Channel in the Regulation of Intracellular Ca2+ Signaling and Mitochondrial Function in the Exocrine Pancreas. Front. Physiol. 2018, 9, 1585. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Muñoz, J.E. Management of pancreatic exocrine insufficiency. Curr. Opin. Gastroenterol. 2019, 35, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Alexandre-Heymann, L.; Mallone, R.; Boitard, C.; Scharfmann, R.; Larger, E. Structure and function of the exocrine pancreas in patients with type 1 diabetes. Rev. Endocr. Metab. Disord. 2019, 20, 129–149. [Google Scholar] [CrossRef]
- Hu, F.; Qiu, X.; Bu, S. Pancreatic islet dysfunction in type 2 diabetes mellitus. Arch. Physiol. Biochem. 2020, 126, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Melton, D.A. Pancreas regeneration. Nature 2018, 557, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Cesmebasi, A.; Malefant, J.; Patel, S.D.; Du Plessis, M.; Renna, S.; Tubbs, R.S.; Loukas, M. The surgical anatomy of the lymphatic system of the pancreas. Clin. Anat. 2015, 28, 527–537. [Google Scholar] [CrossRef] [PubMed]
- de Pretis, N.; Amodio, A.; Frulloni, L. Hypertriglyceridemic pancreatitis: Epidemiology, pathophysiology and clinical management. United Eur. Gastroenterol. J. 2018, 6, 649–655. [Google Scholar] [CrossRef]
- Türkvatan, A.; Erden, A.; Tǔrkoǧlu, M.A.; Yener, Ö. Congenital variants and anomalies of the pancreas and pancreatic duct: Imaging by magnetic resonance cholangiopancreaticography and multidetector computed tomography. Korean J. Radiol. 2013, 14, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Gonoi, W.; Akai, H.; Hagiwara, K.; Akahane, M.; Hayashi, N.; Maeda, E.; Yoshikawa, T.; Kiryu, S.; Tada, M.; Uno, K.; et al. Santorinicele without pancreas divisum pathophysiology: Initial clinical and radiographic investigations. BMC Gastroenterol. 2013, 13, 62–63. [Google Scholar] [CrossRef]
- Pan, F.C.; Brissova, M. Pancreas development in humans. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 77–82. [Google Scholar] [CrossRef]
- Singer, M.V.; Gyr, K.; Sarles, H. Revised classification of pancreatitis. Report of the Second International Symposium on the classification of pancreatitis in Marseille, France, March 28–30, 1984. Gastroenterology 1985, 89, 683–685. [Google Scholar] [CrossRef]
- Machicado, J.D.; Yadav, D. Epidemiology of Recurrent Acute and Chronic Pancreatitis: Similarities and Differences. Dig. Dis. Sci. 2017, 62, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Peery, A.F.; Crockett, S.; Barritt, A.; Dellon, E.S.; Eluri, S.; Gangarosa, L.M.; Jensen, E.T.; Lund, J.L.; Pasricha, S.; Runge, T.; et al. Burden of Gastrointestinal, Liver, and Pancreatic Diseases in the United States. Gastroenterology 2015, 149, 1731–1741.e3. [Google Scholar] [CrossRef]
- Greenberg, J.A.; Hsu, J.; Bawazeer, M.; Marshall, J.; Friedrich, J.O.; Nathens, A.; Coburn, N.; May, G.R.; Pearsall, E.; McLeod, R.S. Clinical practice guideline: Management of acute pancreatitis. Can. J. Surg. 2016, 59, 128–140. [Google Scholar] [CrossRef]
- Gao, Y.J.; Li, Y.Q.; Wang, Q.; Li, S.L.; Li, G.Q.; Ma, J.; Zeng, X.Z.; Huang, L.Y.; Yuan, S.A.; Liu, C.A.; et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J. Gastroenterol. 2006, 41, 681–685. [Google Scholar] [CrossRef]
- Yadav, D.; O’Connell, M.; Papachristou, G.I. Natural history following the first attack of acute pancreatitis. Am. J. Gastroenterol. 2012, 107, 1096–1103. [Google Scholar] [CrossRef]
- Ali, U.A.; Issa, Y.; Hagenaars, J.C.; Bakker, O.J.; van Goor, H.; Nieuwenhuijs, V.B.; Bollen, T.L.; van Ramshorst, B.; Witteman, B.J.; Brink, M.A.; et al. Risk of Recurrent Pancreatitis and Progression to Chronic Pancreatitis After a First Episode of Acute Pancreatitis. Clin. Gastroenterol. Hepatol. 2016, 14, 738–746. [Google Scholar]
- Chen, Y.N.; Zhao, M.W.; Cui, J.; Yao, H.-B. Acute pancreatitis may be a pathogenic factor of fulminant type 1 diabetes mellitus. Chin. Med. J. 2021, 134, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, A.; Hanafusa, T.; Miyagawa, J.I.; Matsuzawa, Y. A Novel Subtype of Type 1 Diabetes Mellitus Characterized by a Rapid Onset and an Absence of Diabetes-Related Antibodies. N. Engl. J. Med. 2000, 342, 301–307. [Google Scholar] [CrossRef]
- Yadav, D.; Timmons, L.; Benson, J.T.; Dierkhising, R.A.; Chari, S.T. Incidence, prevalence, and survival of chronic pancreatitis: A population-based study. Am. J. Gastroenterol. 2011, 106, 2192–2199. [Google Scholar] [CrossRef] [PubMed]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S.; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef]
- Leppäniemi, A.; Tolonen, M.; Tarasconi, A.; Lohse, H.A.S.; Gamberini, E.; Kirkpatrick, A.W.; Ball, C.G.; Parry, N.; Sartelli, M.; Wolbrink, D.R.J.; et al. 2019 WSES guidelines for the management of severe acute pancreatitis. World J. Emerg. Surg. 2019, 14, 27. [Google Scholar]
- Working Group IAP/APA Acute Pancreatitis Guidelines. IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology 2013, 13 (Suppl. S2), e1–e15. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Gaillard, F. Ranson criteria. In Radiopaedia; StatPearls: Treasure Island, FL, USA, 2009. [Google Scholar]
- Balthazar, E.J.; Robinson, D.L.; Megibow, A.J.; Ranson, J.H.C. Acute pancreatitis: Value of CT in establishing prognosis. Radiology 1990, 174, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Mortelé, K.J.; Mergo, P.J.; Taylor, H.M.; Wiesner, W.; Cantisani, V.; Ernst, M.D.; Kalantari, B.N.; Ros, P.R. Peripancreatic vascular abnormalities complicating acute pancreatitis: Contrast-enhanced helical CT findings. Eur. J. Radiol. 2004, 52, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Arif, A.; Jaleel, F.; Rashid, K. Accuracy of BISAP score in prediction of severe acute pancreatitis. Pak. J. Med. Sci. 2019, 35, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, L.; Jin, T.; Xia, Q. Harmless acute pancreatitis score on admission can accurately predict mild acute pancreatitis. J. South Med. Univ. 2020, 40, 190–195. [Google Scholar]
- Sayraç, A.V.; Cete, Y.; Yiğit, Ö.; Aydın, A.G.; Sayrac, N. Utility of HAPS for predicting prognosis in acute pancreatitis. Ulus Travma Acil Cerrahi Derg. 2018, 24, 4. [Google Scholar] [CrossRef]
- Al-Qahtani, H.H.; Alam, M.K.; Waheed, M. Comparison of harmless acute pancreatitis score with Ranson’s score in predicting the severity of acute pancreatitis. J. Coll. Physicians Surg. Pak. 2017, 27, 75–79. [Google Scholar]
- Kui, B.; Pintér, J.; Molontay, R.; Nagy, M.; Farkas, N.; Gede, N.; Vincze, Á.; Bajor, J.; Gódi, S.; Czimmer, J.; et al. EASY-APP: An artificial intelligence model and application for early and easy prediction of severity in acute pancreatitis. Clin. Transl. Med. 2022, 12, e842. [Google Scholar] [CrossRef]
- Sankaran, S.J.; Xiao, A.Y.; Wu, L.M.; Windsor, J.A.; Forsmark, C.E.; Petrov, M.S. Frequency of Progression from Acute to Chronic Pancreatitis and Risk Factors: A Meta-analysis. Gastroenterology 2015, 149, 1490–1500.e1. [Google Scholar] [CrossRef]
- Wilcox, M.C.; Sandhu, B.S.; Singh, V.; Gelrud, A.; Abberbock, J.N.; Sherman, S.; Cote, G.A.; Al-Kaade, S.; Anderson, M.A.; Gardner, T.B.; et al. Racial Differences in the Clinical Profile, Causes, and Outcome of Chronic Pancreatitis. Am. J. Gastroenterol. 2016, 111, 1488–1496. [Google Scholar] [CrossRef]
- Wilcox, C.M.; Yadav, D.; Ye, T.; Gardner, T.B.; Gelrud, A.; Sandhu, B.S.; Lewis, M.D.; Al-Kaade, S.; Cote, G.A.; Forsmark, C.E.; et al. Chronic pancreatitis pain pattern and severity are independent of abdominal imaging findings. Clin. Gastroenterol. Hepatol. 2015, 13, 552–560. [Google Scholar] [CrossRef]
- Lankisch, P.G.; Assmus, C.; Lehnick, D.; Maisonneuve, P.; Lowenfels, A.B. Acute pancreatitis: Does gender matter? Dig. Dis. Sci. 2001, 46, 2470–2474. [Google Scholar] [CrossRef] [PubMed]
- Eichelberger, M.R.; Chatten, J.; Bruce, D.A.; Garcia, V.F.; Goldman, M.; Koop, C.E. Acute pancreatitis and increased intracranial pressure. J. Pediatr. Surg. 1981, 16 (Suppl. S1), 562–570. [Google Scholar] [CrossRef]
- Saluja, A.; Saluja, M.; Villa, A.; Leli, U.; Rutledge, P.; Meldolesi, J.; Steer, M. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J. Clin. Investig. 1989, 84, 1260–1266. [Google Scholar] [CrossRef]
- Samuel, I.; Toriumi, Y.; Zaheer, A.; Joehl, R.J. Mechanism of acute pancreatitis exacerbation by enteral bile-pancreatic juice exclusion. Pancreatology 2004, 4, 527–532. [Google Scholar] [CrossRef]
- Spanier, B.W.M.; Dijkgraaf, M.G.W.; Bruno, M.J. Epidemiology, aetiology and outcome of acute and chronic pancreatitis: An update. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Lankisch, P.G.; Lowenfels, A.B.; Maisonneuve, P.; Oría, A. What is the risk of alcoholic pancreatitis in heavy drinkers? Pancreas 2002, 25, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.P.; Taylor, T.V.; Torrance, H.B. Pressure, volume and the pancreas. Gut 1985, 26, 615–624. [Google Scholar] [CrossRef]
- Wang, G.J.; Gao, C.F.; Wei, D.; Wang, C.; Ding, S.Q. Acute pancreatitis: Etiology and common pathogenesis. World J. Gastroenterol. 2009, 15, 1427–1430. [Google Scholar] [CrossRef]
- Steinle, A.U.; Weidenbach, H.; Wagner, M.; Adler, G.; Schmid, R.M. NF-κB/Rel activation in cerulein pancreatitis. Gastroenterology 1999, 116, 420–430. [Google Scholar] [CrossRef]
- Habtezion, A. Inflammation in acute and chronic pancreatitis. Curr. Opin. Gastroenterol. 2015, 31, 395–399. [Google Scholar] [CrossRef]
- Gukovsky, I.; Li, N.; Todoric, J.; Gukovskaya, A.; Karin, M. Inflammation, autophagy, and obesity: Common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Völzke, H.; Baumeister, S.E.; Alte, D.; Hoffmann, W.; Schwahn, C.; Simon, P.; John, U.; Lerch, M.M. Independent risk factors for gallstone formation in a region with high cholelithiasis prevalence. Digestion 2005, 71, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Buch, S.; Schafmayer, C.; Völzke, H.; Seeger, M.; Miquel, J.F.; Sookoian, S.C.; Egberts, J.H.; Arlt, A.; Pirola, C.J.; Lerch, M.M.; et al. Loci from a genome-wide analysis of bilirubin levels are associated with gallstone risk and composition. Gastroenterology 2010, 139, 1942–1951.e2. [Google Scholar] [CrossRef] [PubMed]
- Lammert, F.; Acalovschi, M.; Ercolani, G.; van Erpecum, K.J.; Gurusamy, K.; van Laarhoven, C.J.; Portincasa, P. EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J. Hepatol. 2016, 65, 146–181. [Google Scholar] [CrossRef]
- Meyerholz, D.K.; Samuel, I. Morphologic characterization of early ligation-induced acute pancreatitis in rats. Am. J. Surg. 2007, 194, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yu, Y.H.; Zhong, M.; Zhang, G.W. Analyzing and identifying risk factors for acute pancreatitis with different etiologies in pregnancy. J. Matern.-Fetal Neonatal Med. 2015, 28, 267–271. [Google Scholar] [CrossRef]
- Linares, C.L.; Pelletier, A.L.; Czernichow, S.; Vergnaud, A.C.; Bonnefont-Rousselot, D.; Levy, P.; Ruszniewski, P.; Bruckert, E. Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia. Pancreas 2008, 37, 13-2. [Google Scholar] [CrossRef]
- Kochar, B.; Akshintala, V.S.; Afghani, E.; Elmunzer, B.J.; Kim, K.J.; Lennon, A.M.; Khashab, M.A.; Kalloo, A.N.; Singh, V.K. Incidence, severity, and mortality of post-ERCP pancreatitis: A systematic review by using randomized, controlled trials. Gastrointest. Endosc. 2015, 81, 143–149.e9. [Google Scholar] [CrossRef]
- Chandrasekhara, V.; Khashab, M.A.; Muthusamy, V.R.; Acosta, R.D.; Agrawal, D.; Bruining, D.H.; Eloubeidi, M.A.; Fanelli, R.D.; Faulx, A.L.; Gurudu, S.R.; et al. Adverse events associated with ERCP. Gastrointest. Endosc. 2017, 85, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Fluhr, G.; Mayerle, J.; Weber, E.; Aghdassi, A.; Simon, P.; Gress, T.; Seufferlein, T.; Mössner, J.; Stallmach, A.; Rösch, T.; et al. Pre-Study protocol MagPEP: A multicentre randomized controlled trial of magnesium sulphate in the prevention of post-ERCP pancreatitis. BMC Gastroenterol. 2013, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.D.; Romac, J.; Vigna, S.R.; Liddle, R.A. A pH-sensitive, neurogenic pathway mediates disease severity in a model of post-ERCP pancreatitis. Gut 2008, 57, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Blaho, M.; Dítě, P.; Kunovský, L.; Kianička, B. Autoimmune pancreatitis—An ongoing challenge. Adv. Med. Sci. 2020, 65, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, A.; Inoue, D.; Takahashi, N. Autoimmune pancreatitis: An update. Abdom. Radiol. 2020, 45, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Chari, S.T.; Lerch, M.M.; Kim, M.H.; Gress, T.M.; Shimosegawa, T. Recent advances in autoimmune pancreatitis: Type 1 and type 2. Postgrad. Med. J. 2014, 90, 18–25. [Google Scholar] [CrossRef]
- Pieringer, H.; Parzer, I.; Wöhrer, A.; Reis, P.; Oppl, B.; Zwerina, J. IgG4- related disease: An orphan disease with many faces. Orphanet J. Rare Dis. 2014, 9, 110. [Google Scholar] [CrossRef]
- Hirabayashi, K.; Zamboni, G. IgG4-related disease. Pathologica 2012, 104, 43–55. [Google Scholar]
- Pitz, S. IgG4-related disease. Ophthalmologe 2021, 118, 787–793. [Google Scholar] [CrossRef]
- Hart, P.A.; Zen, Y.; Chari, S.T. Recent Advances in Autoimmune Pancreatitis. Gastroenterology 2015, 149, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyama, T.; Uchida, K.; Sumimoto, K.; Fukui, Y.; Ikeura, T.; Fukui, T.; Nishio, A.; Shikata, N.; Uemura, Y.; Satoi, S.; et al. Comparison of neutrophil infiltration between type 1 and type 2 autoimmune pancreatitis. Pancreatology 2015, 15, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Shiokawa, M.; Kodama, Y.; Yoshimura, K.; Kawanami, C.; Mimura, J.; Yamashita, Y.; Asada, M.; Kikuyama, M.; Okabe, Y.; Inokuma, T.; et al. Risk of cancer in patients with autoimmune pancreatitis. Am. J. Gastroenterol. 2013, 108, 610–617. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takahashi, H.; Tabeya, T.; Suzuki, C.; Naishiro, Y.; Ishigami, K.; Yajima, H.; Shimizu, Y.; Obara, M.; Yamamoto, H.; et al. Risk of malignancies in IgG4-related disease. Mod. Rheumatol. 2012, 22, 414–418. [Google Scholar] [CrossRef]
- Fukui, T.; Mitsuyama, T.; Takaoka, M.; Uchida, K.; Matsushita, M.; Okazaki, K. Pancreatic cancer associated with autoimmune pancreatitis in remission. Intern. Med. 2008, 47, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Hirth, M.; Münch, M.; Weiss, C.; Löhr, J.M.; Ebert, M.P.; Pfützer, R.H. Risk of Cancer in Patients with Autoimmune Pancreatitis: A Single-Center Experience from Germany. Digestion 2017, 95, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Tada, M.; Sasahira, N.; Isayama, H.; Mizuno, S.; Takagi, K.; Watanabe, T.; Saito, T.; Kawahata, S.; Uchino, R.; et al. Incidence of malignancies in patients with IgG4-related disease. Intern. Med. 2014, 53, 171–176. [Google Scholar] [CrossRef]
- Watanabe, T.; Kudo, M.; Strober, W. Immunopathogenesis of pancreatitis. Mucosal Immunol. 2017, 10, 283–298. [Google Scholar] [CrossRef]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Tóth, M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 2019, 56, 1951–1968.e1. [Google Scholar] [CrossRef]
- Rinderknecht, H. Activation of pancreatic zymogens—Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig. Dis. Sci. 1986, 31, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Szmola, R.; Sahin-Tóth, M. Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: Identity with Rinderknecht’s enzyme Y. Proc. Natl. Acad. Sci. USA. 2007, 104, 11227–11232. [Google Scholar] [CrossRef]
- Wartmann, T.; Mayerle, J.; Kähne, T.; Sahin-Tóth, M.; Ruthenbürger, M.; Matthias, R.; Kruse, A.; Reinheckel, T.; Peters, C.; Weiss, F.U.; et al. Cathepsin L Inactivates Human Trypsinogen, Whereas Cathepsin L-Deletion Reduces the Severity of Pancreatitis in Mice. Gastroenterology 2010, 138, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, E.; Sahin-Tóth, M. Genetic Risk in Chronic Pancreatitis: The Trypsin-Dependent Pathway. Dig. Dis. Sci. 2017, 62, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Szabó, A.; Sahin-Tóth, M. Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J. Biol. Chem. 2012, 287, 20701–20710. [Google Scholar] [CrossRef] [PubMed]
- Aoun, E.; Chang, C.C.H.; Greer, J.B.; Papachristou, G.I.; Barmada, M.M.; Whitcomb, D.C. Pathways to injury in chronic pancreatitis: Decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS ONE 2008, 3, e2003. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, J.; Witt, H.; Szmola, R.; Bhatia, E.; Ózsvári, B.; Landt, O.; Schulz, H.-U.; Gress, T.M.; Pfützer, R.; Löhr, M.; et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 2008, 40, 78–82. [Google Scholar] [CrossRef]
- Beer, S.; Zhou, J.; Szabó, A.; Keiles, S.; Chandak, G.R.; Witt, H.; Sahin-Tóth, M. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 2013, 62, 1616–1624. [Google Scholar] [CrossRef]
- Rosendahl, J.; Kirsten, H.; Hegyi, E.; Kovacs, P.; Weiss, F.U.; Laumen, H.; Lichtner, P.; Ruffert, C.; Chen, J.-M.; Masson, E.; et al. Genome-wide association study identifies inversion in the CTRB1-CTRB2 locus to modify risk for alcoholic and non-alcoholic chronic pancreatitis. Gut 2018, 67, 1855–1863. [Google Scholar] [CrossRef]
- Witt, H.; Sahin-Tóth, M.; Landt, O.; Chen, J.M.; Kähne, T.; Drenth, J.P.H.; Kukor, Z.; Szepessy, E.; Halangk, W.; Dahm, S.; et al. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat. Genet. 2006, 38, 668–673. [Google Scholar] [CrossRef]
- Bess, M.A.; Edis, A.J.; van Heerden, J.A. Hyperparathyroidism and Pancreatitis: Chance or a Causal Association? J. Am. Med. Assoc. 1980, 243, 246–247. [Google Scholar] [CrossRef]
- Jones, M.R.; Hall, O.M.; Kaye, A.M.; Kaye, A.D. Drug-induced acute pancreatitis: A review. Ochsner J. 2015, 15, 45–51. [Google Scholar]
- Aldehuelo, R.S.; de Paredes, A.G.G.; Lázaro, D.R.; Ortega, A.M.; de la Filia Molina, I.G.; López-Durán, S.; Rodríguez-Gandía, M.Á.; López-Sanromán, A.; Albillos, A. Outcomes of drug-induced acute pancreatitis: A ten-year experience of an academic center. Rev. Esp. Enferm. Dig. 2021, 113, 276–279. [Google Scholar]
- Párniczky, A.; Kui, B.; Szentesi, A.; Balázs, A.; Szûcs, Á.; Mosztbacher, D.; Czimmer, J.; Sarlós, P.; Bajor, J.; Gódi, S.; et al. Prospective, Multicentre, Nationwide Clinical Data from 600 Cases of Acute Pancreatitis. PLoS ONE 2016, 11, e0165309. [Google Scholar] [CrossRef]
- Badalov, N.; Baradarian, R.; Iswara, K.; Li, J.; Steinberg, W.; Tenner, S. Drug-Induced Acute Pancreatitis: An Evidence-Based Review. Clin. Gastroenterol. Hepatol. 2007, 5, 648–661.e3. [Google Scholar]
- Spanier, M.B.; Tuynman, H.A.; van der Hulst, R.W.; Dijkgraaf, M.G.; Bruno, M.J. Acute pancreatitis and concomitant use of pancreatitis-associated drugs. Am. J. Gastroenterol. 2011, 106, 2183–2188. [Google Scholar] [PubMed]
- Weissman, S.; Aziz, M.; Perumpail, R.B.; Mehta, T.I.; Patel, R.; Tabibian, J.H. Ever-increasing diversity of drug-induced pancreatitis. World J. Gastroenterol. 2020, 26, 2902–2915. [Google Scholar] [CrossRef] [PubMed]
- Thisted, H.; Jacobsen, J.; Munk, E.M.; Norgaard, B.; Friis, S.; McLaughlin, J.K.; Sørensen, H.T.; Johnsen, S.P. Statins and the risk of acute pancreatitis: A population-based case-control study. Aliment. Pharmacol. Ther. 2006, 23, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.; Batel-Marques, F.; Macedo, A.F. A meta-analysis of serious adverse events reported with exenatide and liraglutide: Acute pancreatitis and cancer. Diabetes Res. Clin. Pract. 2012, 98, 271–284. [Google Scholar] [CrossRef]
- Nitsche, C.; Maertin, S.; Scheiber, J.; Ritter, C.A.; Lerch, M.M.; Mayerle, J. Drug-induced pancreatitis. Curr. Gastroenterol. Rep. 2012, 14, 131–138. [Google Scholar] [CrossRef]
- Wolfe, D.; Kanji, S.; Yazdi, F.; Barbeau, P.; Rice, D.; Beck, A.; Butler, C.; Esmaeilisaraji, L.; Skidmore, B.; Moher, D.; et al. Drug induced pancreatitis: A systematic review of case reports to determine potential drug associations. PLoS ONE 2020, 15, e0231883. [Google Scholar]
- Sotoudehmanesh, R.; Khatibian, M.; Kolahdoozan, S.; Ainechi, S.; Malboosbaf, R.; Nouraie, M. Indomethacin may reduce the incidence and severity of acute pancreatitis after ERCP. Am. J. Gastroenterol. 2007, 102, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.; Carter, R.; Imrie, C.; Evans, S.; O’Suilleabhain, C. Diclofenac reduces the incidence of acute pancreatitis after endoscopic retrograde cholangiopancreatography. Gastroenterology 2003, 124, 1786–1791. [Google Scholar] [CrossRef]
- Kataria, P.S.C.; Kendre, P.P.; Patel, A.A.; Bohra, M.Z.; Tahiliani, N. Tamoxifen induced pancreatitis: An unusual complication of commonly used drug. J. Clin. Diagn. Res. 2017, 11, XD05–XD06. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yang, Q.J.; Dang, F.T.; Yang, J. Drug-induced pancreatitis: An update. Arab. J. Gastroenterol. 2019, 20, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, C.A.; Busto, U.; Sellers, E.M.; Sandor, P.; Ruiz, I.; Roberts, E.A.; Janecek, E.; Domecq, C.; Greenblatt, D.J. A method for estimating the probability of adverse drug reactions. Clin. Pharmacol. Ther. 1981, 30, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Kottanattu, L.; Lava, S.A.G.; Helbling, R.; Simonetti, G.D.; Bianchetti, M.G.; Milani, G.P. Pancreatitis and cholecystitis in primary acute symptomatic Epstein-Barr virus infection—Systematic review of the literature. J. Clin. Virol. 2016, 82, 51–55. [Google Scholar] [CrossRef]
- Aloysius, M.M.; Thatti, A.; Gupta, A.; Sharma, N.; Bansal, P.; Goyal, H. COVID-19 presenting as acute pancreatitis. Pancreatology 2020, 20, 1026–1027. [Google Scholar] [CrossRef]
- Zádori, N.; Párniczky, A.; Szentesi, A.; Hegyi, P. Insufficient implementation of the IAP/APA guidelines on aetiology in acute pancreatitis: Is there a need for implementation managers in pancreatology? United Eur. Gastroenterol. J. 2020, 8, 246–248. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walkowska, J.; Zielinska, N.; Karauda, P.; Tubbs, R.S.; Kurtys, K.; Olewnik, Ł. The Pancreas and Known Factors of Acute Pancreatitis. J. Clin. Med. 2022, 11, 5565. https://doi.org/10.3390/jcm11195565
Walkowska J, Zielinska N, Karauda P, Tubbs RS, Kurtys K, Olewnik Ł. The Pancreas and Known Factors of Acute Pancreatitis. Journal of Clinical Medicine. 2022; 11(19):5565. https://doi.org/10.3390/jcm11195565
Chicago/Turabian StyleWalkowska, Julia, Nicol Zielinska, Piotr Karauda, R. Shane Tubbs, Konrad Kurtys, and Łukasz Olewnik. 2022. "The Pancreas and Known Factors of Acute Pancreatitis" Journal of Clinical Medicine 11, no. 19: 5565. https://doi.org/10.3390/jcm11195565
APA StyleWalkowska, J., Zielinska, N., Karauda, P., Tubbs, R. S., Kurtys, K., & Olewnik, Ł. (2022). The Pancreas and Known Factors of Acute Pancreatitis. Journal of Clinical Medicine, 11(19), 5565. https://doi.org/10.3390/jcm11195565