
The Expression of Follistatin-like 1 Protein Is Associated with the Activation of the EMT Program in Sjögren’s Syndrome





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Treatment and Antibodies
2.2. Bioptic Samples Collection
2.3. Histochemistry
2.4. FSTL1 Immunohistochemical Analysis and Quantification
2.5. SGEC Culture and Treatment
2.6. Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) and Quantitative Real-Time PCR (q-RT-PCR)
2.7. Data Evaluation and Sequence Analysis
2.8. Western Blot Analysis
2.9. FSTL1 siRNA Transfection
2.10. Statistical Analysis
3. Results
3.1. Aberrant Expression of FSTL1 in SS SGs Is Associated with Inflammatory Grade
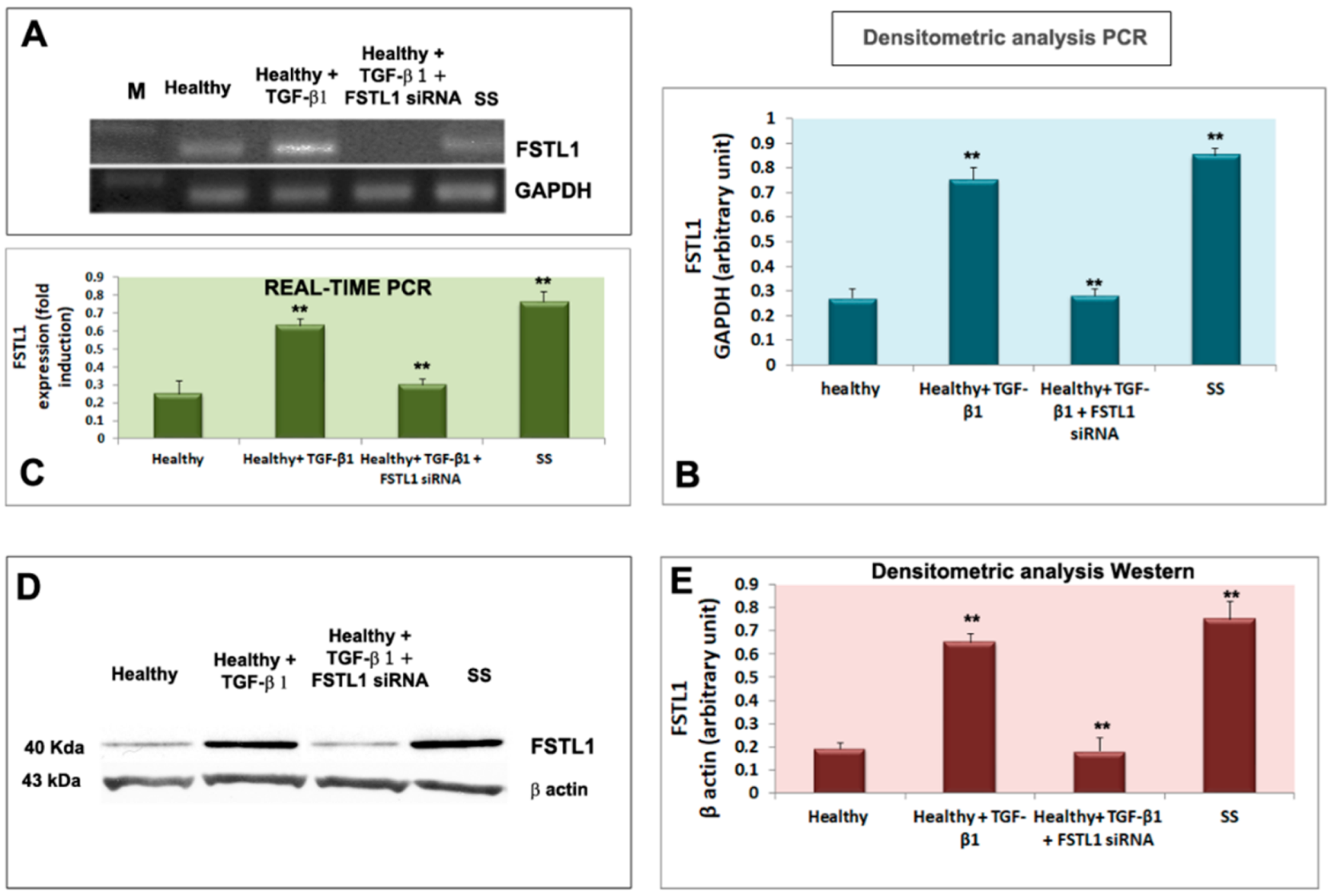
3.2. TGF-β1 Upregulates FSTL1 Expression in Healthy Salivary Glands Epithelial Cells
3.3. FSTL1 Neutralization Attenuates EMT-Related Morphological Changes Induces by TGF-β1 Treatment
3.4. FSTL1 Moderates EMT-Related Mesenchymal Markers Expression in TGF-β1-Treated SGEC
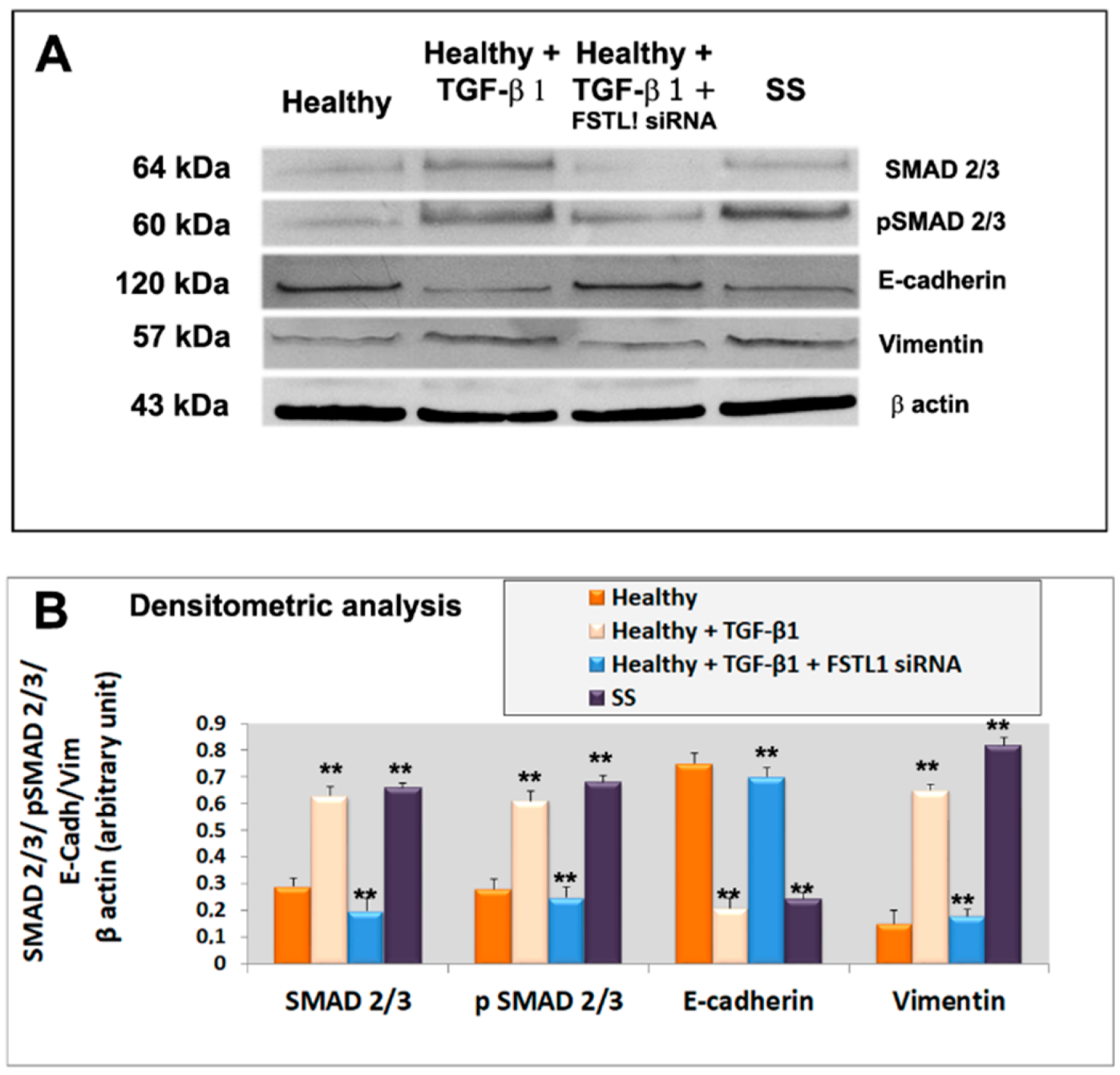
3.5. FSTL1 Modulates EMT by Facilitating TGF-β1 Signaling through SMAD2/3 Phosphorylation and Activation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shibanuma, M.; Mashimo, J.; Mita, A.; Kuroki, T.; Nose, K. Cloning from a mouse osteoblastic cell line of a set of transforming-growth-factor-beta 1-regulated genes, one of which seems to encode a follistatin-related polypeptide. Eur. J. Biochem. 1993, 217, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Mattiotti, A.; Prakash, S.; Barnett, P.; van den Hoff, M.J.B. Follistatin-like 1 in development and human diseases. Cell Mol. Life Sci. 2018, 75, 2339–2354. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Dong, Y.; Yu, M.; Zhang, L.; Yan, X.; Sun, J.; Qiao, L.; Geng, H.; Nakajima, M.; Furuichi, T.; et al. Follistatin-like 1 (Fstl1) is a bone morphogenetic protein (BMP) 4 signaling antagonist in controlling mouse lung development. Proc. Natl. Acad. Sci. USA 2011, 108, 7058–7063. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, L.; Chang, Y.; Ning, W.; Liu, X. Structural and functional study of FK domain of Fstl1. Protein Sci. 2019, 28, 1819–1829. [Google Scholar] [CrossRef]
- Oshima, Y.; Ouchi, N.; Sato, K.; Izumiya, Y.; Pimentel, D.R.; Walsh, K. Follistatin-like 1 is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation 2008, 117, 3099–3108. [Google Scholar] [CrossRef]
- Chaly, Y.; Hostager, B.; Smith, S.; Hirsch, R. Follistatin-like protein 1 and its role in inflammation and inflammatory diseases. Immunol. Res. 2014, 59, 266–272. [Google Scholar] [CrossRef]
- Liu, T.; Liu, Y.; Miller, M.; Cao, L.; Zhao, J.; Wu, J.; Wang, J.; Liu, L.; Li, S.; Zou, M.; et al. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L27–L40. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 6984. [Google Scholar] [CrossRef]
- Zheng, X.; Qi, C.; Zhang, S.; Fang, Y.; Ning, W. TGF-β1 induces Fstl1 via the Smad3-c-Jun pathway in lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L240–L251. [Google Scholar] [CrossRef]
- Dong, Y.; Geng, Y.; Li, L.; Li, X.; Yan, X.; Fang, Y.; Li, X.; Dong, S.; Liu, X.; Li, X.; et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J. Exp. Med. 2015, 212, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Tamm, A.R.; Ribatti, D.; Lisi, S. The TGF-β1 Signaling Pathway as an Attractive Target in the Fibrosis Pathogenesis of Sjögren’s Syndrome. Mediat. Inflamm. 2018, 2018, 1965935. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef]
- Li, X.; Fang, Y.; Jiang, D.; Dong, Y.; Liu, Y.; Zhang, S.; Guo, J.; Qi, C.; Zhao, C.; Jiang, F.; et al. Targeting FSTL1 for Multiple Fibrotic and Systemic Autoimmune Diseases. Mol. Ther. 2021, 29, 347–364. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Xu, N.; Wei, Q.; Wu, M.; Li, X.; Zheng, P.; Sun, S.; Jin, Y.; Zhang, G.; et al. Follistatin-like protein 1 is elevated in systemic autoimmune diseases and correlated with disease activity in patients with rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, R17. [Google Scholar] [CrossRef]
- Mariette, X.; Criswell, L.A. Primary Sjögren’s syndrome. N. Engl. J. Med. 2018, 378, 931–939. [Google Scholar] [CrossRef]
- Franceschini, F.; Cavazzana, I.; Andreoli, L.; Tincani, A. The 2016 classification criteria for primary Sjogren’s syndrome: What’s new? BMC Med. 2017, 15, 69. [Google Scholar] [CrossRef]
- Sens, D.A.; Hintz, D.S.; Rudisill, M.T.; Sens, M.A. Spicer SS Explant culture of human submandibular gland epithelial cells: Evidence for ductal origin. Lab. Investig. 1985, 52, 559–567. [Google Scholar]
- Kapsogeorgou, E.K.; Dimitriou, I.D.; Abu-Helu, R.F.; Moutsopoulos, H.M.; Manoussakis, M.N. Activation of epithelial and myoepithelial cells in the salivary glands of patients with Sjögren’s syndrome: High expression of intercellular adhesion molecule-1 (ICAM.1) in biopsy specimens and cultured cells. Clin. Exp. Immunol. 2001, 124, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Saraux, A.; Devauchelle-Pensec, V. Primary Sjögren’s syndrome: New beginning for evidence-based trials. Lancet 2022, 399, 121–122. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Z.; Zhou, C.; Liu, L.; Huang, C. Epithelial-mesenchymal transition: The history, regulatory mechanism, and cancer therapeutic opportunities. MedComm 2022, 3, e144. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002. [Google Scholar] [CrossRef]
- Hua, W.; ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life Sci. 2020, 77, 2103–2123. [Google Scholar] [CrossRef]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Sisto, M.; Lorusso, L.; Tamma, R.; Ingravallo, G.; Ribatti, D.; Lisi, S. Interleukin-17 and -22 synergy linking inflammation and EMT-dependent fibrosis in Sjögren’s syndrome. Clin. Exp. Immunol. 2019, 198, 261–272. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-Mesenchymal Transition and Its Implications for Fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisto, M.; Ribatti, D.; Lisi, S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in TGF β-Dependent EMT. Biomolecules 2021, 11, 310. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sisto, M.; Ribatti, D.; Ingravallo, G.; Lisi, S. The Expression of Follistatin-like 1 Protein Is Associated with the Activation of the EMT Program in Sjögren’s Syndrome. J. Clin. Med. 2022, 11, 5368. https://doi.org/10.3390/jcm11185368
Sisto M, Ribatti D, Ingravallo G, Lisi S. The Expression of Follistatin-like 1 Protein Is Associated with the Activation of the EMT Program in Sjögren’s Syndrome. Journal of Clinical Medicine. 2022; 11(18):5368. https://doi.org/10.3390/jcm11185368
Chicago/Turabian StyleSisto, Margherita, Domenico Ribatti, Giuseppe Ingravallo, and Sabrina Lisi. 2022. "The Expression of Follistatin-like 1 Protein Is Associated with the Activation of the EMT Program in Sjögren’s Syndrome" Journal of Clinical Medicine 11, no. 18: 5368. https://doi.org/10.3390/jcm11185368
APA StyleSisto, M., Ribatti, D., Ingravallo, G., & Lisi, S. (2022). The Expression of Follistatin-like 1 Protein Is Associated with the Activation of the EMT Program in Sjögren’s Syndrome. Journal of Clinical Medicine, 11(18), 5368. https://doi.org/10.3390/jcm11185368