
Mitochondrial Regulation of the Hypoxia-Inducible Factor in the Development of Pulmonary Hypertension
 , ,
, ,
Abstract
:1. Introduction
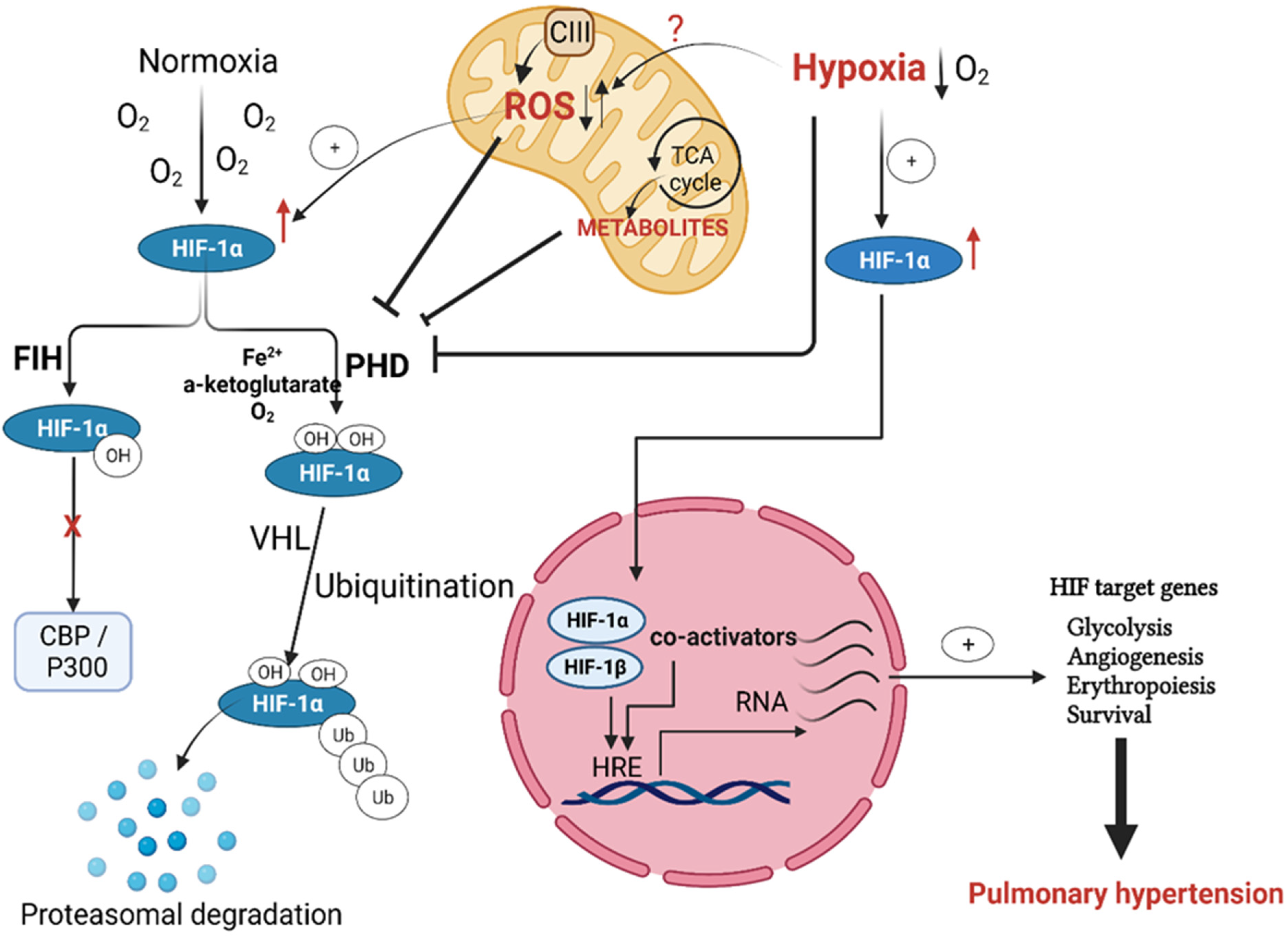
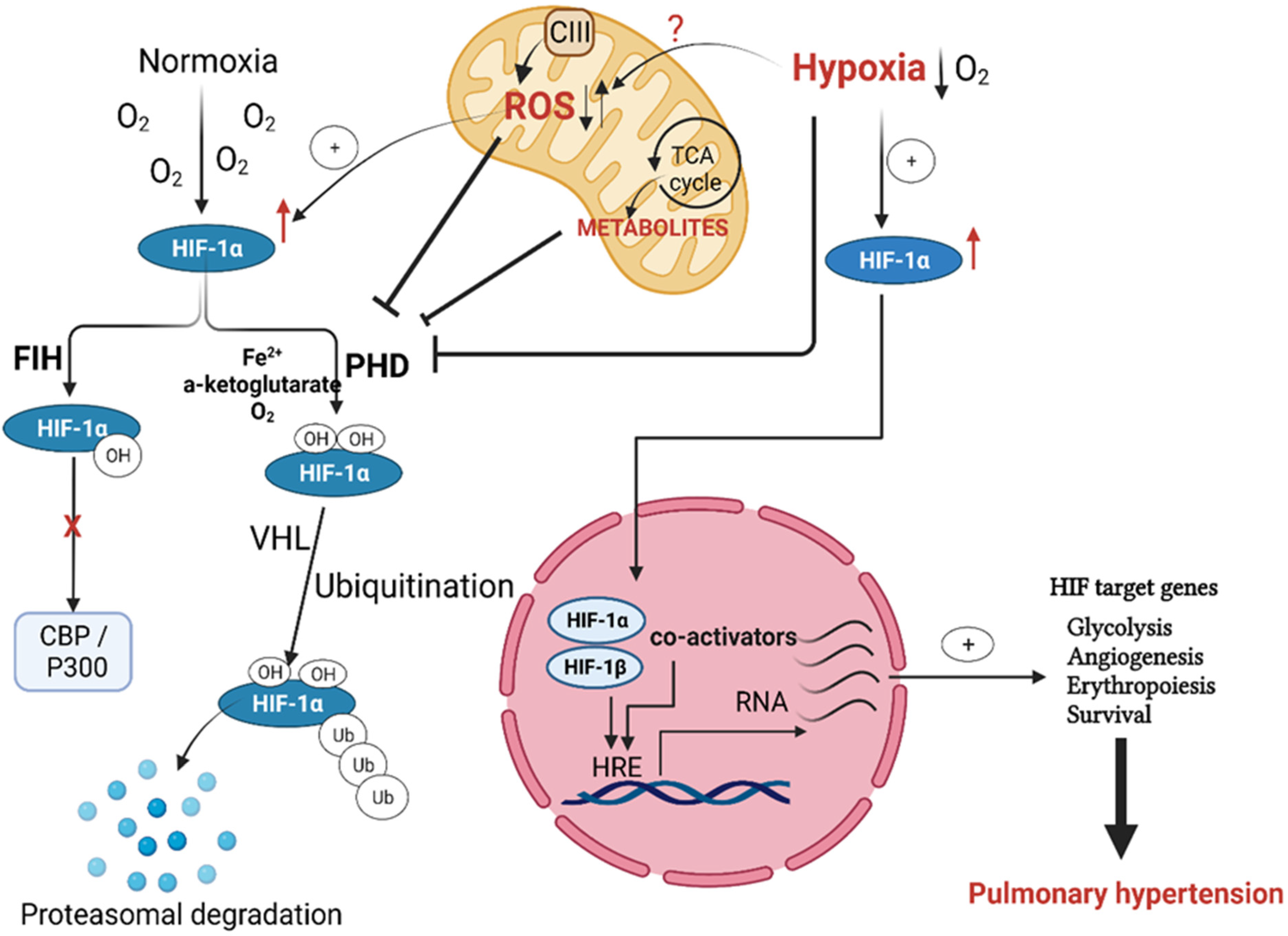
Regulation of Hypoxia Inducible Factor (HIF) in Hypoxia
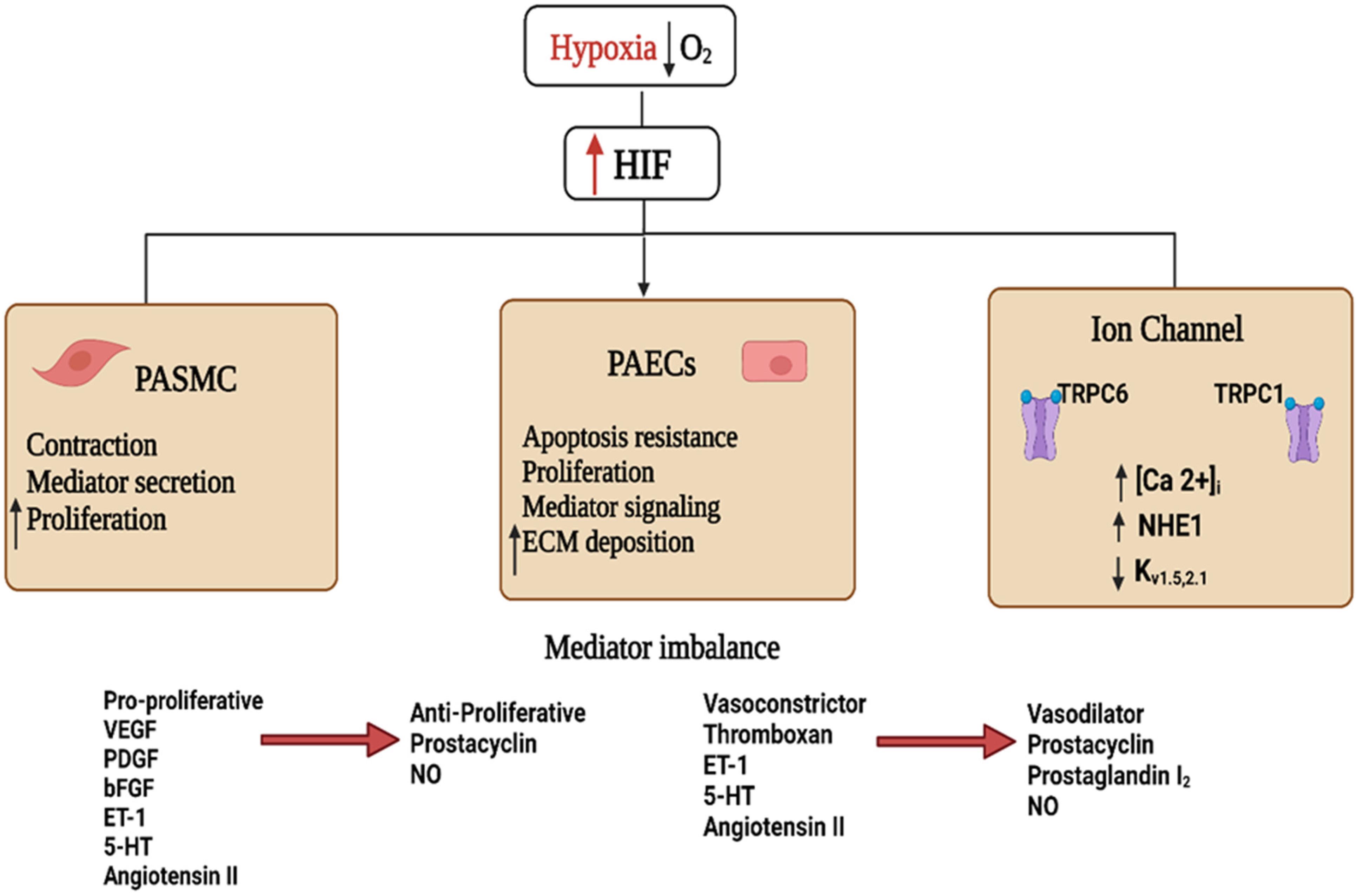
2. HIF Signaling in Hypoxia-Induced PH
3. Mitochondrial Regulation of HIF in Hypoxia-Induced PH
Downstream Signaling of mtROS in Chronic Hypoxia
4. Targeting HIF as a Potential Therapeutic Strategy in Pulmonary Hypertension
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Kardiologia Polska 2015, 73, 1127–1206. [Google Scholar] [CrossRef] [PubMed]
- McGee, M.; Whitehead, N.; Martin, J.; Collins, N. Drug-associated pulmonary arterial hypertension. Clin. Toxicol. 2018, 56, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 2016, 47, 288–303. [Google Scholar] [CrossRef]
- He, M.; Ma, S.; Cai, Q.; Wu, Y.; Shao, C.; Kong, H.; Wang, H.; Zeng, X.; Xie, W. Hypoxia induces the dysfunction of human endothelial colony-forming cells via HIF-1α signaling. Respir. Physiol. Neurobiol. 2018, 247, 87–95. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Investig. 2020, 130, 5638–5651. [Google Scholar] [CrossRef]
- Pak, O.; Aldashev, A.; Welsh, D.; Peacock, A. The effects of hypoxia on the cells of the pulmonary vasculature. Eur. Respir. J. 2007, 30, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Akkayagil, E.; Quanz, K.; Schermuly, R.; Ghofrani, A.; Fink, L.; Hänze, J.; Rose, F.; Seeger, W.; Grimminger, F. Basic features of hypoxic pulmonary vasoconstriction in mice. Respir. Physiol. Neurobiol. 2004, 139, 191–202. [Google Scholar] [CrossRef]
- Weissmann, N.; Grimminger, F.; Walmrath, D.; Seeger, W. Hypoxic vasoconstriction in buffer-perfused rabbit lungs. Respir. Physiol. 1995, 100, 159–169. [Google Scholar] [CrossRef]
- Peake, M.D.; Harabin, A.L.; Brennan, N.J.; Sylvester, J.T. Steady-state vascular responses to graded hypoxia in isolated lungs of five species. J. Appl. Physiol. 1981, 51, 1214–1219. [Google Scholar] [CrossRef]
- Sommer, N.; Hüttemann, M.; Pak, O.; Scheibe, S.; Knoepp, F.; Sinkler, C.; Malczyk, M.; Gierhardt, M.; Esfandiary, A.; Kraut, S.; et al. Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing. Circ. Res. 2017, 121, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, J.S.; Oyelade, T.; Aldhahir, A.M.; Alghamdi, S.M.; Almehmadi, M.; Alqahtani, A.S.; Quaderi, S.; Mandal, S.; Hurst, J.R. Prevalence, Severity and Mortality associated with COPD and Smoking in patients with COVID-19: A Rapid Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0233147. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Expression of hypoxia-inducible factor 1: Mechanisms and consequences. Biochem. Pharmacol. 1999, 59, 47–53. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Frid, M.G.; Mouradian, G.; Li, M.; et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2019, 54, 1900378. [Google Scholar] [CrossRef]
- Schumacker, P.T. Lung Cell Hypoxia: Role of Mitochondrial Reactive Oxygen Species Signaling in Triggering Responses. Proc. Am. Thorac. Soc. 2011, 8, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Nayak, B.; Shim, E.-H.; Livi, C.; Block, K.; Sudarshan, S. The Oncometabolite Fumarate Promotes Pseudohypoxia Through Noncanonical Activation of NF-κB Signaling. J. Biol. Chem. 2014, 289, 24691–24699. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and Maladaptive Cardiorespiratory Responses to Continuous and Intermittent Hypoxia Mediated by Hypoxia-Inducible Factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef]
- Veith, C.; Zakrzewicz, D.; Dahal, B.K.; Bálint, Z.; Murmann, K.; Wygrecka, M.; Seeger, W.; Schermuly, R.T.; Kwapiszewska, G.; Weissmann, N. Hypoxia- or PDGF-BB-dependent paxillin tyrosine phosphorylation in pulmonary hypertension is reversed by HIF-1α depletion or imatinib treatment. Thromb. Haemost. 2014, 112, 1288–1303. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of Hypoxia-inducible Transcription Factor Depends Primarily upon Redox-sensitive Stabilization of Its α Subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1α is mediated by an O 2 -dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Kim, D.; McQuiston, A.S.; Vinh, R.; Rockson, S.G.; Semenza, G.L.; Nicolls, M.R.; Jiang, X. Hypoxia and Hypoxia-Inducible Factors in Lymphedema. Front. Pharmacol. 2022, 13, 851057. [Google Scholar] [CrossRef]
- Martin, D.S.; Khosravi, M.; Grocott, M.P.; Mythen, M.G. Concepts in hypoxia reborn. Crit. Care 2010, 14, 315–317. [Google Scholar] [CrossRef]
- Veith, C.; Schermuly, R.; Brandes, R.; Weissmann, N. Molecular mechanisms of hypoxia-inducible factor-induced pulmonary arterial smooth muscle cell alterations in pulmonary hypertension. J. Physiol. 2015, 594, 1167–1177. [Google Scholar] [CrossRef]
- Xiang, L.; Semenza, G.L. Hypoxia-inducible factors promote breast cancer stem cell specification and maintenance in response to hypoxia or cytotoxic chemotherapy. Adv. Cancer Res. 2018, 141, 175–212. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Laurie, S.S. HIF and pulmonary vascular responses to hypoxia. J. Appl. Physiol. 2014, 116, 867–874. [Google Scholar] [CrossRef]
- Marshall, J.D.; Bazan, I.; Zhang, Y.; Fares, W.H.; Lee, P.J. Mitochondrial dysfunction and pulmonary hypertension: Cause, effect, or both. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L782–L796. [Google Scholar] [CrossRef]
- McElroy, G.; Chandel, N. Mitochondria control acute and chronic responses to hypoxia. Exp. Cell Res. 2017, 356, 217–222. [Google Scholar] [CrossRef]
- Alharbi, K.S.; Fuloria, N.K.; Fuloria, S.; Rahman, S.B.; Al-Malki, W.H.; Shaikh, M.A.J.; Thangavelu, L.; Singh, S.K.; Allam, V.S.R.R.; Jha, N.K.; et al. Nuclear factor-kappa B and its role in inflammatory lung disease. Chem. Interact. 2021, 345, 109568. [Google Scholar] [CrossRef] [PubMed]
- Pagé, E.L.; Robitaille, G.A.; Pouysségur, J.; Richard, D.E. Induction of Hypoxia-inducible Factor-1α by Transcriptional and Translational Mechanisms. J. Biol. Chem. 2002, 277, 48403–48409. [Google Scholar] [CrossRef] [PubMed]
- Pagé, E.L.; Chan, D.A.; Giaccia, A.J.; Levine, M.; Richard, D.E. Hypoxia-inducible Factor-1α Stabilization in Nonhypoxic Conditions: Role of Oxidation and Intracellular Ascorbate Depletion. Mol. Biol. Cell 2008, 19, 86–94. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Swiderska, A.; Coney, A.; Alzahrani, A.; Aldossary, H.; Batis, N.; Ray, C.; Kumar, P.; Holmes, A. Mitochondrial Succinate Metabolism and Reactive Oxygen Species Are Important but Not Essential for Eliciting Carotid Body and Ventilatory Responses to Hypoxia in the Rat. Antioxidants 2021, 10, 840. [Google Scholar] [CrossRef]
- Jernigan, N.L.; Resta, T.C. Calcium Homeostasis and Sensitization in Pulmonary Arterial Smooth Muscle. Microcirculation 2014, 21, 259–271. [Google Scholar] [CrossRef]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P.T. Hypoxic Pulmonary Vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef]
- Ball, M.K.; Waypa, G.B.; Mungai, P.T.; Nielsen, J.M.; Czech, L.; Dudley, V.J.; Beussink, L.; Dettman, R.W.; Berkelhamer, S.K.; Steinhorn, R.H.; et al. Regulation of Hypoxia-induced Pulmonary Hypertension by Vascular Smooth Muscle Hypoxia-Inducible Factor-1α. Am. J. Respir. Crit. Care Med. 2014, 189, 314–324. [Google Scholar] [CrossRef]
- Smith, K.A.; Waypa, G.B.; Dudley, V.J.; Budinger, G.R.S.; Abdala-Valencia, H.; Bartom, E.; Schumacker, P.T. Role of Hypoxia-Inducible Factors in Regulating Right Ventricular Function and Remodeling during Chronic Hypoxia–induced Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 63, 652–664. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Barnes, E.A.; Alvira, C.M.; Ying, L.; Reddy, S.; Cornfield, D.N. Hypoxia-inducible factor-1α in pulmonary artery smooth muscle cells lowers vascular tone by decreasing myosin light chain phosphorylation. Circ. Res. 2013, 112, 1230–1233. [Google Scholar] [CrossRef] [Green Version]
- Shan, F.; Li, J.; Huang, Q.-Y. HIF-1 Alpha-Induced Up-Regulation of miR-9 Contributes to Phenotypic Modulation in Pulmonary Artery Smooth Muscle Cells During Hypoxia. J. Cell. Physiol. 2014, 229, 1511–1520. [Google Scholar] [CrossRef]
- Zeng, Y.; Liu, H.; Kang, K.; Wang, Z.; Hui, G.; Zhang, X.; Zhong, J.; Peng, W.; Ramchandran, R.; Raj, J.U.; et al. Hypoxia inducible factor-1 mediates expression of miR-322: Potential role in proliferation and migration of pulmonary arterial smooth muscle cells. Sci. Rep. 2015, 5, 12098. [Google Scholar] [CrossRef]
- Awad, E.M.; Ahmed, A.-S.F.; El-Daly, M.; Amin, A.H.; El-Tahawy, N.F.; Wagdy, A.; Hollenberg, M.D.; Taye, A. Dihydromyricetin protects against high glucose-induced endothelial dysfunction: Role of HIF-1α/ROR2/NF-κB. Biomed. Pharmacother. 2022, 153, 113308. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, K.; Zheng, Q.; Zhang, C.; Tang, H.; Babicheva, A.; Jiang, Q.; Li, M.; Chen, Y.; Carr, S.G.; et al. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L216–L228. [Google Scholar] [CrossRef]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443. [Google Scholar] [CrossRef]
- Liu, J.; Wang, W.; Wang, L.; Chen, S.; Tian, B.; Huang, K.; Corrigan, C.J.; Ying, S.; Wang, W.; Wang, C. IL-33 Initiates Vascular Remodelling in Hypoxic Pulmonary Hypertension by up-Regulating HIF-1α and VEGF Expression in Vascular Endothelial Cells. eBioMedicine 2018, 33, 196–210. [Google Scholar] [CrossRef]
- Freund-Michel, V.; Khoyrattee, N.; Savineau, J.-P.; Muller, B.; Guibert, C. Mitochondria: Roles in pulmonary hypertension. Int. J. Biochem. Cell Biol. 2014, 55, 93–97. [Google Scholar] [CrossRef]
- Makker, K.; Afolayan, A.J.; Teng, R.-J.; Konduri, G.G. Altered hypoxia-inducible factor-1α (HIF-1α) signaling contributes to impaired angiogenesis in fetal lambs with persistent pulmonary hypertension of the newborn (PPHN). Physiol. Rep. 2019, 7, e13986. [Google Scholar] [CrossRef]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaço, R.D.D.R.; Southwood, M.; Toshner, M.; Alexander, L.E.C.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2α–arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef]
- Wang, J.; Weigand, L.; Lu, W.; Sylvester, J.; Semenza, G.L.; Shimoda, L.A. Hypoxia Inducible Factor 1 Mediates Hypoxia-Induced TRPC Expression and Elevated Intracellular Ca 2+ in Pulmonary Arterial Smooth Muscle Cells. Circ. Res. 2006, 98, 1528–1537. [Google Scholar] [CrossRef] [Green Version]
- Malczyk, M.; Veith, C.; Fuchs, B.; Hofmann, K.; Storch, U.; Schermuly, R.T.; Witzenrath, M.; Ahlbrecht, K.; Fecher-Trost, C.; Flockerzi, V.; et al. Classical Transient Receptor Potential Channel 1 in Hypoxia-induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 1451–1459. [Google Scholar] [CrossRef]
- Wang, J.; Juhaszova, M.; Rubin, L.J.; Yuan, X.J. Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J. Clin. Investig. 1997, 100, 2347–2353. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Fallon, M.; Pisarcik, S.; Wang, J.; Semenza, G.L. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am. J. Physiol. Cell. Mol. Physiol. 2006, 291, L941–L949. [Google Scholar] [CrossRef] [PubMed]
- Rios, E.J.; Fallon, M.; Wang, J.; Shimoda, L.A. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2005, 289, L867–L874. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Mizuno, S.; Bogaard, H.J. The role of hypoxia in pulmonary vascular diseases: A perspective. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L457–L465. [Google Scholar] [CrossRef]
- Farghaly, T.A.; Al-Hasani, W.A.; Abdulwahab, H.G. An updated patent review of VEGFR-2 inhibitors (2017-present). Expert Opin. Ther. Patents 2021, 31, 989–1007. [Google Scholar] [CrossRef]
- Fröhlich, S.; Boylan, J.; McLoughlin, P. Hypoxia-Induced Inflammation in the Lung. Am. J. Respir. Cell Mol. Biol. 2013, 48, 271–279. [Google Scholar] [CrossRef]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.N.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Circ. Physiol. 2008, 294, H570–H578. [Google Scholar] [CrossRef]
- Wujak, M.; Veith, C.; Wu, C.-Y.; Wilke, T.; Kanbagli, Z.I.; Novoyatleva, T.; Guenther, A.; Seeger, W.; Grimminger, F.; Sommer, N.; et al. Adenylate Kinase 4—A Key Regulator of Proliferation and Metabolic Shift in Human Pulmonary Arterial Smooth Muscle Cells via Akt and HIF-1α Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 10371. [Google Scholar] [CrossRef]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.-H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1–Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Clin. Trans. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Chen, K.-H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension. Circulation 2018, 138, 287–304. [Google Scholar] [CrossRef]
- Waypa, G.B.; Schumacker, P.T. Hypoxia-induced changes in pulmonary and systemic vascular resistance: Where is the O2 sensor? Respir. Physiol. Neurobiol. 2010, 174, 201–211. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2015, 61, 199–209. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Bonnet, S.; Michelakis, E.D.; Porter, C.; Andrade, M.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An Abnormal Mitochondrial–Hypoxia Inducible Factor-1α–Kv Channel Pathway Disrupts Oxygen Sensing and Triggers Pulmonary Arterial Hypertension in Fawn Hooded Rats. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos y Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia–reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-Induced Pulmonary Vascular Remodeling. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F.; et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2018, 51, 1701024. [Google Scholar] [CrossRef]
- Weir, E.K.; Archer, S.L. The role of redox changes in oxygen sensing. Respir. Physiol. Neurobiol. 2010, 174, 182–191. [Google Scholar] [CrossRef]
- Gnaiger, E. Bioenergetics at low oxygen: Dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir. Physiol. 2001, 128, 277–297. [Google Scholar] [CrossRef]
- Buckler, K.J.; Turner, P.J. Oxygen sensitivity of mitochondrial function in rat arterial chemoreceptor cells. J. Physiol. 2013, 591, 3549–3563. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Y.; Lu, L.; Wang, L.; Chen, M.; Hu, T. Expression and Correlation of Hypoxia-Inducible Factor-1α (HIF-1α) with Pulmonary Artery Remodeling and Right Ventricular Hypertrophy in Experimental Pulmonary Embolism. Med Sci. Monit. 2017, 23, 2083–2088. [Google Scholar] [CrossRef]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.-C.; Schermuly, R.; Ghofrani, A.; Kwapiszewska, G.; et al. Hypoxia-Dependent Regulation of Nonphagocytic NADPH Oxidase Subunit NOX4 in the Pulmonary Vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef]
- Clerici, C.; Planès, C. Gene regulation in the adaptive process to hypoxia in lung epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L267–L274. [Google Scholar] [CrossRef]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.K.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef]
- Truong, L.; Zheng, Y.-M.; Wang, Y.-X. The Potential Important Role of Mitochondrial Rieske Iron–Sulfur Protein as a Novel Therapeutic Target for Pulmonary Hypertension in Chronic Obstructive Pulmonary Disease. Biomedicines 2022, 10, 957. [Google Scholar] [CrossRef]
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide Generated at Mitochondrial Complex III Triggers Acute Responses to Hypoxia in the Pulmonary Circulation. Am. J. Respir. Crit. Care Med. 2013, 187, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Kang, B.-Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Hart, C.M.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Helbock, H.J.; Beckman, K.B.; Ames, B.N. 8-Hydroxydeoxyguanosine and 8-hydroxyguanine as biomarkers of oxidative DNA damage. Methods Enzym. 1999, 300, 156–166. [Google Scholar] [CrossRef]
- Almalki, W.H.; Alzahrani, A.; El-Daly, M.E.-S.M.; Ahmed, A.S.H.F.F. The emerging potential of SIRT-3 in oxidative stress-inflammatory axis associated increased neuroinflammatory component for metabolically impaired neural cell. Chem. Interactions 2020, 333, 109328. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.; Cardoso, S.M.; Clish, C.; et al. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1α Destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.H.; Guarente, L.P. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef]
- Waypa, G.B.; Osborne, S.W.; Marks, J.D.; Berkelhamer, S.K.; Kondapalli, J.; Schumacker, P.T. Sirtuin 3 Deficiency Does Not Augment Hypoxia-Induced Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 885–891. [Google Scholar] [CrossRef]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of Hypoxia-inducible Factor-1α Protein in Hypoxia Occurs Independently of Mitochondrial Reactive Oxygen Species Production*. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-inducible Factor-1α during Hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible Factor-1 Activation in Nonhypoxic Conditions: The Essential Role of Mitochondrial-derived Reactive Oxygen Species. Mol. Biol. Cell 2010, 21, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Fijalkowska, I.; Xu, W.; Comhair, S.A.; Janocha, A.J.; Mavrakis, L.A.; Krishnamachary, B.; Zhen, L.; Mao, T.; Richter, A.; Erzurum, S.C.; et al. Hypoxia Inducible-Factor1α Regulates the Metabolic Shift of Pulmonary Hypertensive Endothelial Cells. Am. J. Pathol. 2010, 176, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Chandel, N.S. Genetics of Mitochondrial Electron Transport Chain in Regulating Oxygen Sensing. Methods Enzymol. 2007, 435, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef]
- Hewitson, K.S.; Liénard, B.M.; McDonough, M.; Clifton, I.J.; Butler, D.; Soares, A.S.; Oldham, N.J.; McNeill, L.A.; Schofield, C.J. Structural and Mechanistic Studies on the Inhibition of the Hypoxia-inducible Transcription Factor Hydroxylases by Tricarboxylic Acid Cycle Intermediates. J. Biol. Chem. 2007, 282, 3293–3301. [Google Scholar] [CrossRef]
- Koivunen, P.; Hirsilä, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of Hypoxia-inducible Factor (HIF) Hydroxylases by Citric Acid Cycle Intermediates. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef]
- Sommer, N.; Alebrahimdehkordi, N.; Pak, O.; Knoepp, F.; Strielkov, I.; Scheibe, S.; Dufour, E.; Andjelković, A.; Sydykov, A.; Saraji, A.; et al. Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci. Adv. 2020, 6, eaba0694. [Google Scholar] [CrossRef]
- Giordano, L.; Farnham, A.; Dhandapani, P.K.; Salminen, L.; Bhaskaran, J.; Voswinckel, R.; Rauschkolb, P.; Scheibe, S.; Sommer, N.; Beisswenger, C.; et al. Alternative Oxidase Attenuates Cigarette Smoke-induced Lung Dysfunction and Tissue Damage. Am. J. Respir. Cell Mol. Biol. 2019, 60, 515–522. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457.e13–470.e13. [Google Scholar] [CrossRef] [Green Version]
- Szibor, M.; Dhandapani, P.K.; Dufour, E.; Holmström, K.; Zhuang, Y.; Salwig, I.; Wittig, I.; Heidler, J.; Gizatullina, Z.; Gainutdinov, T.; et al. Broad AOX expression in a genetically tractable mouse model does not disturb normal physiology. Dis. Model. Mech. 2016, 10, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M.; Ebert, B.L.; Neil, C.; Brenner, K.; Papaioannou, I.; Melas, A.; Tolliday, N.; Lamb, J.; Pantopoulos, K.; Golub, T.; et al. Small-Molecule Inhibitors of HIF-2a Translation Link Its 5′UTR Iron-Responsive Element to Oxygen Sensing. Mol. Cell 2008, 32, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.-Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2α Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.W.; Kim, S.-Y.; Lee, J.H.; Lee, Y.-S. YC-1 attenuates hypoxia-induced pulmonary arterial hypertension in mice. Pulm. Pharmacol. Ther. 2011, 24, 638–646. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, Y.; Peng, G.; Liu, N.; Tian, H.; Pan, D.; Liu, L.; Yang, X.; Li, C.; Li, W.; et al. Topotecan prevents hypoxia-induced pulmonary arterial hypertension and inhibits hypoxia-inducible factor-1α and TRPC channels. Int. J. Biochem. Cell Biol. 2018, 104, 161–170. [Google Scholar] [CrossRef]
- Kurosawa, R.; Satoh, K.; Kikuchi, N.; Kikuchi, H.; Saigusa, D.; Al-Mamun, E.; Siddique, M.A.; Omura, J.; Satoh, T.; Sunamura, S.; et al. Identification of Celastramycin as a Novel Therapeutic Agent for Pulmonary Arterial Hypertension. Circ. Res. 2019, 125, 309–327. [Google Scholar] [CrossRef]
- Docherty, C.; Nilsen, M.; MacLean, M.R. Influence of 2-Methoxyestradiol and Sex on Hypoxia-Induced Pulmonary Hypertension and Hypoxia-Inducible Factor-1-α. J. Am. Heart Assoc. 2019, 8, e011628. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, Q.; Yuan, Y.; Li, Y.; Gong, X. Effects of 17β-estradiol and 2-methoxyestradiol on the oxidative stress-hypoxia inducible factor-1 pathway in hypoxic pulmonary hypertensive rats. Exp. Ther. Med. 2017, 13, 2537–2543. [Google Scholar] [CrossRef]
- He, Y.; Fang, X.; Shi, J.; Li, X.; Xie, M.; Liu, X. Apigenin attenuates pulmonary hypertension by inducing mitochondria-dependent apoptosis of PASMCs via inhibiting the hypoxia inducible factor 1α–KV1.5 channel pathway. Chem. Interactions 2020, 317, 108942. [Google Scholar] [CrossRef]
- Abud, E.M.; Maylor, J.; Undem, C.; Punjabi, A.; Zaiman, A.L.; Myers, A.C.; Sylvester, J.T.; Semenza, G.L.; Shimoda, L.A. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1239–1244. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Teng, X.; Zhang, L.; Chen, J.; Liu, Z.; Chen, X.; Zhao, S.; Yang, S.; Feng, J.; Yan, X. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat. Commun. 2019, 10, 3551. [Google Scholar] [CrossRef]
- Chen, T.; Zhou, Q.; Tang, H.; Bozkanat, M.; Yuan, J.X.; Raj, J.U.; Zhou, G. miR-17/20 Controls Prolyl Hydroxylase 2 (PHD2)/Hypoxia-Inducible Factor 1 (HIF1) to Regulate Pulmonary Artery Smooth Muscle Cell Proliferation. J. Am. Heart Assoc. 2016, 5, e004510. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Chi, P.-L.; Shen, M.-C.; Shu, C.-W.; Wann, S.-R.; Liu, C.-P.; Tseng, C.-J.; Huang, W.-C. Caffeic Acid Phenethyl Ester Rescues Pulmonary Arterial Hypertension through the Inhibition of AKT/ERK-Dependent PDGF/HIF-1α In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 1468. [Google Scholar] [CrossRef] [PubMed]
- Antoniu, S.A. Targeting PDGF pathway in pulmonary arterial hypertension. Expert Opin. Ther. Targets 2012, 16, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Gou, D.; Ramchandran, R.; Peng, X.; Yao, L.; Kang, K.; Sarkar, J.; Wang, Z.; Zhou, G.; Raj, J.U. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L682–L691. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. J. Cereb. Blood Flow Metab. 2020, 178, 6–30. [Google Scholar] [CrossRef]
- Lachmanová, V.; Hniličková, O.; Povýšilová, V.; Hampl, V.; Herget, J. N-acetylcysteine inhibits hypoxic pulmonary hypertension most effectively in the initial phase of chronic hypoxia. Life Sci. 2005, 77, 175–182. [Google Scholar] [CrossRef]
- Redout, E.M.; Van Der Toorn, A.; Zuidwijk, M.J.; Van De Kolk, C.W.A.; Van Echteld, C.J.A.; Musters, R.J.P.; Van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am. J. Physiol. Circ. Physiol. 2010, 298, H1038–H1047. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Steinhorn, R.H.; Gladwin, M.T. Antioxidant Therapy for the Treatment of Pulmonary Hypertension. Antioxidants Redox Signal. 2013, 18, 1723–1726. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Compound | Experimental Setting | Target | Main Findings | Ref. |
|---|---|---|---|---|
| Compound 76 (C76) | In vitro: PASMCs, PAECs, lung samples from iPAH patients In vivo: MCT and SuHx models of PAH | ⇑ IRP1 to inhibit HIF2α signaling | ⇓ RVSP ⇓ RVH ⇓ RVR | [102,103] |
| 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole (yc-1) | In vitro: hPASMCs exposed to hypoxia In vivo: chronic hypoxia (28 day) PH mouse model | ⇑ sGC signaling to inhibit HIF1α expression | ⇓ PVR ⇓ RVH | [104] |
| Topotecan (TPT) | In vitro: hPASMCs exposed to hypoxia In vivo: chronic hypoxia (28 day) PH rat model PH | ⇓ HIF1α protein accumulation. ⇓ HIF1α target genes expression | ⇓ PASMCs growth. ⇓ PVR | [105] |
| Celastramycin | In vitro: PASMCs from iPAH patients. | ⇓ HIF1α | ⇓ PASMCs growth. ⇓ oxidative stress and inflammation. | [106] |
| 2-methoxyestradiol (2-ME2) | In vitro: hPASMCs exposed to hypoxia In vivo: chronic hypoxia (28 day) PH rat model PH | ⇑ MnSOD activity ⇓ ROS production ⇓ HIF1α expression | ⇓ PASMCs growth ⇓ RVSP ⇓ RVH ⇓ PVR | [107,108] |
| Apigenin | In vivo: chronic hypoxia (28 day) PH rat model PH | ⇓ Akt signaling ⇓ HIF1α expression | ⇓ RVH ⇓ PVR | [109] |
| Digoxin | In vivo: chronic hypoxia (28 day) PH mouse model PH | ⇓ HIF1α transcription and protein synthesis | ⇓ RVP ⇓ RVR | [110] |
| Anti-CD146 mAb AA98 | In vivo: chronic hypoxia (28 day) PH mouse model PH, and MCT mouse model | ⇓ CD146 dimerization and ⇓ HIF1α response | ⇑ cardiac function ⇓ PH development | [111] |
| PHD2 activator (R59949) | In vivo: chronic hypoxia (28 day) PH mouse model PH | ⇑ PHD2 and ⇓ HIF1α levels | ⇓ PVR | [112] |
| Caffeic acid phenethyl ester (CAPE) | In vivo: MCT mouse model | ⇓ AKT/ERK activation and ⇓ HIF1α expression | ⇓ proliferation & apoptosis resistance. ⇓ RVSP ⇓ PVR | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeidan, E.M.; Hossain, M.A.; El-Daly, M.; Abourehab, M.A.S.; Khalifa, M.M.A.; Taye, A. Mitochondrial Regulation of the Hypoxia-Inducible Factor in the Development of Pulmonary Hypertension. J. Clin. Med. 2022, 11, 5219. https://doi.org/10.3390/jcm11175219
Zeidan EM, Hossain MA, El-Daly M, Abourehab MAS, Khalifa MMA, Taye A. Mitochondrial Regulation of the Hypoxia-Inducible Factor in the Development of Pulmonary Hypertension. Journal of Clinical Medicine. 2022; 11(17):5219. https://doi.org/10.3390/jcm11175219
Chicago/Turabian StyleZeidan, Esraa M., Mohammad Akbar Hossain, Mahmoud El-Daly, Mohammed A. S. Abourehab, Mohamed M. A. Khalifa, and Ashraf Taye. 2022. "Mitochondrial Regulation of the Hypoxia-Inducible Factor in the Development of Pulmonary Hypertension" Journal of Clinical Medicine 11, no. 17: 5219. https://doi.org/10.3390/jcm11175219
APA StyleZeidan, E. M., Hossain, M. A., El-Daly, M., Abourehab, M. A. S., Khalifa, M. M. A., & Taye, A. (2022). Mitochondrial Regulation of the Hypoxia-Inducible Factor in the Development of Pulmonary Hypertension. Journal of Clinical Medicine, 11(17), 5219. https://doi.org/10.3390/jcm11175219