
Hajdu-Cheney Syndrome: A Novel NOTCH2 Mutation in a Spanish Child in Treatment with Vibrotherapy: A Case Report

 ,
,  ,
, 
 ,
, 
Abstract
:1. Introduction
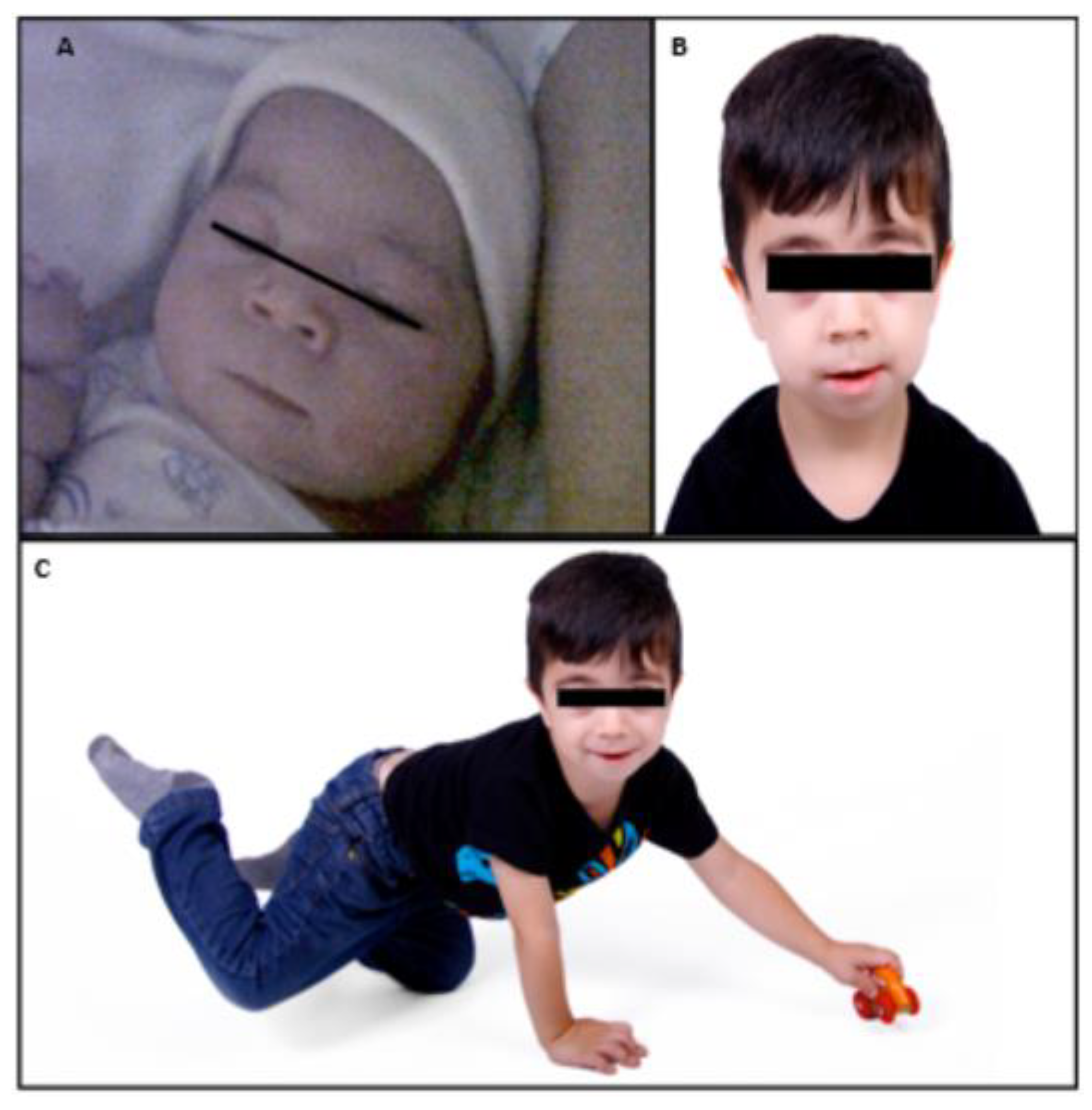
2. Patient Information
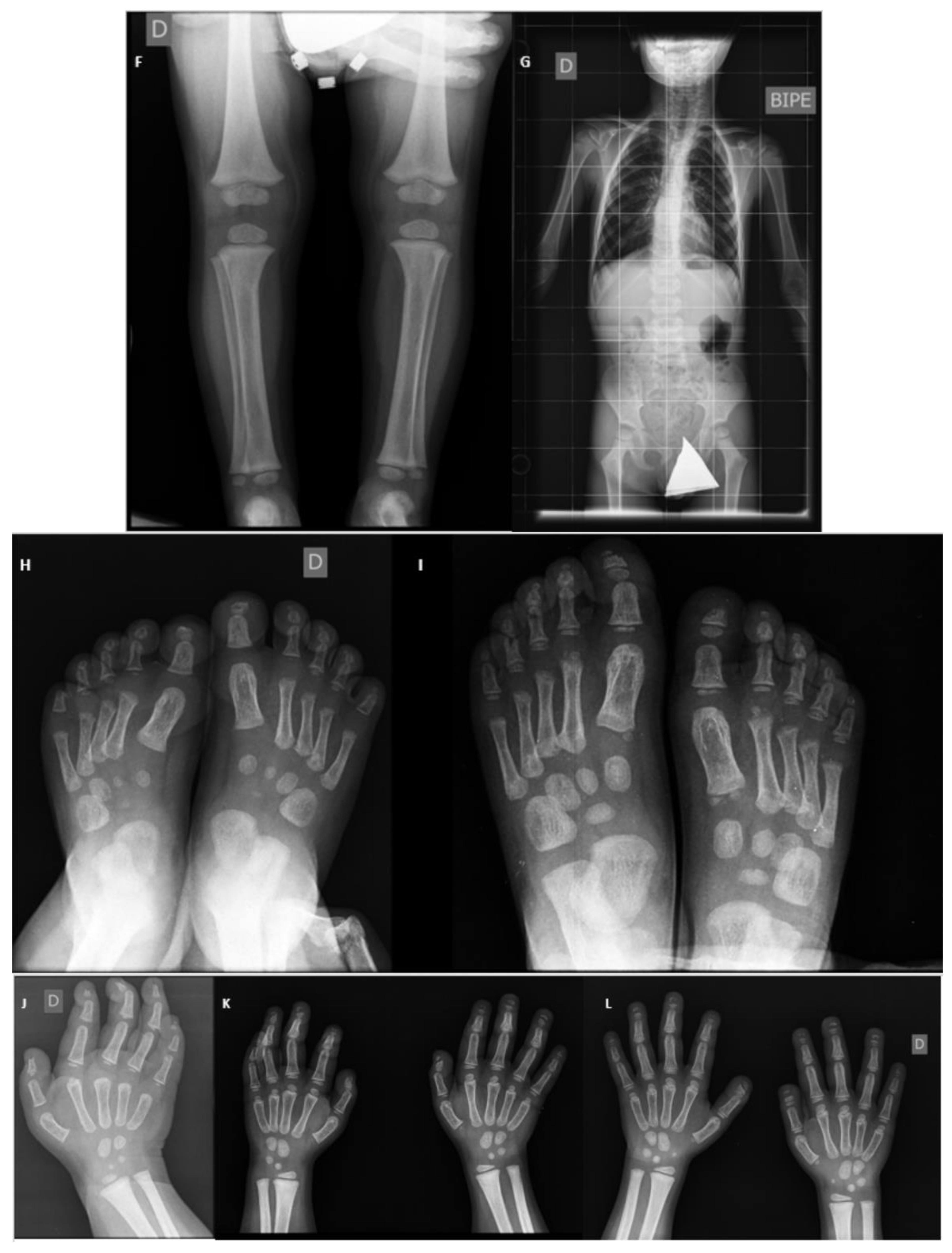
3. Musculoskeletal Features
4. Endocrine Aspects
5. Respiratory Features
6. Cardiological Aspects
7. Neurosurgery Aspects
8. Neurology Aspects
9. Early Care
10. Ophthalmological Aspects
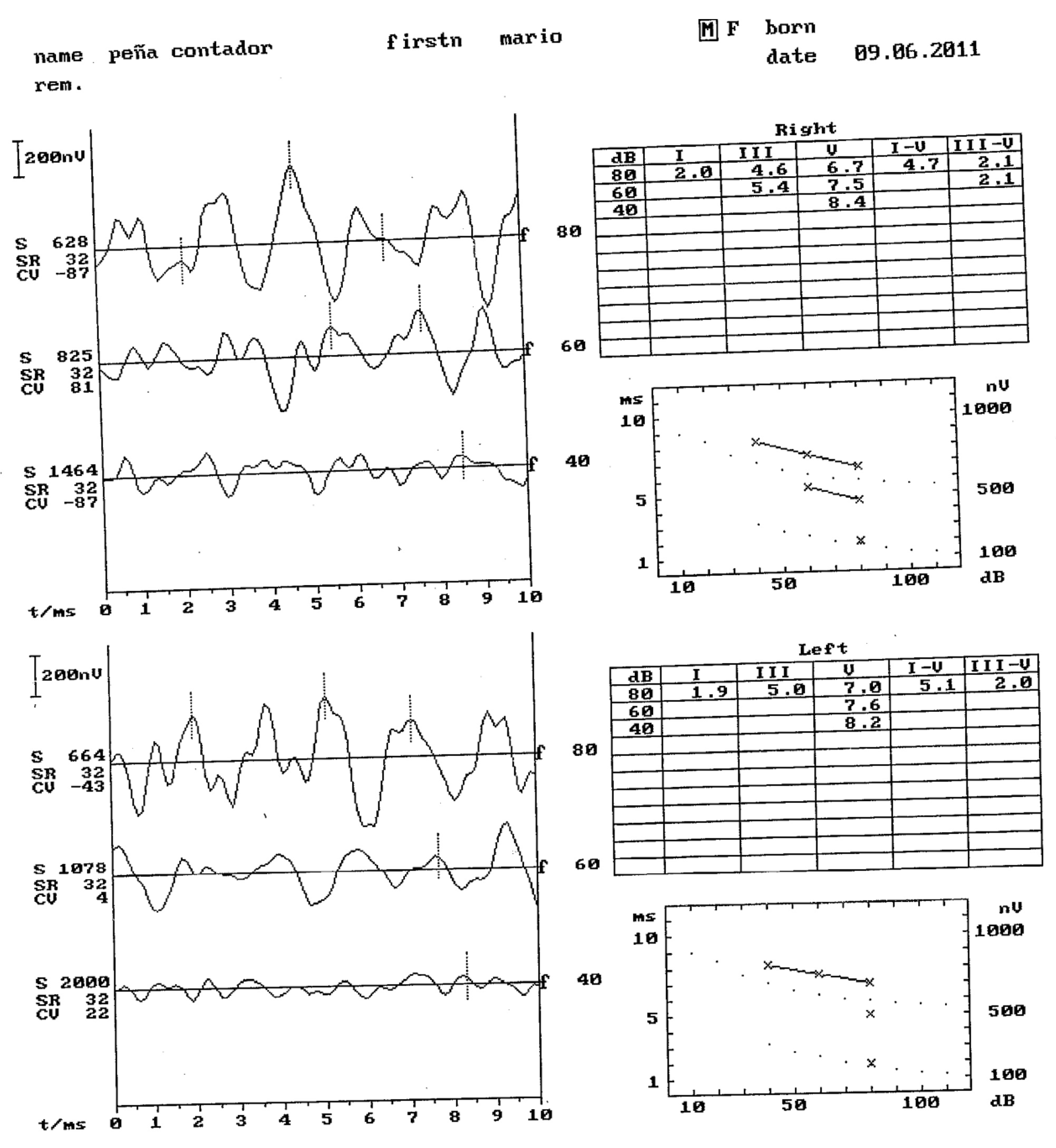
11. Otorrine Aspects
12. Maxillofacial Features
13. Gait Study
14. Psychosocial Aspects and Lifestyle
15. Milestones
16. Prognosis
17. Therapeutic Intervention
18. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Enfermedades Raras: Comprender Esta Prioridad de Salud Pública; Organización Europea de Enfermedades Raras de París: Paris, France, 2005.
- Orphanet. Available online: https://www.orpha.net/consor/cgi-bin/Disease.php?lng=ES (accessed on 31 July 2022).
- OMIM. Available online: https://www.omim.org/entry/102500?search=%22hajdu-cheney syndrome%22&highlight=0%2C spanNear%28%5BspanOr%28%5Bhajducheney%2C%7CspanNear%28%5Bhajdu%2C%7Ccheney%5D%2C%7C0%2C%7Ctrue%29%5D%29%2CspanOr%28%5Bsyndromic%2C%7Csyndrome%5D%29%5D%2C true%29 (accessed on 31 July 2022).
- Singh, J.A.; Williams, C.B.; McAlister, W.H. Talo-patello-scaphoid osteolysis, synovitis, and short fourth metacarpals in sisters: A new syndrome? Am. J. Med. Genet. 2003, 121A, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Orphanet: Acroosteolisis Tipo Dominante. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=ES&data_id=1276&Disease_Disease_Search_diseaseGroup=Hajdu-cheney&Disease_Disease_Search_diseaseType=Pat&Enfermedad(es)/grupo%20de%20enfermedad-es=Acroosteolisis-tipo-dominante&title=Acroosteolisis%20tip (accessed on 31 July 2022).
- Simpson, M.A.; Irving, M.D.; Asilmaz, E.; Gray, M.J.; Dafou, D.; Elmslie, F.V.; Mansour, S.; Holder, S.E.; Brain, C.E.; Burton, B.K.; et al. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011, 43, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Descartes, M.; Rojnueangnit, K.; Cole, L.; Sutton, A.; Morgan, S.L.; Patry, L.; Samuels, M.E. Hajdu-Cheney syndrome: Phenotypical progression with de-novo NOTCH2 mutation. Clin. Dysmorphol. 2014, 23, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Majewski, J.; Schwartzentruber, J.A.; Caqueret, A.; Patry, L.; Marcadier, J.; Fryns, J.P.; Boycott, K.M.; Ste-Marie, L.G.; Mckiernan, F.E.; Marik, I.; et al. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 2011, 32, 1114–1117. [Google Scholar] [CrossRef]
- Hajdu, N.; Kauntze, R. Cranio-skeletal dysplasia. Br. J. Radiol. 1948, 21, 42–48. [Google Scholar] [CrossRef]
- Cheney, W. Acro-Osteolysis. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1965, 94, 595–607. [Google Scholar]
- Brennan, A.M.; Pauli, R.M. Hajdu-Cheney syndrome: Evolution of phenotype and clinical problems. Am. J. Med. Genet. 2001, 100, 292–310. [Google Scholar] [CrossRef]
- Regev, M.; Pode-Shakked, B.; Jacobson, J.M.; Raas-Rothschild, A.; Goldstein, D.B.; Anikster, Y. Phenotype variability in Hajdu-Cheney syndrome. Eur. J. Med. Genet. 2019, 62, 35–38. [Google Scholar] [CrossRef]
- Brown, D.M.; Bradford, D.S.; Gorlin, R.J.; Desnick, R.J.; Langer, L.O.; Jowsey, J.; Sauk, J.J. The acro-osteolysis syndrome: Morphologic and biochemical studies. J. Pediatr. 1976, 88, 573–580. [Google Scholar] [CrossRef]
- Letchumanan, P.; Thumboo, J.; Leong, R.T.K. A patient with progressive shortening of the fingers. J. Rheumatol. 2009, 36, 198–199. [Google Scholar] [CrossRef]
- Bazopoulou-Kyrkanidou, E.; Vrahopoulos, T.P.; Eliades, G.; Vastardis, H.; Tosios, K.; Vrotsos, I.A. Periodontitis Associated with Hajdu-Cheney Syndrome. J. Periodontol. 2007, 78, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Siklar, Z.; Tanyer, G.; Dallar, Y.; Aksoy, F.G. Hajdu-Cheney syndrome with growth hormone deficiency and neuropathy. J. Pediatr. Endocrinol. Metab. 2000, 13, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.J.; Saba, C.; Rennert, O.M. Kidney abnormalities in Hajdu-Cheney syndrome. Pediatr. Nephrol. 1996, 10, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Sargin, G.; Cildag, S.; Senturk, T. Hajdu-Cheney syndrome with ventricular septal defect. Kaohsiung J. Med. Sci. 2013, 29, 343–344. [Google Scholar] [CrossRef]
- Currarino, G. Hajdu-Cheney syndrome associated with serpentine fibulae and polycystic kidney disease. Pediatr. Radiol. 2009, 39, 47–52. [Google Scholar] [CrossRef]
- Sawin, P.D.; Menezes, A.H. Basilar invagination in osteogenesis imperfecta and related osteochondrodysplasias: Medical and surgical management. J. Neurosurg. 1997, 86, 950–960. [Google Scholar] [CrossRef]
- Nishimura, G.; Aoaki, K.; Haga, N.; Hasegawa, T.; Aoki, K.; Haga, N.; Hasegawa, T. Syringohydromyelia in Hajdu-Cheney syndrome. Pediatr. Radiol. 1996, 26, 59–61. [Google Scholar] [CrossRef]
- Schawo, S.; Weber, M.A.; Libicher, M. Junge frau mit rückenschmerzen und akroosteolysen. Radiologe 2006, 46, 901–904. [Google Scholar] [CrossRef]
- O’Reilly, M.A.R.; Shaw, D.G. Hajdu-Cheney syndrome. Ann. Rheum. Dis. 1994, 53, 276–279. [Google Scholar] [CrossRef]
- Gibofsky, A. Genetics of the hajdu-cheney syndrome. Arthritis Rheum. 1987, 30, 718. [Google Scholar] [CrossRef]
- Cortés-Martín, J.; Díaz-Rodríguez, L.; Piqueras-Sola, B.; Sánchez-García, J.C.; Menor-Rodríguez, M.J.; Rodríguez-Blanque, R. Nursing Care Plan for Patients with Hajdu-Cheney Syndrome. Int. J. Environ. Res. Public Health 2022, 19, 7489. [Google Scholar] [CrossRef] [PubMed]
- Pittaway, J.F.H.; Harrison, C.; Rhee, Y.; Holder-Espinasse, M.; Fryer, A.E.; Cundy, T.; Drake, W.M.; Irving, M.D. Bisphosphonate therapy for spinal osteoporosis in Hajdu-Cheney syndrome—New data and literature review. Orphanet J. Rare Dis. 2018, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Moraleda-Barreno, E.; Romero-López, M.; Cayetano-Menéndez, M.J. La prueba de cribado del inventario de desarrollo de Battelle para la detección precoz de alteraciones del desarrollo en parálisis cerebral. An. Pediatr. 2011, 75, 372–379. [Google Scholar] [CrossRef] [PubMed]
- García, E.; Padilla, I.; Franco, A. Vibroterapia en la inhibición de la espasticidad asociada a la enfermedad motriz cerebral. Rev. Iberoam. Fisioter. Kinesiol. 2003, 4, 66–74. [Google Scholar]
- Evans, W.R.H. Dare to think rare: Diagnostic delay and rare diseases. Br. J. Gen. Pract. 2018, 68, 224–225. [Google Scholar] [CrossRef]
- Feder. Enfermedades Raras. Available online: https://www.enfermedades-raras.org/enfermedades-raras/conoce-mas-sobre-er (accessed on 31 July 2022).
- Herrmann, J.; Zugibe, F.T.; Gilbert, E.F.; Opitz, J.M. Arthro-Dento-Osteo Dysplasia (Hajdu-Cheney Syndrome). Z. Kinderheilk. 1973, 114, 93–110. [Google Scholar] [CrossRef]
- Elmore, S.M. Pycnodysostosis: A review. J. Bone Jt. Surg. Am. 2019, 49, 153–163. [Google Scholar] [CrossRef]
- Narumi, Y.; Min, B.J.; Shimizu, K.; Kazukawa, I.; Sameshima, K.; Nakamura, K.; Kosho, T.; Rhee, Y.; Chung, Y.S.; Kim, O.H.; et al. Clinical consequences in truncating mutations in exon 34 of NOTCH2: Report of six patients with Hajdu-Cheney syndrome and a patient with serpentine fibula polycystic kidney syndrome. Am. J. Med. Genet. Part A 2013, 161, 518–526. [Google Scholar] [CrossRef]
- Ahmad, A.; Deeb, H.; Alasmar, D. Hajdu Cheney syndrome; A novel NOTCH2 mutation in a Syrian child, and treatment with zolidronic acid: A case report and a literature review of treatments. Ann. Med. Surg. 2021, 71, 103023. [Google Scholar] [CrossRef]
- Swan, L.; Gole, G.; Sabesan, V.; Cardinal, J.; Coman, D. Congenital Glaucoma: A Novel Ocular Manifestation of Hajdu-Cheney Syndrome. Case Rep. Genet. 2018, 2018, 2508345. [Google Scholar] [CrossRef]
- Ades, L.C.; Morris, L.L.; Haan, E.A. Hydrocephalus in Hajdu-Cheney syndrome. J. Med. Genet. 1993, 30, 175. [Google Scholar] [CrossRef] [PubMed]
- Takatani, R.; Someya, T.; Kazukawa, I.; Nishimura, G.; Minagawa, M.; Kohno, Y. Hajdu-Cheney syndrome: Infantile onset of hydrocephalus and serpentine fibulae. Pediatr. Int. 2009, 51, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Harnasch, H. Die Akroosteolysis, ein neues Krankheitsbild. RöFo-Fortschr. Geb. Röntgenstrahlen Bildgeb. Verfahr. 1949, 72, 352–359. [Google Scholar] [CrossRef]
- Rosenmann, E.; Penchas, S.; Cohen, T.; Aviad, I. Sporadic idiopathic acro-osteolysis with cranio-skeletal dysplasia, polycystic kidneys and glomerulonephritis: A case of the hajdu-cheney syndrome. Pediatr. Radiol. 1977, 6, 116–120. [Google Scholar] [CrossRef]
- Elias, A.N.; Pinals, R.S.; Clarke Anderson, H.; Gould, L.V.; Streeten, D.H.P. Hereditary osteodysplasia with acro-osteolysis (the Hajdu-Cheney syndrome). Am. J. Med. 1978, 65, 627–636. [Google Scholar] [CrossRef]
- Leidig-Bruckner, G.; Pfeilschifter, J.; Penning, N.; Limberg, B.; Priemel, M.; Delling, G.; Ziegler, R. Severe osteoporosis in familial Hajdu-Cheney syndrome: Progression of acro-osteolysis and osteoporosis during long-term follow-up. J. Bone Miner. Res. 1999, 14, 2036–2041. [Google Scholar] [CrossRef]
- Stathopoulos, I.P.; Trovas, G.; Lampropoulou-Adamidou, K.; Koromila, T.; Kollia, P.; Papaioannou, N.A.; Lyritis, G. Severe osteoporosis and mutation in NOTCH2 gene in a woman with Hajdu-Cheney syndrome. Bone 2013, 52, 366–371. [Google Scholar] [CrossRef]
- Nunziata, V.; di Giovanni, G.; Ballanti, P.; Bonucci, E. High turnover osteoporosis in acro-osteolysis (Hajdu-Cheney syndrome). J. Endocrinol. Investig. Off. J. Ital. Soc. Endocrinol. 1990, 13, 251–255. [Google Scholar] [CrossRef]
- Vissarionov, S.V.; Filippova, A.N.; Zhurbitskaia, M.V.; Khusainov, N.O.; Belyanchikov, S.M. Surgical treatment of a patient with multiple fractures of the thoracic and lumbar vertebrae associated with Hajdu-Cheney syndrome. Hir. Pozvonochnika 2019, 16, 25–31. [Google Scholar] [CrossRef]
- Chawla, B.S. Cranio-skeletal dysplasia with acro-osteolysis. Br. J. Radiol. 1964, 37, 702–705. [Google Scholar] [CrossRef]
- Jiménez, I.; Medina-Gontier, J.; Caballero, J.; Medina, J. Hand Deformities in Hajdu-Cheney Syndrome: A Case Series of 3 Patients Across 3 Consecutive Generations. J. Hand Surg. Am. 2020, 46, P73.E1–P73.E5. [Google Scholar] [CrossRef] [PubMed]
- Shurtleff, D.B.; Sparkes, R.S.; Clawson, D.K.; Guntheroth, W.G.; Mottet, N.K. Hereditary Osteolysis with Hypertension and Nephropathy. J. Am. Med. Assoc. 1964, 188, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Ventosa, P.; Gorina, N.; Balaguer Santamaria, A.; Riera, L.; Casals, I. Acroosteólisis en un paciente de 4 años: Datos clínicos del síndrome de Hajdu-Cheney. An. Pediatr. 2013, 79, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.E.; Street, D.M. Idiopathic Non-Familial Aero-Osteolysis. Radiology 1957, 69, 259–262. [Google Scholar] [CrossRef]
- Colmenares Roldán, L.; de la Calle Rodríguez, N. Síndrome de Hajdu Cheney, una enfermedad poco frecuente. Rev. CES Med. 2013, 27, 101–106. [Google Scholar] [CrossRef]
- Williams, B. Foramen magnum impaction in a case of acro-osteolysis. Br. J. Surg. 1977, 64, 70–73. [Google Scholar] [CrossRef]
- Weleber, R.G.; Beals, R.K. The Hajdu-Cheney syndrome. Report of two cases and review of the literature. J. Pediatr. 1976, 88, 243–249. [Google Scholar] [CrossRef]
- Sasaki, K.; Ito, Y.; Kawame, H.; Kikuchi, A.; Tanaka, H. Fatal case of Hajdu-Cheney syndrome with idiopathic pulmonary hemosiderosis. Pediatr. Int. 2019, 61, 190–192. [Google Scholar] [CrossRef]
- Shaw, D.G. Acro-osteolysis and bone fragility. Br. J. Radiol. 1969, 42, 934–936. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, Y.J.; Kang, J.; Shin, T.J.; Hyun, H.-K.; Kim, Y.-J.; Lee, S.-H.; Lee, Z.H.; Kim, J.-W. Dental implications in Hajdu-Cheney syndrome: A novel case report and review of the literature. Oral Dis. 2018, 24, 1037–1041. [Google Scholar] [CrossRef]
- Antoniades, K.; Kaklamanos, E.; Kavadia, S.; Hatzistilianou, M.; Antoniades, V. Hajdu-Cheney syndrome (acro-osteolysis): A case report of dental interest. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2003, 95, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Vingerhoedt, E.; Bailleul-Forestier, I.; Fellus, P.; Schoenaers, J.; Frijns, J.P.; Carels, C. Syndrome of Hajdu-Cheney: Three case reports of orofacial interest. Cleft Palate-Craniofacial J. 2010, 47, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Sakka, S.; Gafni, R.I.; Davies, J.H.; Clarke, B.; Tebben, P.; Samuels, M.; Saraff, V.; Klaushofer, K.; Fratzl-Zelman, N.; Roschger, P.; et al. Bone Structural Characteristics and Response to Bisphosphonate Treatment in Children with Hajdu-Cheney Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 4163–4172. [Google Scholar] [CrossRef]
- Galli-Tsinopoulou, A.; Kyrgios, I.; Giza, S.; Giannopoulou, E.M.; Maggana, I.; Laliotis, N. Two-year cyclic infusion of pamidronate improves bone mass density and eliminates risk of fractures in a girl with osteoporosis due to Hajdu-Cheney syndrome. Minerva Endocrinol. 2012, 37, 283–289. [Google Scholar]
- Al-Mayouf, S.M.; Madi, S.M.; Bin-Abbas, B.S. Cyclic intravenous pamidronate treatment in children with nodulosis, arthropathy and osteolysis syndrome. Ann. Rheum. Dis. 2006, 65, 1672–1673. [Google Scholar] [CrossRef]
- Murtagh-Schaffer, C.; Moquin, R.R. Spinal reconstruction in Hajdu-Cheney syndrome. JAAPA 2008, 21, 29–33. [Google Scholar] [CrossRef]
- Zeman, J.; Houstkova, H.; Kozlowski, K. Hajdu-Cheney syndrome in a 3 1/2 year old girl. Australas. Radiol. 1994, 38, 228–230. [Google Scholar] [CrossRef]
- August, D.A.; Ramos, D.C. Anesthesia for a child with Hajdu-Cheney syndrome. Paediatr. Anaesth. 2009, 19, 649–650. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Nakamura, K.; Takahashi, Y. A case report of anesthesia for a child with Hajdu-Cheney syndrome. J. Anesth. 2013, 27, 949–950. [Google Scholar] [CrossRef]
- Battelino, N.; Writzl, K.; Bratanič, N.; Irving, M.D.; Novljan, G. End-Stage Renal Disease in an Infant With Hajdu-Cheney Syndrome. Ther. Apher. Dial. 2016, 20, 318–321. [Google Scholar] [CrossRef]
- Gong, R.L.; Wu, J.; Chen, T.X. A Novel Mutation of Notch homolog protein 2 gene in a Chinese Family with Hajdu-Cheney Syndrome. Chin. Med. J. (Engl.) 2017, 130, 2883–2884. [Google Scholar] [CrossRef] [PubMed]
- Graversen, L.; Handrup, M.M.; Irving, M.; Hove, H.; Diness, B.R.; Risom, L.; Svaneby, D.; Aagaard, M.M.; Vogel, I.; Gjørup, H.; et al. Phenotypic presentations of Hajdu-Cheney syndrome according to age—5 distinct clinical presentations. Eur. J. Med. Genet. 2019, 63, 103650. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DATE | WEIGHT kg | SIZE cm |
|---|---|---|
| Newborn | 4.340 | 54 |
| 2 years and 6 months | 12 | 89.7 |
| 5 years | 15.5 | 105 |
| 6 years and 8 months | 18 | 117 |
| 10 years and 1 month | 24.6 | 131.9 |
| 10 years and 8 months | 26.4 | 134 |
| 11 years and 1 month | 26.3 | 135.4 |
| Personal-Social | Adaptive | Gross Motor | Fine Motor | Total Motor | Receptive | Expressive | Total Comunication | Cognitive | Total Score | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age equivalent in months | 23 | 27 | 27 | 24 | 25 | 24 | 17 | 20 | 32 | 24 |
| Birth | 2011 |
|---|---|
| Diagnosis | 2013 |
| Beginning of therapy with early care | 2014 |
| Use of glasses | 2014 |
| Use of corrective insoles | 2015 |
| Initiation of bisphosphonate therapy | 2016 |
| Detection of PDA | 2018 |
| Focal vibration treatment | 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortés-Martín, J.; Díaz-Rodríguez, L.; Piqueras-Sola, B.; Sánchez-García, J.C.; González, A.L.; Rodríguez-Blanque, R. Hajdu-Cheney Syndrome: A Novel NOTCH2 Mutation in a Spanish Child in Treatment with Vibrotherapy: A Case Report. J. Clin. Med. 2022, 11, 5205. https://doi.org/10.3390/jcm11175205
Cortés-Martín J, Díaz-Rodríguez L, Piqueras-Sola B, Sánchez-García JC, González AL, Rodríguez-Blanque R. Hajdu-Cheney Syndrome: A Novel NOTCH2 Mutation in a Spanish Child in Treatment with Vibrotherapy: A Case Report. Journal of Clinical Medicine. 2022; 11(17):5205. https://doi.org/10.3390/jcm11175205
Chicago/Turabian StyleCortés-Martín, Jonathan, Lourdes Díaz-Rodríguez, Beatriz Piqueras-Sola, Juan Carlos Sánchez-García, Antonio Liñán González, and Raquel Rodríguez-Blanque. 2022. "Hajdu-Cheney Syndrome: A Novel NOTCH2 Mutation in a Spanish Child in Treatment with Vibrotherapy: A Case Report" Journal of Clinical Medicine 11, no. 17: 5205. https://doi.org/10.3390/jcm11175205
APA StyleCortés-Martín, J., Díaz-Rodríguez, L., Piqueras-Sola, B., Sánchez-García, J. C., González, A. L., & Rodríguez-Blanque, R. (2022). Hajdu-Cheney Syndrome: A Novel NOTCH2 Mutation in a Spanish Child in Treatment with Vibrotherapy: A Case Report. Journal of Clinical Medicine, 11(17), 5205. https://doi.org/10.3390/jcm11175205