
Relevance of Pathogenetic Mechanisms to Clinical Effectiveness of B-Cell-Depleting Monoclonal Antibodies in Multiple Sclerosis



Abstract
1. Introduction
2. Insights into MS Pathogenesis: Onset of Autoimmunity
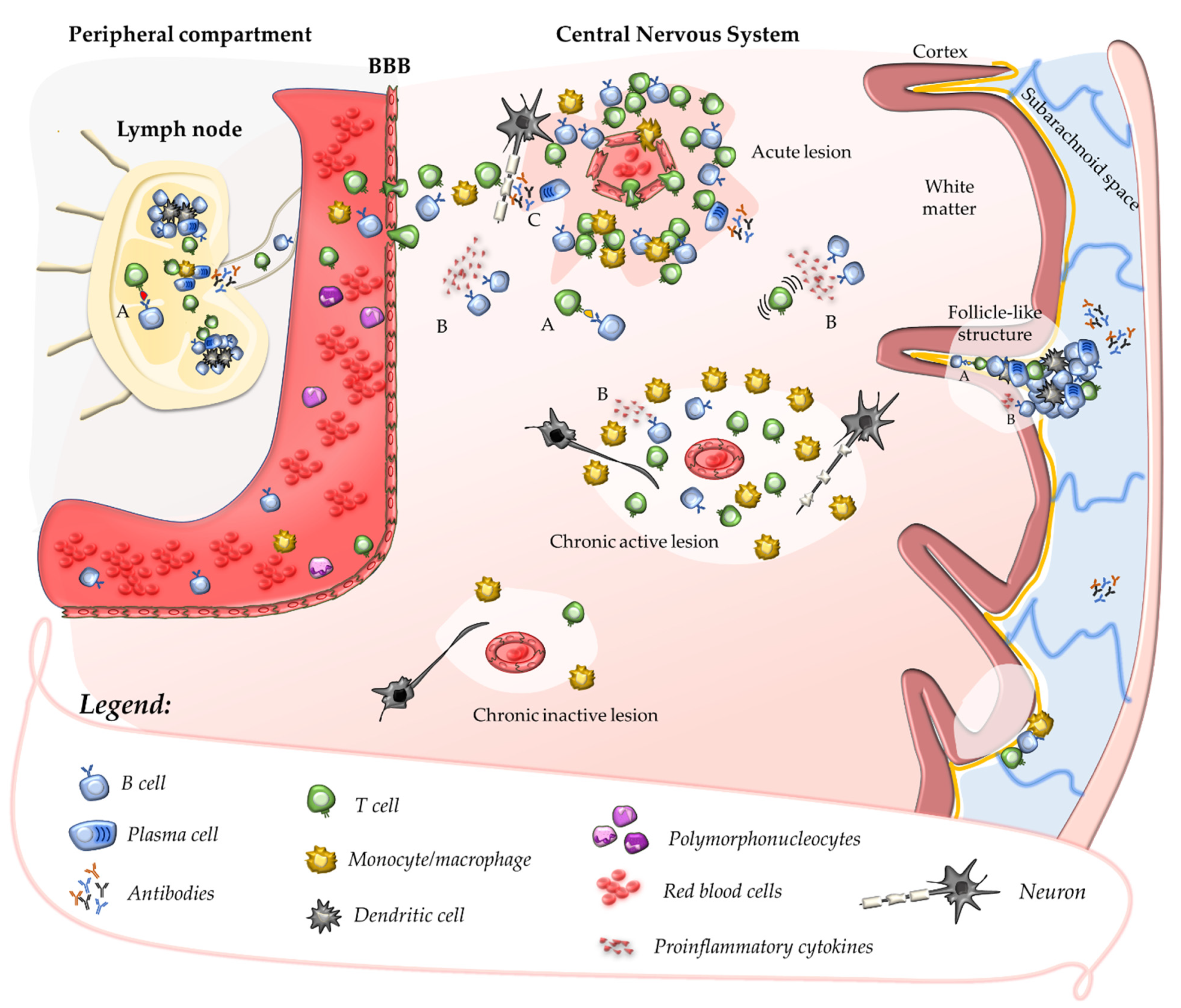
2.1. Primary Autoimmune Response in the Peripheral Compartment
2.2. Secondary Autoimmune Response in the Central Nervous System
2.3. Contribution of B Cells and Humoral Response to Acute CNS Injury
3. Compartmentalisation of the Inflammatory Response within the CNS
3.1. Chronic Perivascular Cuffs and Smouldering Lesions
3.2. MRI Markers of Compartmentalised Inflammation
3.3. Follicle-like Structures in the Leptomeninges
3.4. Innate Immunity and Degenerative Phenomena
4. Relevance of Pathogenetic Mechanisms to the Effectiveness of Disease-Modifying Treatments
5. B Cells and B-Cell-Depleting Antibodies
5.1. B-Cell Maturation and Surface Markers
5.2. Mechanisms of Lymphocyte Depletion Induced by Anti-CD20 Monoclonal Antibodies
5.3. Immunological Effects of B-Cell Depletion
5.4. Impact of CD20-Depleting Monoclonal Antibodies on the Peripheral Autoimmune Response and Compartmentalised Inflammation

{kind=link}
| Rituximab | Ocrelizumab | Ofatumumab | |
|---|---|---|---|
| Binding site of the CD20 protein | intermediate-distal portion of the extracellular loop between TM3 and TM4 (amino acids 170–172) | intermediate-distal portion of the extracellular loop between TM3 and TM4 (amino acids 170–172 + 162–166) | between the first extracellular loop and the proximal portion of the second extracellular loop |
| Acute focal inflammation | Depletion of potentially pathogenetic B and CD20+ T cells. Impairment of antigen-presentation in peripheral lymphoid organs (primary autoimmune response) and within the CNS (secondary autoimmune response). Reduced production of B-cell-derived pro-inflammatory cytokines. | ||
| Chronic inflammation compartmentalised within the CNS | Reduced replenishment of encephalitogenic cells from peripheral blood. Impairment of antigen-presentation within the CNS (maintenance of the secondary autoimmune response and epitope-spreading). Reduced production of B-cell-derived pro-inflammatory cytokines. Reduced activation of microglia and astrocytes. a Depletion of B cells from follicle-like structures in the meninges. a | ||
6. Conclusions
Funding
Conflicts of Interest
References
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2018, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 2013, 333, 1–4. [Google Scholar] [CrossRef]
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schroder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404. [Google Scholar] [CrossRef]
- Junker, A.; Ivanidze, J.; Malotka, J.; Eiglmeier, I.; Lassmann, H.; Wekerle, H.; Meinl, E.; Hohlfeld, R.; Dornmair, K. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain 2007, 130, 2789–2799. [Google Scholar] [CrossRef]
- Skulina, C.; Schmidt, S.; Dornmair, K.; Babbe, H.; Roers, A.; Rajewsky, K.; Wekerle, H.; Hohlfeld, R.; Goebels, N. Multiple sclerosis: Brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc. Natl. Acad. Sci. USA 2004, 101, 2428–2433. [Google Scholar] [CrossRef]
- Markovic-Plese, S. Degenerate T-cell receptor recognition, autoreactive cells, and the autoimmune response in multiple sclerosis. Neuroscientist 2009, 15, 225–231. [Google Scholar] [CrossRef]
- Montes, M.; Zhang, X.; Berthelot, L.; Laplaud, D.A.; Brouard, S.; Jin, J.; Rogan, S.; Armao, D.; Jewells, V.; Soulillou, J.P.; et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin. Immunol. 2009, 130, 133–144. [Google Scholar] [CrossRef]
- Saxena, A.; Martin-Blondel, G.; Mars, L.T.; Liblau, R.S. Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett. 2011, 585, 3758–3763. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Amoriello, R.; Chernigovskaya, M.; Greiff, V.; Carnasciali, A.; Massacesi, L.; Barilaro, A.; Repice, A.M.; Biagioli, T.; Aldinucci, A.; Muraro, P.A.; et al. TCR repertoire diversity in Multiple Sclerosis: High-dimensional bioinformatics analysis of sequences from brain, cerebrospinal fluid and peripheral blood. EBioMedicine 2021, 68, 103429. [Google Scholar] [CrossRef] [PubMed]
- Amoriello, R.; Greiff, V.; Aldinucci, A.; Bonechi, E.; Carnasciali, A.; Peruzzi, B.; Repice, A.M.; Mariottini, A.; Saccardi, R.; Mazzanti, B.; et al. The TCR Repertoire Reconstitution in Multiple Sclerosis: Comparing One-Shot and Continuous Immunosuppressive Therapies. Front Immunol. 2020, 11, 559. [Google Scholar] [CrossRef]
- Gestri, D.; Baldacci, L.; Taiuti, R.; Galli, E.; Maggi, E.; Piccinni, M.P.; Vergelli, M.; Massacesi, L. Oligoclonal T cell repertoire in cerebrospinal fluid of patients with inflammatory diseases of the nervous system. J. Neurol. Neurosurg. Psychiatry 2001, 70, 767–772. [Google Scholar] [CrossRef][Green Version]
- Martin, R.; Jaraquemada, D.; Flerlage, M.; Richert, J.; Whitaker, J.; Long, E.O.; McFarlin, D.E.; McFarland, H.F. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J. Immunol. 1990, 145, 540–548. [Google Scholar]
- Bielekova, B.; Sung, M.H.; Kadom, N.; Simon, R.; McFarland, H.; Martin, R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J. Immunol. 2004, 172, 3893–3904. [Google Scholar] [CrossRef]
- Hellings, N.; Baree, M.; Verhoeven, C.; D’Hooghe, M.B.; Medaer, R.; Bernard, C.C.; Raus, J.; Stinissen, P. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J. Neurosci. Res. 2001, 63, 290–302. [Google Scholar] [CrossRef]
- del Pilar Martin, M.; Cravens, P.D.; Winger, R.; Kieseier, B.C.; Cepok, S.; Eagar, T.N.; Zamvil, S.S.; Weber, M.S.; Frohman, E.M.; Kleinschmidt-DeMasters, B.K. Depletion of B lymphocytes from cerebral perivascular spaces by rituximab. Arch. Neurol. 2009, 66, 1016–1020. [Google Scholar]
- Sawcer, S.; Franklin, R.J.; Ban, M. Multiple sclerosis genetics. Lancet Neurol. 2014, 13, 700–709. [Google Scholar] [CrossRef]
- Amato, M.P.; Derfuss, T.; Hemmer, B.; Liblau, R.; Montalban, X.; Soelberg Sorensen, P.; Miller, D.H.; Group, E.F.W. Environmental modifiable risk factors for multiple sclerosis: Report from the 2016 ECTRIMS focused workshop. Mult. Scler. 2017, 24, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Jersild, C.; Svejgaard, A.; Fog, T. HL-A antigens and multiple sclerosis. Lancet 1972, 1, 1240–1241. [Google Scholar] [CrossRef]
- Ryan, S.O.; Cobb, B.A. Roles for major histocompatibility complex glycosylation in immune function. Semin. Immunopathol. 2012, 34, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics, Consortium; ANZgene; IIBDGC; WTCCC2. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365. [Google Scholar] [CrossRef]
- Titus, H.E.; Chen, Y.; Podojil, J.R.; Robinson, A.P.; Balabanov, R.; Popko, B.; Miller, S.D. Pre-clinical and Clinical Implications of “Inside-Out” vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Front. Cell. Neurosci. 2020, 14, 599717. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Harkiolaki, M.; Holmes, S.L.; Svendsen, P.; Gregersen, J.W.; Jensen, L.T.; McMahon, R.; Friese, M.A.; van Boxel, G.; Etzensperger, R.; Tzartos, J.S.; et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity 2009, 30, 348–357. [Google Scholar] [CrossRef]
- Haring, J.S.; Pewe, L.L.; Perlman, S. Bystander CD8 T cell-mediated demyelination after viral infection of the central nervous system. J. Immunol. 2002, 169, 1550–1555. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- ’t Hart, B.A.; Luchicchi, A.; Schenk, G.J.; Stys, P.K.; Geurts, J.J.G. Mechanistic underpinning of an inside–out concept for autoimmunity in multiple sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 1709–1719. [Google Scholar] [CrossRef]
- Laman, J.D.; Weller, R.O. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J. Neuroimmune Pharmacol. 2013, 8, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Mundt, S.; Greter, M.; Flugel, A.; Becher, B. The CNS Immune Landscape from the Viewpoint of a T Cell. Trends Neurosci. 2019, 42, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Mapunda, J.A.; Tibar, H.; Regragui, W.; Engelhardt, B. How Does the Immune System Enter the Brain? Front. Immunol. 2022, 13, 805657. [Google Scholar] [CrossRef]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef]
- Kawakami, N.; Flügel, A. Knocking at the brain’s door: Intravital two-photon imaging of autoreactive T cell interactions with CNS structures. Semin. Immunopathol. 2010, 32, 275–287. [Google Scholar] [CrossRef]
- Croxford, J.L.; Olson, J.K.; Miller, S.D. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler’s virus-induced demyelinating disease model of multiple sclerosis. Autoimmun. Rev. 2002, 1, 251–260. [Google Scholar] [CrossRef]
- Scalfari, A.; Neuhaus, A.; Degenhardt, A.; Rice, G.P.; Muraro, P.A.; Daumer, M.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study 10: Relapses and long-term disability. Brain 2010, 133, 1914–1929. [Google Scholar] [CrossRef]
- Filippi, M.; Rocca, M.A.; Ciccarelli, O.; De Stefano, N.; Evangelou, N.; Kappos, L.; Rovira, A.; Sastre-Garriga, J.; Tintorè, M.; Frederiksen, J.L. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol. 2016, 15, 292–303. [Google Scholar] [CrossRef]
- Absinta, M.; Sati, P.; Schindler, M.; Leibovitch, E.C.; Ohayon, J.; Wu, T.; Meani, A.; Filippi, M.; Jacobson, S.; Cortese, I.C.; et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J. Clin. Investig. 2016, 126, 2597–2609. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Hemmer, B.; Cepok, S. The role of antibodies in multiple sclerosis. Biochim. Biophy. Acta 2011, 1812, 239–245. [Google Scholar] [CrossRef]
- Li, R.; Patterson, K.R.; Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 2018, 19, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Kabat, E.A.; Moore, D.H.; Landow, H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J. Clin. Investig. 1942, 21, 571–577. [Google Scholar] [CrossRef]
- Correale, J.; de los Milagros Bassani Molinas, M. Oligoclonal bands and antibody responses in multiple sclerosis. J. Neurol. 2002, 249, 375–389. [Google Scholar] [CrossRef]
- Gaitan, M.I.; Maggi, P.; Wohler, J.; Leibovitch, E.; Sati, P.; Calandri, I.L.; Merkle, H.; Massacesi, L.; Silva, A.C.; Jacobson, S.; et al. Perivenular brain lesions in a primate multiple sclerosis model at 7-tesla magnetic resonance imaging. Mult. Scler. 2014, 20, 64–71. [Google Scholar] [CrossRef]
- Hart, B.A.; Massacesi, L. Clinical, pathological, and immunologic aspects of the multiple sclerosis model in common marmosets (Callithrix jacchus). J. Neuropathol. Exp. Neurol. 2009, 68, 341–355. [Google Scholar] [CrossRef]
- Genain, C.P.; Cannella, B.; Hauser, S.L.; Raine, C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999, 5, 170–175. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Cencioni, M.T.; Mattoscio, M.; Magliozzi, R.; Bar-Or, A.; Muraro, P.A. B cells in multiple sclerosis-from targeted depletion to immune reconstitution therapies. Nat. Rev. Neurol. 2021, 17, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Lovato, L.; Willis, S.N.; Rodig, S.J.; Caron, T.; Almendinger, S.E.; Howell, O.W.; Reynolds, R.; O’Connor, K.C.; Hafler, D.A. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 2011, 134, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jelcic, I.; Muhlenbruch, L.; Haunerdinger, V.; Toussaint, N.C.; Zhao, Y.; Cruciani, C.; Faigle, W.; Naghavian, R.; Foege, M.; et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell 2020, 183, 1264–1281.e20. [Google Scholar] [CrossRef] [PubMed]
- Chari, D.M. Remyelination in multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 589–620. [Google Scholar] [CrossRef]
- Meinl, E.; Krumbholz, M.; Derfuss, T.; Junker, A.; Hohlfeld, R. Compartmentalization of inflammation in the CNS: A major mechanism driving progressive multiple sclerosis. J. Neurol. Sci. 2008, 274, 42–44. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Bruck, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Bruck, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Luchetti, S.; Fransen, N.L.; van Eden, C.G.; Ramaglia, V.; Mason, M.; Huitinga, I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: A retrospective autopsy cohort analysis. Acta Neuropathol. 2018, 135, 511–528. [Google Scholar] [CrossRef]
- Massacesi, L. Compartmentalization of the immune response in the central nervous system and natural history of multiple sclerosis. Implications for therapy. Clin. Neurol. Neurosurg. 2002, 104, 177–181. [Google Scholar] [CrossRef]
- University of California, San Francisco MS-EPIC Team; Cree, B.A.C.; Hollenbach, J.A.; Bove, R.; Kirkish, G.; Sacco, S.; Caverzasi, E.; Bischof, A.; Gundel, T.; Zhu, A.H.; et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann. Neurol. 2019, 85, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Hoftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H.; et al. Slow expansion of multiple sclerosis iron rim lesions: Pathology and 7 T magnetic resonance imaging. Acta Neuropathol. 2017, 133, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Sati, P.; Masuzzo, F.; Nair, G.; Sethi, V.; Kolb, H.; Ohayon, J.; Wu, T.; Cortese, I.C.M.; Reich, D.S. Association of Chronic Active Multiple Sclerosis Lesions with Disability In Vivo. JAMA Neurol. 2019, 76, 1474–1483. [Google Scholar] [CrossRef]
- Absinta, M.; Sati, P.; Fechner, A.; Schindler, M.K.; Nair, G.; Reich, D.S. Identification of Chronic Active Multiple Sclerosis Lesions on 3T MRI. AJNR Am. J. Neuroradiol. 2018, 39, 1233–1238. [Google Scholar] [CrossRef]
- Bitsch, A.; Kuhlmann, T.; Stadelmann, C.; Lassmann, H.; Lucchinetti, C.; Bruck, W. A longitudinal MRI study of histopathologically defined hypointense multiple sclerosis lesions. Ann. Neurol. 2001, 49, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Truyen, L.; van Waesberghe, J.H.; van Walderveen, M.A.; van Oosten, B.W.; Polman, C.H.; Hommes, O.R.; Ader, H.J.; Barkhof, F. Accumulation of hypointense lesions (“black holes”) on T1 spin-echo MRI correlates with disease progression in multiple sclerosis. Neurology 1996, 47, 1469–1476. [Google Scholar] [CrossRef]
- Elliott, C.; Wolinsky, J.S.; Hauser, S.L.; Kappos, L.; Barkhof, F.; Bernasconi, C.; Wei, W.; Belachew, S.; Arnold, D.L. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult. Scler. 2019, 25, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Bø, L.; Vedeler, C.A.; Nyland, H.I.; Trapp, B.D.; Mørk, S.J. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J. Neuropathol. Exp. Neurol. 2003, 62, 723–732. [Google Scholar] [CrossRef]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef]
- Bevan, R.J.; Evans, R.; Griffiths, L.; Watkins, L.M.; Rees, M.I.; Magliozzi, R.; Allen, I.; McDonnell, G.; Kee, R.; Naughton, M. Meningeal inflammation and cortical demyelination in acute multiple sclerosis. Ann. Neurol. 2018, 84, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Serafini, B.; Aloisi, F.; Reynolds, R. A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010, 68, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Vuolo, L.; Rao, A.; Nair, G.; Sati, P.; Cortese, I.C.; Ohayon, J.; Fenton, K.; Reyes-Mantilla, M.I.; Maric, D.; et al. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology 2015, 85, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Latour, L.L.; Kang, D.W.; Ezzeddine, M.A.; Chalela, J.A.; Warach, S. Early blood–brain barrier disruption in human focal brain ischemia. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 56, 468–477. [Google Scholar] [CrossRef]
- Harrison, D.M.; Wang, K.Y.; Fiol, J.; Naunton, K.; Royal, W., 3rd; Hua, J.; Izbudak, I. Leptomeningeal Enhancement at 7T in Multiple Sclerosis: Frequency, Morphology, and Relationship to Cortical Volume. J. Neuroimaging 2017, 27, 461–468. [Google Scholar] [CrossRef]
- Zivadinov, R.; Ramasamy, D.P.; Vaneckova, M.; Gandhi, S.; Chandra, A.; Hagemeier, J.; Bergsland, N.; Polak, P.; Benedict, R.H.; Hojnacki, D.; et al. Leptomeningeal contrast enhancement is associated with progression of cortical atrophy in MS: A retrospective, pilot, observational longitudinal study. Mult. Scler. 2017, 23, 1336–1345. [Google Scholar] [CrossRef]
- Zurawski, J.; Tauhid, S.; Chu, R.; Khalid, F.; Healy, B.C.; Weiner, H.L.; Bakshi, R. 7T MRI cerebral leptomeningeal enhancement is common in relapsing-remitting multiple sclerosis and is associated with cortical and thalamic lesions. Mult. Scler. J. 2019, 26, 177–187. [Google Scholar] [CrossRef]
- Ighani, M.; Jonas, S.; Izbudak, I.; Choi, S.; Lema-Dopico, A.; Hua, J.; O’Connor, E.E.; Harrison, D.M. No association between cortical lesions and leptomeningeal enhancement on 7-Tesla MRI in multiple sclerosis. Mult. Scler. J. 2020, 26, 165–176. [Google Scholar] [CrossRef]
- Ineichen, B.V.; Tsagkas, C.; Absinta, M.; Reich, D.S. Leptomeningeal enhancement in multiple sclerosis and other neurological diseases: A systematic review and Meta-Analysis. NeuroImage Clin. 2022, 33, 102939. [Google Scholar] [CrossRef]
- Tillack, K.; Breiden, P.; Martin, R.; Sospedra, M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 2012, 188, 3150–3159. [Google Scholar] [CrossRef]
- Gandhi, R.; Laroni, A.; Weiner, H.L. Role of the innate immune system in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2010, 221, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011, 585, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M. Neurodegeneration in multiple sclerosis is a process separate from inflammation: No. Mult. Scler. 2015, 21, 1628–1631. [Google Scholar] [CrossRef]
- Louapre, C.; Lubetzki, C. Neurodegeneration in multiple sclerosis is a process separate from inflammation: Yes. Mult. Scler. 2015, 21, 1626–1628. [Google Scholar] [CrossRef]
- Cross, A.H.; Naismith, R.T. Established and novel disease-modifying treatments in multiple sclerosis. J. Intern. Med. 2014, 275, 350–363. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Burrows, P.D.; Wang, J.Y. B Cell Development and Maturation. Adv. Exp. Med. Biol. 2020, 1254, 1–22. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Cree, B.A.C.; Hauser, S.L. Ocrelizumab and Other CD20(+) B-Cell-Depleting Therapies in Multiple Sclerosis. Neurotherapeutics 2017, 14, 835–841. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R.; et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): A double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: A randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020, 19, 402–412. [Google Scholar] [CrossRef]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Lammens, A.; Schafer, W.; Georges, G.; Schwaiger, M.; Mossner, E.; Hopfner, K.P.; Umana, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs 2013, 5, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Terui, Y.; Mishima, Y.; Sugimura, N.; Kojima, K.; Sakurai, T.; Mishima, Y.; Kuniyoshi, R.; Taniyama, A.; Yokoyama, M.; Sakajiri, S.; et al. Identification of CD20 C-terminal deletion mutations associated with loss of CD20 expression in non-Hodgkin’s lymphoma. Clin. Cancer Res. 2009, 15, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Tomita, A. Genetic and Epigenetic Modulation of CD20 Expression in B-Cell Malignancies: Molecular Mechanisms and Significance to Rituximab Resistance. J. Clin. Exp. Hematop. 2016, 56, 89–99. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef]
- Lee, D.S.W.; Rojas, O.L.; Gommerman, J.L. B cell depletion therapies in autoimmune disease: Advances and mechanistic insights. Nat. Rev. Drug Discov. 2021, 20, 179–199. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef]
- Dunn, N.; Juto, A.; Ryner, M.; Manouchehrinia, A.; Piccoli, L.; Fink, K.; Piehl, F.; Fogdell-Hahn, A. Rituximab in multiple sclerosis: Frequency and clinical relevance of anti-drug antibodies. Mult. Scler. 2018, 24, 1224–1233. [Google Scholar] [CrossRef]
- Alvarez, E.; Nair, K.V.; Sillau, S.; Shelton, I.; Seale, R.; Selva, S.; Corboy, J.; Vollmer, T.L. Tolerability and Safety of Switching from Rituximab to Ocrelizumab: Evaluating Factors Associated with Infusion Related Reactions. Mult. Scler. J.-Exp. Transl. Clin. 2022, 8, 20552173211069359. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.H.; Stark, J.L.; Lauber, J.; Ramsbottom, M.J.; Lyons, J.-A. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 2006, 180, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Khosroshahi, A.; Bloch, D.B.; Deshpande, V.; Stone, J.H. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010, 62, 1755–1762. [Google Scholar] [CrossRef]
- Jelcic, I.; Al Nimer, F.; Wang, J.; Lentsch, V.; Planas, R.; Jelcic, I.; Madjovski, A.; Ruhrmann, S.; Faigle, W.; Frauenknecht, K.; et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4(+) T Cells in Multiple Sclerosis. Cell 2018, 175, 85–100.e23. [Google Scholar] [CrossRef]
- Cinamon, G.; Zachariah, M.A.; Lam, O.M.; Foss, F.W., Jr.; Cyster, J.G. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008, 9, 54–62. [Google Scholar] [CrossRef]
- Schuh, E.; Berer, K.; Mulazzani, M.; Feil, K.; Meinl, I.; Lahm, H.; Krane, M.; Lange, R.; Pfannes, K.; Subklewe, M. Features of human CD3+ CD20+ T cells. J. Immunol. 2016, 197, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.N.; Yaari, G.; Vander Heiden, J.A.; Church, G.; Donahue, W.F.; Hintzen, R.Q.; Huttner, A.J.; Laman, J.D.; Nagra, R.M.; Nylander, A.; et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 2014, 6, 248ra107. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Avasarala, J. It’s Time For Combination Therapies: In Multiple Sclerosis. Innov. Clin. Neurosci. 2017, 14, 28–30. [Google Scholar]
- Rubenstein, J.; Rosenberg, J.; Damon, L. High-dose methotrexate plus rituximab (Anti-CD20) monoclonal antibody in the treatment of primary CNS lymphoma. In Proceedings of the Society for Neuro-Oncology Fourth Annual Meeting, Scottsdale, AZ, USA, 17–21 November 1999. [Google Scholar]
- Batchelor, T.T.; Grossman, S.A.; Mikkelsen, T.; Ye, X.; Desideri, S.; Lesser, G.J. Rituximab monotherapy for patients with recurrent primary CNS lymphoma. Neurology 2011, 76, 929–930. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Belachew, S.; Wolinsky, J.S.; Hauser, S.L.; Kappos, L.; Barkhof, F.; Bernasconi, C.; Fecker, J.; Model, F.; Wei, W.; et al. Chronic white matter lesion activity predicts clinical progression in primary progressive multiple sclerosis. Brain 2019, 142, 2787–2799. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.P.; Simonsen, H.; Frederiksen, J.L.; Rostrup, E.; Larsson, H.B.W. Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clin. 2013, 4, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Komori, M.; Lin, Y.C.; Cortese, I.; Blake, A.; Ohayon, J.; Cherup, J.; Maric, D.; Kosa, P.; Wu, T.; Bielekova, B. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann. Clin. Transl. Neurol. 2016, 3, 166–179. [Google Scholar] [CrossRef]
- Bonnan, M.; Ferrari, S.; Courtade, H.; Money, P.; Desblache, P.; Barroso, B.; Debeugny, S. No Early Effect of Intrathecal Rituximab in Progressive Multiple Sclerosis (EFFRITE Clinical Trial). Mult. Scler. Int. 2021, 2021, 8813498. [Google Scholar] [CrossRef]
- Anthony, D.C.; Dickens, A.M.; Seneca, N.; Couch, Y.; Campbell, S.; Checa, B.; Kersemans, V.; Warren, E.A.; Tredwell, M.; Sibson, N.R.; et al. Anti-CD20 inhibits T cell-mediated pathology and microgliosis in the rat brain. Ann. Clin. Transl. Neurol. 2014, 1, 659–669. [Google Scholar] [CrossRef]
- Roodselaar, J.; Zhou, Y.; Leppert, D.; Hauser, A.E.; Urich, E.; Anthony, D.C. Anti-CD20 Disrupts Meningeal B-Cell Aggregates in a Model of Secondary Progressive Multiple Sclerosis. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8. [Google Scholar] [CrossRef]
- Brand, R.M.; Friedrich, V.; Diddens, J.; Pfaller, M.; Romana de Franchis, F.; Radbruch, H.; Hemmer, B.; Steiger, K.; Lehmann-Horn, K. Anti-CD20 Depletes Meningeal B Cells but Does Not Halt the Formation of Meningeal Ectopic Lymphoid Tissue. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8, e1012. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massacesi, L.; Mariottini, A.; Nicoletti, F. Relevance of Pathogenetic Mechanisms to Clinical Effectiveness of B-Cell-Depleting Monoclonal Antibodies in Multiple Sclerosis. J. Clin. Med. 2022, 11, 4288. https://doi.org/10.3390/jcm11154288
Massacesi L, Mariottini A, Nicoletti F. Relevance of Pathogenetic Mechanisms to Clinical Effectiveness of B-Cell-Depleting Monoclonal Antibodies in Multiple Sclerosis. Journal of Clinical Medicine. 2022; 11(15):4288. https://doi.org/10.3390/jcm11154288
Chicago/Turabian StyleMassacesi, Luca, Alice Mariottini, and Ferdinando Nicoletti. 2022. "Relevance of Pathogenetic Mechanisms to Clinical Effectiveness of B-Cell-Depleting Monoclonal Antibodies in Multiple Sclerosis" Journal of Clinical Medicine 11, no. 15: 4288. https://doi.org/10.3390/jcm11154288
APA StyleMassacesi, L., Mariottini, A., & Nicoletti, F. (2022). Relevance of Pathogenetic Mechanisms to Clinical Effectiveness of B-Cell-Depleting Monoclonal Antibodies in Multiple Sclerosis. Journal of Clinical Medicine, 11(15), 4288. https://doi.org/10.3390/jcm11154288