
Hypercapnia in COPD: Causes, Consequences, and Therapy

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
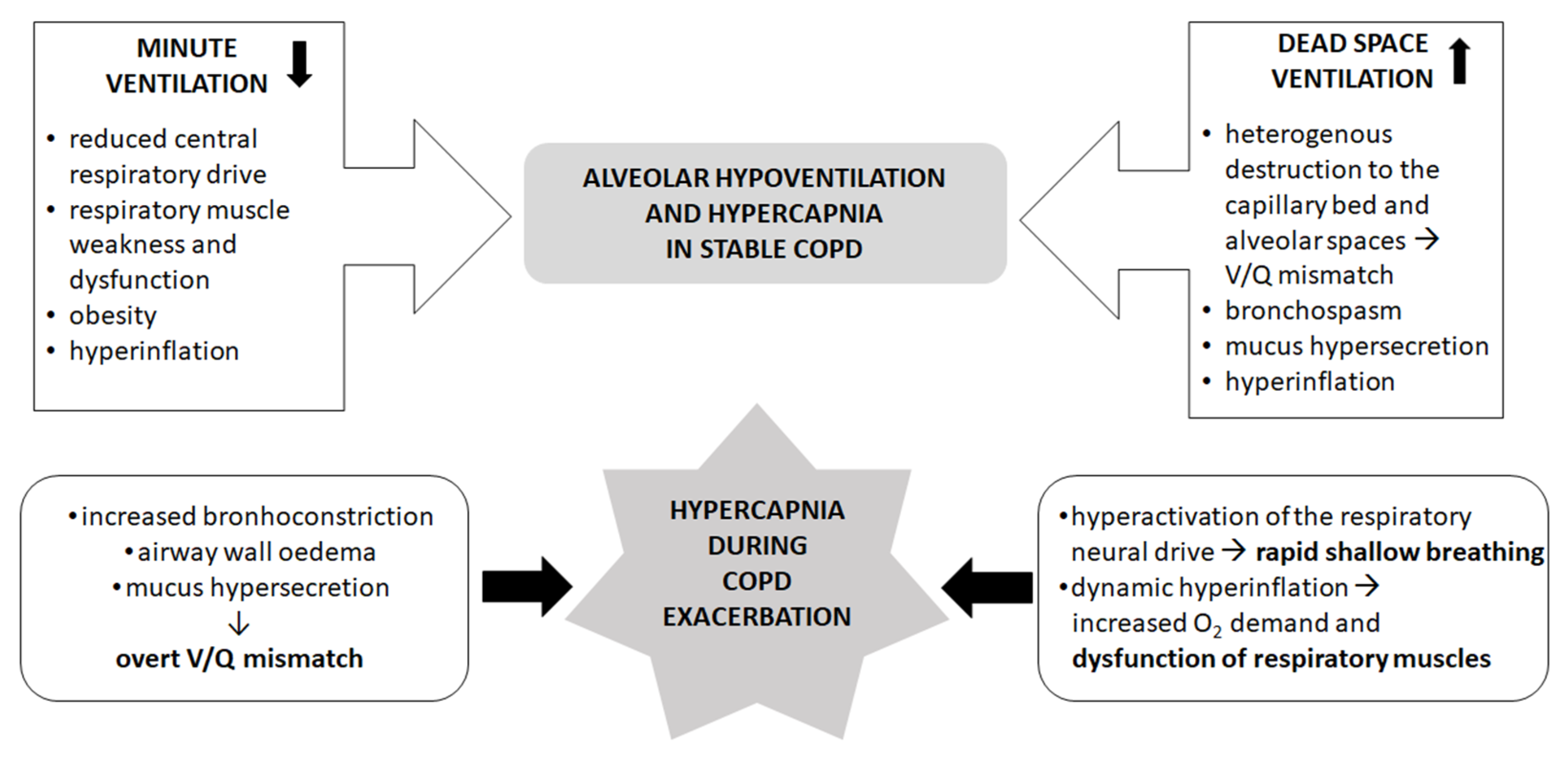
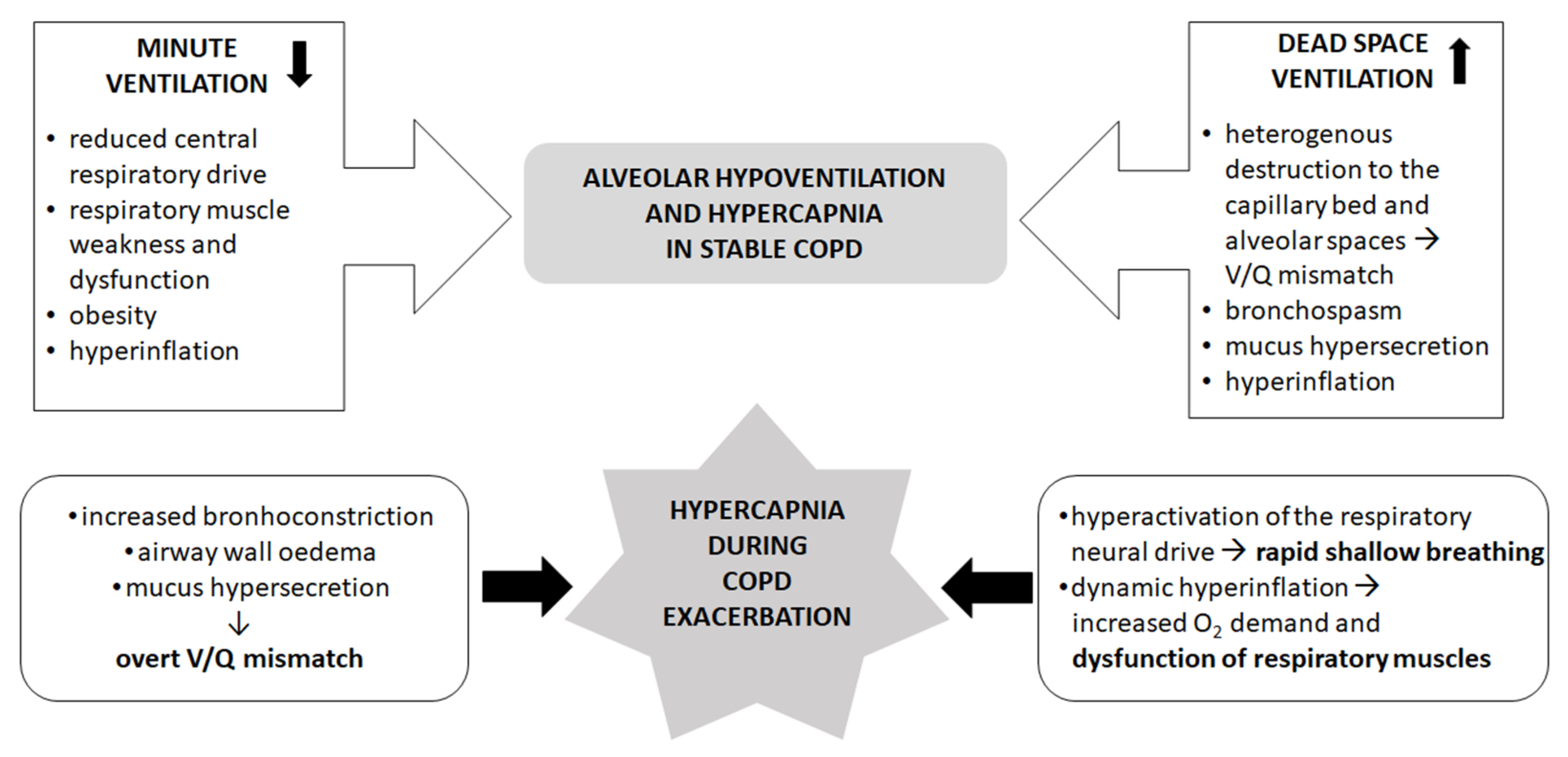
2. The Mechanisms of Hypercapnia in Stable COPD
3. The Mechanisms of Hypercapnia during Exacerbation of COPD
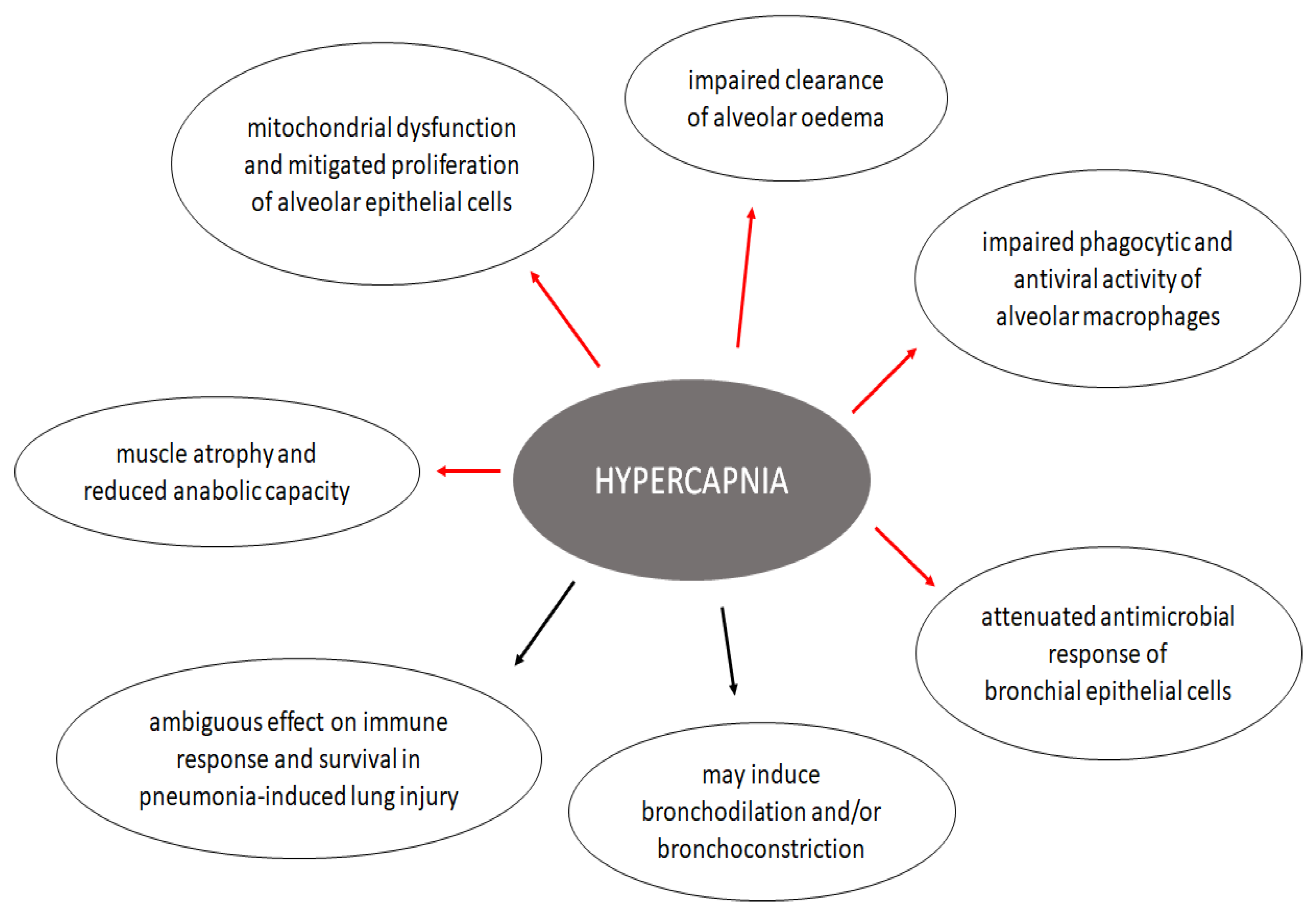
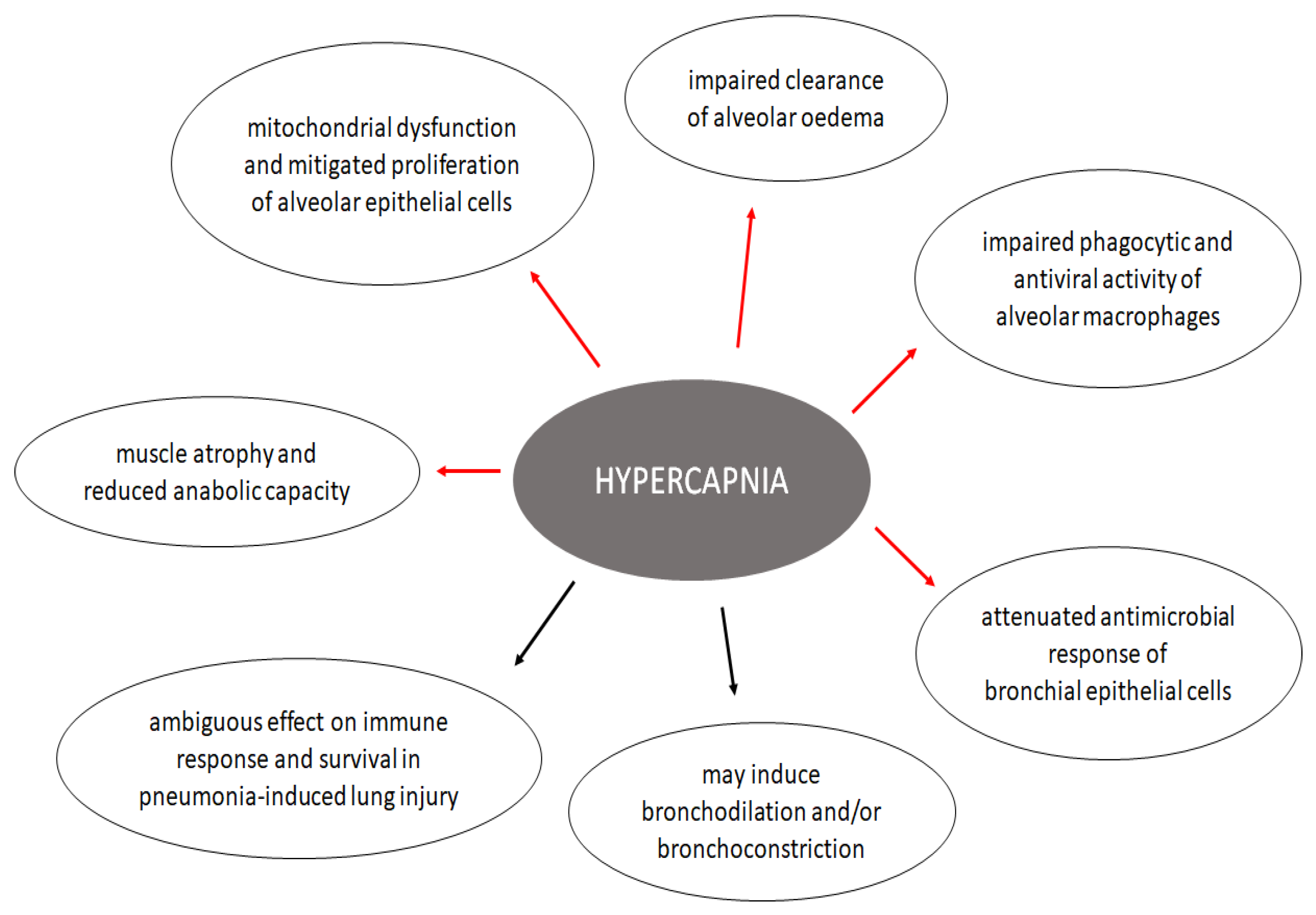
4. Effects of Hypercapnia on the Lung
4.1. Effects on Alveolar Epithelium
4.2. Effects on Immunity and Inflammatory Response of the Respiratory System
4.3. Effects on Airway Mechanics
4.4. Effects on Pulmonary Circulation
5. Systemic Effects of Hypercapnia
5.1. Cardiovascular Effects
5.2. Musculoskeletal Effects
6. Noninvasive Ventilation in COPD
6.1. NIV in Stable Hypercapnic COPD
6.2. NIV during Acute Exacerbations of COPD
6.3. NIV after an Acute Exacerbation
7. Discussion
7.1. Clinical Recommendations
7.2. Future Research Directions
- Observational studies are needed to understand the impact and stability of the hypercapnia exacerbator phenotype.
- Phase III long-term NIV trials with physiological and biological outcomes are needed in patients with chronic hypercapnic respiratory failure.
- Studies designed to understand the impact of long-term NIV on cardiovascular outcomes are needed.
- Studies designed to understand factors predicting treatment response to long-term NIV are needed.
- Studies designed to establish minimal hours of usage necessary with long-term NIV are needed.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease (2021 Report). Available online: https://goldcopd.org/wp-content/uploads/2020/11/GOLD-REPORT-2021-v1.1-25Nov20_WMV.pdf (accessed on 9 September 2021).
- Gibson, G.J.; Loddenkemper, R.; Lundbäck, B.; Sibille, Y. Respiratory health and disease in Europe: The new European Lung White Book. Eur. Respir. J. 2013, 42, 559–563. [Google Scholar] [CrossRef]
- Dreher, M.; Neuzeret, P.C.; Windisch, W.; Martens, D.; Hoheisel, G.; Groschel, A.; Woehrle, H.; Fetsch, T.; Graml, A.; Kohnlein, T. Prevalence of Chronic Hypercapnia in Severe Chronic Obstructive Pulmonary Disease: Data from the HOmeVent Registry. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 2377–2384. [Google Scholar] [CrossRef] [Green Version]
- Saure, E.W.; Eagan, T.M.; Jensen, R.L.; Voll-Aanerud, M.; Aukrust, P.; Bakke, P.S.; Hardie, J.A. Explained variance for blood gases in a population with COPD. Clin. Respir. J. 2012, 6, 72–80. [Google Scholar] [CrossRef]
- Rodriguez-Roisin, R.; Drakulovic, M.; Rodriguez, D.A.; Roca, J.; Barbera, J.A.; Wagner, P.D. Ventilation-perfusion imbalance and chronic obstructive pulmonary disease staging severity. J. Appl. Physiol. (1985) 2009, 106, 1902–1908. [Google Scholar] [CrossRef]
- The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Briva, A.; Vadasz, I.; Lecuona, E.; Welch, L.C.; Chen, J.; Dada, L.A.; Trejo, H.E.; Dumasius, V.; Azzam, Z.S.; Myrianthefs, P.M.; et al. High CO2 levels impair alveolar epithelial function independently of pH. PLoS ONE 2007, 2, e1238. [Google Scholar] [CrossRef]
- Ceco, E.; Celli, D.; Weinberg, S.; Shigemura, M.; Welch, L.C.; Volpe, L.; Chandel, N.S.; Bharat, A.; Lecuona, E.; Sznajder, J.I. Elevated CO2 Levels Delay Skeletal Muscle Repair by Increasing Fatty Acid Oxidation. Front. Physiol. 2020, 11, 630910. [Google Scholar] [CrossRef]
- Wang, N.; Gates, K.L.; Trejo, H.; Favoreto, S., Jr.; Schleimer, R.P.; Sznajder, J.I.; Beitel, G.J.; Sporn, P.H. Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J. 2010, 24, 2178–2190. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, Z.; Bornefalk-Hermansson, A.; Franklin, K.A.; Midgren, B.; Ekstrom, M.P. Hypo- and hypercapnia predict mortality in oxygen-dependent chronic obstructive pulmonary disease: A population-based prospective study. Respir. Res. 2014, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Lecuona, E.; Trejo, H.E.; Sznajder, J.I. Regulation of Na,K-ATPase during acute lung injury. J. Bioenerg. Biomembr. 2007, 39, 391–395. [Google Scholar] [CrossRef]
- Vohwinkel, C.U.; Lecuona, E.; Sun, H.; Sommer, N.; Vadasz, I.; Chandel, N.S.; Sznajder, J.I. Elevated CO(2) levels cause mitochondrial dysfunction and impair cell proliferation. J. Biol. Chem. 2011, 286, 37067–37076. [Google Scholar] [CrossRef] [Green Version]
- Agusti, A.; Bel, E.; Thomas, M.; Vogelmeier, C.; Brusselle, G.; Holgate, S.; Humbert, M.; Jones, P.; Gibson, P.G.; Vestbo, J.; et al. Treatable traits: Toward precision medicine of chronic airway diseases. Eur. Respir. J. 2016, 47, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Burrows, B.; Earle, R.H. Course and prognosis of chronic obstructive lung disease. A prospective study of 200 patients. N. Engl. J. Med. 1969, 280, 397–404. [Google Scholar] [CrossRef]
- Neff, T.A.; Petty, T.L. Tolerance and survival in severe chronic hypercapnia. Arch. Intern. Med. 1972, 129, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Nizet, T.A.; van den Elshout, F.J.; Heijdra, Y.F.; van de Ven, M.J.; Mulder, P.G.; Folgering, H.T. Survival of chronic hypercapnic COPD patients is predicted by smoking habits, comorbidity, and hypoxemia. Chest 2005, 127, 1904–1910. [Google Scholar] [CrossRef] [Green Version]
- Osadnik, C.R.; Tee, V.S.; Carson-Chahhoud, K.V.; Picot, J.; Wedzicha, J.A.; Smith, B.J. Non-invasive ventilation for the management of acute hypercapnic respiratory failure due to exacerbation of chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2017, 7, Cd004104. [Google Scholar] [CrossRef] [Green Version]
- Ergan, B.; Oczkowski, S.; Rochwerg, B.; Carlucci, A.; Chatwin, M.; Clini, E.; Elliott, M.; Gonzalez-Bermejo, J.; Hart, N.; Lujan, M.; et al. European Respiratory Society guidelines on long-term home non-invasive ventilation for management of COPD. Eur. Respir. J. 2019, 54, 1901003. [Google Scholar] [CrossRef] [Green Version]
- Macrea, M.; Oczkowski, S.; Rochwerg, B.; Branson, R.D.; Celli, B.; Coleman, J.M.; Hess, D.R.; Knight, S.L.; Ohar, J.A.; Orr, J.E.; et al. Long-Term Noninvasive Ventilation in Chronic Stable Hypercapnic Chronic Obstructive Pulmonary Disease. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e74–e87. [Google Scholar] [CrossRef]
- Wagner, P.D.; Dantzker, D.R.; Dueck, R.; Clausen, J.L.; West, J.B. Ventilation-perfusion inequality in chronic obstructive pulmonary disease. J. Clin. Investig. 1977, 59, 203–216. [Google Scholar] [CrossRef]
- Gorini, M.; Misuri, G.; Corrado, A.; Duranti, R.; Iandelli, I.; De Paola, E.; Scano, G. Breathing pattern and carbon dioxide retention in severe chronic obstructive pulmonary disease. Thorax 1996, 51, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, D.A.; Jover, L.; Drakulovic, M.B.; Gómez, F.P.; Roca, J.; Barberà, J.A.; Wagner, P.D.; Rodríguez-Roisin, R. Below what FEV1 should arterial blood be routinely taken to detect chronic respiratory failure in COPD? Arch. Bronconeumol. 2011, 47, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Mathews, A.M.; Wysham, N.G.; Xie, J.; Qin, X.; Giovacchini, C.X.; Ekström, M.; MacIntyre, N.R. Hypercapnia in Advanced Chronic Obstructive Pulmonary Disease: A Secondary Analysis of the National Emphysema Treatment Trial. Chronic Obstr. Pulm. Dis. 2020, 7, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.S.; Tin, C.; Song, G. Submissive hypercapnia: Why COPD patients are more prone to CO2 retention than heart failure patients. Respir. Physiol. Neurobiol. 2015, 216, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussos, C.; Fixley, M.; Gross, D.; Macklem, P.T. Fatigue of inspiratory muscles and their synergic behavior. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1979, 46, 897–904. [Google Scholar] [CrossRef]
- Calverley, P.M.; Walker, P. Chronic obstructive pulmonary disease. Lancet 2003, 362, 1053–1061. [Google Scholar] [CrossRef]
- O’Donnell, D.E.; D’Arsigny, C.; Fitzpatrick, M.; Webb, K.A. Exercise hypercapnia in advanced chronic obstructive pulmonary disease: The role of lung hyperinflation. Am. J. Respir. Crit. Care Med. 2002, 166, 663–668. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, D.E.; Webb, K.A.; Neder, J.A. Lung hyperinflation in COPD: Applying physiology to clinical practice. COPD Res. Pract. 2015, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Decramer, M. Hyperinflation and respiratory muscle interaction. Eur. Respir. J. 1997, 10, 934–941. [Google Scholar]
- Dodd, D.S.; Yarom, J.; Loring, S.H.; Engel, L.A. O2 cost of inspiratory and expiratory resistive breathing in humans. J. Appl. Physiol. (1985) 1988, 65, 2518–2523. [Google Scholar] [CrossRef]
- Bégin, P.; Grassino, A. Inspiratory muscle dysfunction and chronic hypercapnia in chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1991, 143, 905–912. [Google Scholar] [CrossRef]
- Bellemare, F.; Grassino, A. Force reserve of the diaphragm in patients with chronic obstructive pulmonary disease. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.A.; Zagelbaum, G.; Gross, D.; Roussos, C.; Macklem, P.T. Clinical manifestations of inspiratory muscle fatigue. Am. J. Med. 1982, 73, 308–316. [Google Scholar] [CrossRef]
- Gea, J.; Sancho-Muñoz, A.; Chalela, R. Nutritional status and muscle dysfunction in chronic respiratory diseases: Stable phase versus acute exacerbations. J. Thorac. Dis. 2018, 10, S1332–S1354. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.K.; Cho, E.N.; Chang, J.; Ahn, C.M.; Kim, H.J. Sarcopenia correlates with systemic inflammation in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topeli, A.; Laghi, F.; Tobin, M.J. The voluntary drive to breathe is not decreased in hypercapnic patients with severe COPD. Eur. Respir. J. 2001, 18, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, R.; Keighley, J.; Auchincloss, J.H. Mechanisms of Chronic Carbon Dioxide Retention in Patients with Obstructive Pulmonary Disease. Am. J. Med. 1965, 38, 217–225. [Google Scholar] [CrossRef]
- Lourenço, R.V.; Miranda, J.M. Drive and performance of the ventilatory apparatus in chronic obstructive lung disease. N. Engl. J. Med. 1968, 279, 53–59. [Google Scholar] [CrossRef]
- Richter, D.W.; Ballanyi, K.; Schwarzacher, S. Mechanisms of respiratory rhythm generation. Curr. Opin. Neurobiol. 1992, 2, 788–793. [Google Scholar] [CrossRef]
- Neubauer, J.A.; Melton, J.E.; Edelman, N.H. Modulation of respiration during brain hypoxia. J. Appl. Physiol. (1985) 1990, 68, 441–451. [Google Scholar] [CrossRef]
- Abdo, W.F.; Heunks, L.M. Oxygen-induced hypercapnia in COPD: Myths and facts. Crit. Care 2012, 16, 323. [Google Scholar] [CrossRef] [Green Version]
- Kaw, R.; Hernandez, A.V.; Walker, E.; Aboussouan, L.; Mokhlesi, B. Determinants of hypercapnia in obese patients with obstructive sleep apnea: A systematic review and metaanalysis of cohort studies. Chest 2009, 136, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Kjensli, A.; Falch, J.A.; Ryg, M.; Blenk, T.; Armbrecht, G.; Diep, L.M.; Ellingsen, I. High prevalence of vertebral deformities in COPD patients: Relationship to disease severity. Eur. Respir. J. 2009, 33, 1018–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gea, J.; Agusti, A.; Roca, J. Pathophysiology of muscle dysfunction in COPD. J. Appl. Physiol. (1985) 2013, 114, 1222–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, H.; Dodic, S.; Goh, S.S.; Green, C.; Wang, W.C.; Kaul, S.; Tiruvoipati, R. Factors associated with hospital mortality in critically ill patients with exacerbation of COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 2361–2366. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.F.; Sheng, Y.; Zhu, N.; Tan, Y.; Xie, X.H.; Wang, S.Y.; Cai, J.F. The v-DECAF score can predict 90-day all-cause mortality in patients with COPD exacerbation requiring invasive mechanical ventilation. Clin. Respir. J. 2019, 13, 438–445. [Google Scholar] [CrossRef]
- Chu, C.M.; Chan, V.L.; Lin, A.W.; Wong, I.W.; Leung, W.S.; Lai, C.K. Readmission rates and life threatening events in COPD survivors treated with non-invasive ventilation for acute hypercapnic respiratory failure. Thorax 2004, 59, 1020–1025. [Google Scholar] [CrossRef] [Green Version]
- Hartl, S.; Lopez-Campos, J.L.; Pozo-Rodriguez, F.; Castro-Acosta, A.; Studnicka, M.; Kaiser, B.; Roberts, C.M. Risk of death and readmission of hospital-admitted COPD exacerbations: European COPD Audit. Eur. Respir. J. 2016, 47, 113–121. [Google Scholar] [CrossRef]
- Csoma, B.; Bikov, A.; Tóth, F.; Losonczy, G.; Müller, V.; Lázár, Z. Blood eosinophils on hospital admission for COPD exacerbation do not predict the recurrence of moderate and severe relapses. ERJ Open Res. 2021, 7, 00543-2020. [Google Scholar] [CrossRef]
- Seneff, M.G.; Wagner, D.P.; Wagner, R.P.; Zimmerman, J.E.; Knaus, W.A. Hospital and 1-year survival of patients admitted to intensive care units with acute exacerbation of chronic obstructive pulmonary disease. JAMA 1995, 274, 1852–1857. [Google Scholar] [CrossRef]
- Marthan, R.; Castaing, Y.; Manier, G.; Guenard, H. Gas exchange alterations in patients with chronic obstructive lung disease. Chest 1985, 87, 470–475. [Google Scholar] [CrossRef]
- Torres, A.; Reyes, A.; Roca, J.; Wagner, P.D.; Rodriguez-Roisin, R. Ventilation-perfusion mismatching in chronic obstructive pulmonary disease during ventilator weaning. Am. Rev. Respir. Dis. 1989, 140, 1246–1250. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, K.; Bosken, C.H.; Paré, P.D.; Bai, T.R.; Wiggs, B.R.; Hogg, J.C. Small airways dimensions in asthma and in chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1993, 148, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Fleury, B.; Murciano, D.; Talamo, C.; Aubier, M.; Pariente, R.; Milic-Emili, J. Work of breathing in patients with chronic obstructive pulmonary disease in acute respiratory failure. Am. Rev. Respir. Dis. 1985, 131, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Barberà, J.A.; Roca, J.; Ferrer, A.; Félez, M.A.; Díaz, O.; Roger, N.; Rodriguez-Roisin, R. Mechanisms of worsening gas exchange during acute exacerbations of chronic obstructive pulmonary disease. Eur. Respir. J. 1997, 10, 1285–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calverley, P.M.; Koulouris, N.G. Flow limitation and dynamic hyperinflation: Key concepts in modern respiratory physiology. Eur. Respir. J. 2005, 25, 186–199. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, D.E.; Parker, C.M. COPD exacerbations·3: Pathophysiology. Thorax 2006, 61, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Haluszka, J.; Chartrand, D.A.; Grassino, A.E.; Milic-Emili, J. Intrinsic PEEP and arterial PCO2 in stable patients with chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1990, 141, 1194–1197. [Google Scholar] [CrossRef]
- Orozco-Levi, M.; Lloreta, J.; Minguella, J.; Serrano, S.; Broquetas, J.M.; Gea, J. Injury of the human diaphragm associated with exertion and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 1734–1739. [Google Scholar] [CrossRef]
- Ram, F.S.F.; Picot, J.; Lightowler, J.; Wedzicha, J.A. Non-invasive positive pressure ventilation for treatment of respiratory failure due to exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2004, 3, CD004104. [Google Scholar] [CrossRef]
- Rochwerg, B.; Brochard, L.; Elliott, M.W.; Hess, D.; Hill, N.S.; Nava, S.; Navalesi, P.; Antonelli, M.; Brozek, J.; Conti, G.; et al. Official ERS/ATS clinical practice guidelines: Noninvasive ventilation for acute respiratory failure. Eur. Respir. J. 2017, 50, 1602426. [Google Scholar] [CrossRef]
- Cummins, E.P.; Strowitzki, M.J.; Taylor, C.T. Mechanisms and Consequences of Oxygen and Carbon Dioxide Sensing in Mammals. Physiol. Rev. 2020, 100, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Saint-Criq, V.; Patel, W.; Ibrahim, S.H.; Verdon, B.; Ward, C.; Garnett, J.P.; Tarran, R.; Cann, M.J.; Gray, M.A. Hypercapnia modulates cAMP signalling and cystic fibrosis transmembrane conductance regulator-dependent anion and fluid secretion in airway epithelia. J. Physiol. 2016, 594, 1643–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadasz, I.; Dada, L.A.; Briva, A.; Trejo, H.E.; Welch, L.C.; Chen, J.; Toth, P.T.; Lecuona, E.; Witters, L.A.; Schumacker, P.T.; et al. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J. Clin. Investig. 2008, 118, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Shigemura, M.; Sznajder, J.I. Elevated CO2 modulates airway contractility. Interface Focus 2021, 11, 20200021. [Google Scholar] [CrossRef]
- Shigemura, M.; Lecuona, E.; Sznajder, J.I. Effects of hypercapnia on the lung. J. Physiol. 2017, 595, 2431–2437. [Google Scholar] [CrossRef] [Green Version]
- Lecuona, E.; Sun, H.; Chen, J.; Trejo, H.E.; Baker, M.A.; Sznajder, J.I. Protein kinase A-Ialpha regulates Na,K-ATPase endocytosis in alveolar epithelial cells exposed to high CO(2) concentrations. Am. J. Respir. Cell Mol. Biol. 2013, 48, 626–634. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, D.; Hassett, P.; Contreras, M.; Higgins, B.D.; McKeown, S.T.; McAuley, D.F.; O’Brien, T.; Laffey, J.G. Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-kappaB dependent mechanism. Thorax 2009, 64, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Doerr, C.H.; Gajic, O.; Berrios, J.C.; Caples, S.; Abdel, M.; Lymp, J.F.; Hubmayr, R.D. Hypercapnic acidosis impairs plasma membrane wound resealing in ventilator-injured lungs. Am. J. Respir. Crit. Care Med. 2005, 171, 1371–1377. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Puentes, G.A.; Westerly, B.; Schiavo, D.; Wang, S.; Stroetz, R.; Walters, B.; Hubmayr, R.D.; Oeckler, R.A. Hypercapnia Alters Alveolar Epithelial Repair by a pH-Dependent and Adenylate Cyclase-Mediated Mechanism. Sci. Rep. 2019, 9, 349. [Google Scholar] [CrossRef] [Green Version]
- Casalino-Matsuda, S.M.; Wang, N.; Ruhoff, P.T.; Matsuda, H.; Nlend, M.C.; Nair, A.; Szleifer, I.; Beitel, G.J.; Sznajder, J.I.; Sporn, P.H.S. Hypercapnia Alters Expression of Immune Response, Nucleosome Assembly and Lipid Metabolism Genes in Differentiated Human Bronchial Epithelial Cells. Sci. Rep. 2018, 8, 13508. [Google Scholar] [CrossRef]
- Masterson, C.; Horie, S.; McCarthy, S.D.; Gonzalez, H.; Byrnes, D.; Brady, J.; Fandino, J.; Laffey, J.G.; O’Toole, D. Hypercapnia in the critically ill: Insights from the bench to the bedside. Interface Focus 2021, 11, 20200032. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Oliver, K.M.; Lenihan, C.R.; Fitzpatrick, S.F.; Bruning, U.; Scholz, C.C.; Slattery, C.; Leonard, M.O.; McLoughlin, P.; Taylor, C.T. NF-kappaB links CO2 sensing to innate immunity and inflammation in mammalian cells. J. Immunol. 2010, 185, 4439–4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keogh, C.E.; Scholz, C.C.; Rodriguez, J.; Selfridge, A.C.; von Kriegsheim, A.; Cummins, E.P. Carbon dioxide-dependent regulation of NF-kappaB family members RelB and p100 gives molecular insight into CO2-dependent immune regulation. J. Biol. Chem. 2017, 292, 11561–11571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masterson, C.; O’Toole, D.; Leo, A.; McHale, P.; Horie, S.; Devaney, J.; Laffey, J.G. Effects and Mechanisms by Which Hypercapnic Acidosis Inhibits Sepsis-Induced Canonical Nuclear Factor-kappaB Signaling in the Lung. Crit. Care Med. 2016, 44, e207–e217. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.J.; Dong, P.; Hosszu, E.K.; Doyle, I.R. Effect of CO2 on LPS-induced cytokine responses in rat alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L96–L103. [Google Scholar] [CrossRef]
- Casalino-Matsuda, S.M.; Chen, F.; Gonzalez-Gonzalez, F.J.; Nair, A.; Dib, S.; Yemelyanov, A.; Gates, K.L.; Budinger, G.R.S.; Beitel, G.J.; Sporn, P.H.S. Hypercapnia Suppresses Macrophage Antiviral Activity and Increases Mortality of Influenza A Infection via Akt1. J. Immunol. 2020, 205, 489–501. [Google Scholar] [CrossRef]
- Chonghaile, M.N.; Higgins, B.D.; Costello, J.; Laffey, J.G. Hypercapnic acidosis attenuates lung injury induced by established bacterial pneumonia. Anesthesiology 2008, 109, 837–848. [Google Scholar] [CrossRef] [Green Version]
- O’Croinin, D.F.; Hopkins, N.O.; Moore, M.M.; Boylan, J.F.; McLoughlin, P.; Laffey, J.G. Hypercapnic acidosis does not modulate the severity of bacterial pneumonia-induced lung injury. Crit. Care Med. 2005, 33, 2606–2612. [Google Scholar] [CrossRef]
- O’Croinin, D.F.; Nichol, A.D.; Hopkins, N.; Boylan, J.; O’Brien, S.; O’Connor, C.; Laffey, J.G.; McLoughlin, P. Sustained hypercapnic acidosis during pulmonary infection increases bacterial load and worsens lung injury. Crit. Care Med. 2008, 36, 2128–2135. [Google Scholar] [CrossRef]
- Shigemura, M.; Lecuona, E.; Angulo, M.; Homma, T.; Rodriguez, D.A.; Gonzalez-Gonzalez, F.J.; Welch, L.C.; Amarelle, L.; Kim, S.J.; Kaminski, N.; et al. Hypercapnia increases airway smooth muscle contractility via caspase-7-mediated miR-133a-RhoA signaling. Sci. Transl. Med. 2018, 10, aat1662. [Google Scholar] [CrossRef] [Green Version]
- Rodarte, J.R.; Hyatt, R.E. Effect of acute exposure to CO2 on lung mechanics in normal man. Respir. Physiol. 1973, 17, 135–145. [Google Scholar] [CrossRef]
- Uno, T.; Homma, T.; Shigemura, M.; Fukuda, Y.; Kimura, T.; Onitsuka, C.; Kawahara, T.; Sato, H.; Akimoto, K.; Suganuma, H.; et al. Correlation of Arterial CO2 and Respiratory Impedance Values among Subjects with COPD. J. Clin. Med. 2020, 9, 2819. [Google Scholar] [CrossRef] [PubMed]
- El Mays, T.Y.; Saifeddine, M.; Choudhury, P.; Hollenberg, M.D.; Green, F.H. Carbon dioxide enhances substance P-induced epithelium-dependent bronchial smooth muscle relaxation in Sprague-Dawley rats. Can. J. Physiol. Pharmacol. 2011, 89, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Astin, T.W.; Barer, G.R.; Shaw, J.W.; Warren, P.M. The action of carbon dioxide on constricted airways. J. Physiol. 1973, 235, 607–623. [Google Scholar] [CrossRef]
- Croxton, T.L.; Lande, B.; Hirshman, C.A. Role of intracellular pH in relaxation of porcine tracheal smooth muscle by respiratory gases. Am. J. Physiol. 1995, 268, L207–L213. [Google Scholar] [CrossRef]
- Tisi, G.M.; Wolfe, W.G.; Fallat, R.J.; Nadel, J.A. Effects of O2 and CO2 on airway smooth muscle following pulmonary vascular occlusion. J. Appl. Physiol. 1970, 28, 570–573. [Google Scholar] [CrossRef]
- Twort, C.H.; Cameron, I.R. Effects of PCO2, pH and extracellular calcium on contraction of airway smooth muscle from rats. Respir. Physiol. 1986, 66, 259–267. [Google Scholar] [CrossRef]
- Yamakage, M.; Lindeman, K.S.; Hirshman, C.A.; Croxton, T.L. Intracellular pH regulates voltage-dependent Ca2+ channels in porcine tracheal smooth muscle cells. Am. J. Physiol. 1995, 268, L642–L646. [Google Scholar] [CrossRef]
- Sterling, G.M.; Holst, P.E.; Nadel, J.A. Effect of CO2 and pH on bronchoconstriction caused by serotonin vs. acetylcholine. J. Appl. Physiol. 1972, 32, 39–43. [Google Scholar] [CrossRef]
- Lutter, J.I.; Jorres, R.A.; Kahnert, K.; Schwarzkopf, L.; Studnicka, M.; Karrasch, S.; Schulz, H.; Vogelmeier, C.F.; Holle, R.; for the COSYCONET Study Group. Health-related quality of life associates with change in FEV1 in COPD: Results from the COSYCONET cohort. BMC Pulm. Med. 2020, 20, 148. [Google Scholar] [CrossRef]
- Budweiser, S.; Hitzl, A.P.; Jorres, R.A.; Schmidbauer, K.; Heinemann, F.; Pfeifer, M. Health-related quality of life and long-term prognosis in chronic hypercapnic respiratory failure: A prospective survival analysis. Respir. Res. 2007, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohnlein, T.; Windisch, W.; Kohler, D.; Drabik, A.; Geiseler, J.; Hartl, S.; Karg, O.; Laier-Groeneveld, G.; Nava, S.; Schonhofer, B.; et al. Non-invasive positive pressure ventilation for the treatment of severe stable chronic obstructive pulmonary disease: A prospective, multicentre, randomised, controlled clinical trial. Lancet Respir. Med. 2014, 2, 698–705. [Google Scholar] [CrossRef]
- Windisch, W.; on behalf of the Quality of Life in Home Mechanical Ventilation Study Group. Impact of home mechanical ventilation on health-related quality of life. Eur. Respir. J. 2008, 32, 1328–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaouat, A.; Bugnet, A.S.; Kadaoui, N.; Schott, R.; Enache, I.; Ducolone, A.; Ehrhart, M.; Kessler, R.; Weitzenblum, E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 172, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.H.; Iversen, M.; Kjaergaard, J.; Mortensen, J.; Nielsen-Kudsk, J.E.; Bendstrup, E.; Videbaek, R.; Carlsen, J. Prevalence, predictors, and survival in pulmonary hypertension related to end-stage chronic obstructive pulmonary disease. J. Heart Lung Transplant. 2012, 31, 373–380. [Google Scholar] [CrossRef]
- Kovacs, G.; Avian, A.; Bachmaier, G.; Troester, N.; Tornyos, A.; Douschan, P.; Foris, V.; Sassmann, T.; Zeder, K.; Lindenmann, J.; et al. Severe Pulmonary Hypertension in COPD: Impact on Survival and Diagnostic Approach. Chest 2022. [Google Scholar] [CrossRef]
- McGuire, M.; Bradford, A. Chronic intermittent hypercapnic hypoxia increases pulmonary arterial pressure and haematocrit in rats. Eur. Respir. J. 2001, 18, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Foex, P.; Prys-Roberts, C. Effects of changes in PaCO2 on pulmonary input impedance. J. Appl. Physiol. 1975, 38, 52–57. [Google Scholar] [CrossRef]
- Viitanen, A.; Salmenpera, M.; Heinonen, J. Right ventricular response to hypercarbia after cardiac surgery. Anesthesiology 1990, 73, 393–400. [Google Scholar] [CrossRef]
- Dessap, A.M.; Charron, C.; Devaquet, J.; Aboab, J.; Jardin, F.; Brochard, L.; Vieillard-Baron, A. Impact of acute hypercapnia and augmented positive end-expiratory pressure on right ventricle function in severe acute respiratory distress syndrome. Intensive Care Med. 2009, 35, 1850–1858. [Google Scholar] [CrossRef]
- Brimioulle, S.; Lejeune, P.; Vachiery, J.L.; Leeman, M.; Melot, C.; Naeije, R. Effects of acidosis and alkalosis on hypoxic pulmonary vasoconstriction in dogs. Am. J. Physiol. 1990, 258, H347–H353. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.E., Jr.; Van Benthuysen, K.; Jackson, J.T.; Tucker, C.E.; Kaiser, D.L.; Grover, R.F.; Weil, J.V. Right ventricular performance during increased afterload impaired by hypercapnic acidosis in conscious dogs. Circ. Res. 1983, 52, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Held, M.; Walthelm, J.; Baron, S.; Roth, C.; Jany, B. Functional impact of pulmonary hypertension due to hypoventilation and changes under noninvasive ventilation. Eur. Respir. J. 2014, 43, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Crystal, G.J. Carbon Dioxide and the Heart: Physiology and Clinical Implications. Anesth. Analg. 2015, 121, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Korponay, T.C.; Balnis, J.; Vincent, C.E.; Singer, D.V.; Chopra, A.; Adam, A.P.; Ginnan, R.; Singer, H.A.; Jaitovich, A. High CO2 Downregulates Skeletal Muscle Protein Anabolism via AMP-activated Protein Kinase alpha2-mediated Depressed Ribosomal Biogenesis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Corfield, D.R.; McKay, L.C. Regional Cerebrovascular Responses to Hypercapnia and Hypoxia. Adv. Exp. Med. Biol. 2016, 903, 157–167. [Google Scholar] [CrossRef]
- Chen, W.; Thomas, J.; Sadatsafavi, M.; FitzGerald, J.M. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: A systematic review and meta-analysis. Lancet Respir. Med. 2015, 3, 631–639. [Google Scholar] [CrossRef]
- Curley, G.; Laffey, J.G.; Kavanagh, B.P. Bench-to-bedside review: Carbon dioxide. Crit. Care 2010, 14, 220. [Google Scholar] [CrossRef] [Green Version]
- Pelletier-Galarneau, M.; deKemp, R.A.; Hunter, C.; Klein, R.; Klein, M.; Ironstone, J.; Fisher, J.A.; Ruddy, T.D. Effects of Hypercapnia on Myocardial Blood Flow in Healthy Human Subjects. J. Nucl. Med. 2018, 59, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.J.; Dey, D.; Sykes, J.; Klein, M.; Butler, J.; Kovacs, M.S.; Sobczyk, O.; Sharif, B.; Bi, X.; Kali, A.; et al. Arterial CO2 as a Potent Coronary Vasodilator: A Preclinical PET/MR Validation Study with Implications for Cardiac Stress Testing. J. Nucl. Med. 2017, 58, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Turek, Z.; Kreuzer, F. Effect of shifts of the O2 dissociation curve upon alveolar-arterial O2 gradients in computer models of the lung with ventilation-perfusion mismatching. Respir. Physiol. 1981, 45, 133–139. [Google Scholar] [CrossRef]
- Brogan, T.V.; Robertson, H.T.; Lamm, W.J.; Souders, J.E.; Swenson, E.R. Carbon dioxide added late in inspiration reduces ventilation-perfusion heterogeneity without causing respiratory acidosis. J. Appl. Physiol. (1985) 2004, 96, 1894–1898. [Google Scholar] [CrossRef] [PubMed]
- Swenson, E.R.; Robertson, H.T.; Hlastala, M.P. Effects of inspired carbon dioxide on ventilation-perfusion matching in normoxia, hypoxia, and hyperoxia. Am. J. Respir. Crit. Care Med. 1994, 149, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Cullen, D.J.; Eger, E.I. Cardiovascular effects of carbon dioxide in man. Anesthesiology 1974, 41, 345–349. [Google Scholar] [CrossRef]
- Akca, O.; Doufas, A.G.; Morioka, N.; Iscoe, S.; Fisher, J.; Sessler, D.I. Hypercapnia improves tissue oxygenation. Anesthesiology 2002, 97, 801–806. [Google Scholar] [CrossRef]
- Balnis, J.; Korponay, T.C.; Jaitovich, A. AMP-Activated Protein Kinase (AMPK) at the Crossroads between CO2 Retention and Skeletal Muscle Dysfunction in Chronic Obstructive Pulmonary Disease (COPD). Int. J. Mol. Sci. 2020, 21, 955. [Google Scholar] [CrossRef] [Green Version]
- Marquis, K.; Debigare, R.; Lacasse, Y.; LeBlanc, P.; Jobin, J.; Carrier, G.; Maltais, F. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 166, 809–813. [Google Scholar] [CrossRef]
- Schols, A.M.; Broekhuizen, R.; Weling-Scheepers, C.A.; Wouters, E.F. Body composition and mortality in chronic obstructive pulmonary disease. Am. J. Clin. Nutr. 2005, 82, 53–59. [Google Scholar] [CrossRef]
- Swallow, E.B.; Reyes, D.; Hopkinson, N.S.; Man, W.D.; Porcher, R.; Cetti, E.J.; Moore, A.J.; Moxham, J.; Polkey, M.I. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax 2007, 62, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Jaitovich, A.; Angulo, M.; Lecuona, E.; Dada, L.A.; Welch, L.C.; Cheng, Y.; Gusarova, G.; Ceco, E.; Liu, C.; Shigemura, M.; et al. High CO2 levels cause skeletal muscle atrophy via AMP-activated kinase (AMPK), FoxO3a protein, and muscle-specific Ring finger protein 1 (MuRF1). J. Biol. Chem. 2015, 290, 9183–9194. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- British Thoracic Society Standards of Care Committee. Non-invasive ventilation in acute respiratory failure. Thorax 2002, 57, 192–211. [Google Scholar] [CrossRef] [Green Version]
- Corrado, A.; Gorini, M. Negative-pressure ventilation: Is there still a role? Eur. Respir. J. 2002, 20, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, C.M.; Stone, R.A.; Buckingham, R.J.; Pursey, N.A.; Lowe, D.; On behalf of the National Chronic Obstructive Pulmonary Disease Resources and Outcomes Project (NCROP) Implementation Group. Acidosis, non-invasive ventilation and mortality in hospitalised COPD exacerbations. Thorax 2011, 66, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.W. Noninvasive ventilation in acute exacerbations of COPD. Eur. Respir. Rev. 2005, 14, 39–42. [Google Scholar] [CrossRef]
- Casanova, C.; Celli, B.R.; Tost, L.; Soriano, E.; Abreu, J.; Velasco, V.; Santolaria, F. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000, 118, 1582–1590. [Google Scholar] [CrossRef] [Green Version]
- Clini, E.; Sturani, C.; Rossi, A.; Viaggi, S.; Corrado, A.; Donner, C.F.; Ambrosino, N. The Italian multicentre study on noninvasive ventilation in chronic obstructive pulmonary disease patients. Eur. Respir. J. 2002, 20, 529–538. [Google Scholar] [CrossRef]
- McEvoy, R.D.; Pierce, R.J.; Hillman, D.; Esterman, A.; Ellis, E.E.; Catcheside, P.G.; O’Donoghue, F.J.; Barnes, D.J.; Grunstein, R.R.; on behalf of the Australian Trial of Non-Invasive Ventilation in Chronic Airflow Limitation (AVCAL) Study Group. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: A randomised controlled trial. Thorax 2009, 64, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Windisch, W.; Haenel, M.; Storre, J.H.; Dreher, M. High-intensity non-invasive positive pressure ventilation for stable hypercapnic COPD. Int. J. Med. Sci. 2009, 6, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Chiang, L.L.; Liu, C.Y.; Ho, S.C.; Sheng, T.F.; Yu, C.T.; Lin, H.C.; Kuo, H.P. Efficacy of nocturnal nasal positive pressure ventilation in hypercapnic patients with severe obstructive lung diseases. Chang. Gung Med. J. 2004, 27, 98–106. [Google Scholar]
- Zhou, L.; Li, X.; Guan, L.; Chen, J.; Guo, B.; Wu, W.; Huo, Y.; Zhou, Z.; Liang, Z.; Zhou, Y.; et al. Home noninvasive positive pressure ventilation with built-in software in stable hypercapnic COPD: A short-term prospective, multicenter, randomized, controlled trial. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1279–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, S.P.; Peterson, M.W.; Wilson, J.S.; Durairaj, L. Noninvasive positive pressure ventilation in subjects with stable COPD: A randomized trial. Int. J. Chronic Obstr. Pulm. Dis. 2013, 8, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Duiverman, M.L.; Wempe, J.B.; Bladder, G.; Jansen, D.F.; Kerstjens, H.A.; Zijlstra, J.G.; Wijkstra, P.J. Nocturnal non-invasive ventilation in addition to rehabilitation in hypercapnic patients with COPD. Thorax 2008, 63, 1052–1057. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Martin, E.; Ruiz, F.O.; Ramos, P.C.; Lopez-Campos, J.L.; Azcona, B.V.; Cortes, E.B. Randomized trial of non-invasive ventilation combined with exercise training in patients with chronic hypercapnic failure due to chronic obstructive pulmonary disease. Respir. Med. 2014, 108, 1741–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eman Shebl, R.; Abderaboh, M.M. Bi-level positive airway pressure ventilation for patients with stable hypercapnic chronic obstructive pulmonary disease. Egypt. J. Chest Dis. Tuberc. 2015, 64, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Dreher, M.; Storre, J.H.; Schmoor, C.; Windisch, W. High-intensity versus low-intensity non-invasive ventilation in patients with stable hypercapnic COPD: A randomised crossover trial. Thorax 2010, 65, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrod, R.; Mikelsons, C.; Paul, E.A.; Wedzicha, J.A. Randomized controlled trial of domiciliary noninvasive positive pressure ventilation and physical training in severe chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2000, 162, 1335–1341. [Google Scholar] [CrossRef]
- Tsolaki, V.; Pastaka, C.; Karetsi, E.; Zygoulis, P.; Koutsokera, A.; Gourgoulianis, K.I.; Kostikas, K. One-year non-invasive ventilation in chronic hypercapnic COPD: Effect on quality of life. Respir. Med. 2008, 102, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.E.; Dobler, C.C.; Morrow, A.S.; Beuschel, B.; Alsawas, M.; Benkhadra, R.; Seisa, M.; Mittal, A.; Sanchez, M.; Daraz, L.; et al. Association of Home Noninvasive Positive Pressure Ventilation With Clinical Outcomes in Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-analysis. JAMA 2020, 323, 455–465. [Google Scholar] [CrossRef]
- Mathioudakis, A.G.; Abroug, F.; Agusti, A.; Ananth, S.; Bakke, P.; Bartziokas, K.; Beghe, B.; Bikov, A.; Bradbury, T.; Brusselle, G.; et al. ERS Statement: A core outcome set for clinical trials evaluating the management of chronic obstructive pulmonary disease (COPD) exacerbations. Eur. Respir. J. 2021, 59, 2102006. [Google Scholar] [CrossRef]
- Donaldson, G.C.; Seemungal, T.A.; Bhowmik, A.; Wedzicha, J.A. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002, 57, 847–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, J.R.; Vestbo, J.; Anzueto, A.; Locantore, N.; Mullerova, H.; Tal-Singer, R.; Miller, B.; Lomas, D.A.; Agusti, A.; Macnee, W.; et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N. Engl. J. Med. 2010, 363, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Pierson, D.J. Complications associated with mechanical ventilation. Crit. Care Clin. 1990, 6, 711–724. [Google Scholar] [CrossRef]
- Scheinhorn, D.J.; Hassenpflug, M.S.; Votto, J.J.; Chao, D.C.; Epstein, S.K.; Doig, G.S.; Knight, E.B.; Petrak, R.A.; Ventilation Outcomes Study, G. Post-ICU mechanical ventilation at 23 long-term care hospitals: A multicenter outcomes study. Chest 2007, 131, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasta, J.F.; McLaughlin, T.P.; Mody, S.H.; Piech, C.T. Daily cost of an intensive care unit day: The contribution of mechanical ventilation. Crit. Care Med. 2005, 33, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Conoscenti, C.C.; Menashe, P.; Nair, S. Noninvasive face mask ventilation in patients with acute respiratory failure. Chest 1989, 95, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.P.; Chan, V.L.; Liong, J.T.; Lam, J.Y.; Leung, W.S.; Lin, A.; Chu, C.M. A pilot trial of non-invasive home ventilation after acidotic respiratory failure in chronic obstructive pulmonary disease. Int. J. Tuberc. Lung Dis. 2010, 14, 642–649. [Google Scholar] [PubMed]
- De Backer, L.; Vos, W.; Dieriks, B.; Daems, D.; Verhulst, S.; Vinchurkar, S.; Ides, K.; De Backer, J.; Germonpre, P.; De Backer, W. The effects of long-term noninvasive ventilation in hypercapnic COPD patients: A randomized controlled pilot study. Int. J. Chronic Obstr. Pulm. Dis. 2011, 6, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Murphy, P.B.; Rehal, S.; Arbane, G.; Bourke, S.; Calverley, P.M.A.; Crook, A.M.; Dowson, L.; Duffy, N.; Gibson, G.J.; Hughes, P.D.; et al. Effect of Home Noninvasive Ventilation With Oxygen Therapy vs. Oxygen Therapy Alone on Hospital Readmission or Death After an Acute COPD Exacerbation: A Randomized Clinical Trial. JAMA 2017, 317, 2177–2186. [Google Scholar] [CrossRef]
- Struik, F.M.; Sprooten, R.T.; Kerstjens, H.A.; Bladder, G.; Zijnen, M.; Asin, J.; Cobben, N.A.; Vonk, J.M.; Wijkstra, P.J. Nocturnal non-invasive ventilation in COPD patients with prolonged hypercapnia after ventilatory support for acute respiratory failure: A randomised, controlled, parallel-group study. Thorax 2014, 69, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Costello, R.; Deegan, P.; Fitzpatrick, M.; McNicholas, W.T. Reversible hypercapnia in chronic obstructive pulmonary disease: A distinct pattern of respiratory failure with a favorable prognosis. Am. J. Med. 1997, 102, 239–244. [Google Scholar] [CrossRef]
- Funk, G.C.; Breyer, M.K.; Burghuber, O.C.; Kink, E.; Kirchheiner, K.; Kohansal, R.; Schmidt, I.; Hartl, S. Long-term non-invasive ventilation in COPD after acute-on-chronic respiratory failure. Respir. Med. 2011, 105, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Study Name | Population | Primary Outcome | Favors NIV | Baseline PaCO2, kPa | AE Frequency at Baseline | BMI | OSA | Normalizing Hypercapnia, Yes/No | NIV Mode |
|---|---|---|---|---|---|---|---|---|---|
| Casanova 2000 [127] | Stable | Number of AEs | No | 6.8 ± 1.1 | No data | 25 ± 4 | Excluded | No | Nasal BiPAP, S mode, EPAP: 4 cmH2O; IPAP: 12 cmH2O |
| Clini 2002 [128] | Stable | Arterial blood gas values, hospital and ICU admissions, total hospital and ICU length of stay, HRQL | Partly | 7.2 ± 0.6 | No data | 26 ± 5 | Excluded | Yes (5% decrease) | Nasal BiPAP, S/T mode, backup frequency: 8/min; EPAP: 2–5 cmH2O; IPAP: maximal tolerated pressure |
| Duiverman 2008 [134] | Stable | HRQL, functional status and gas exchange parameters | Yes | 6.89 ± 0.68 | No data | 27.1 ± 6.4 | Excluded | Yes (PaCO2 < 6.0 kPa) | BiPAP, S/T mode; IPAP: maximal tolerated pressure titrated towards an optimal correction of nocturnal arterial blood gases (PaCO2 6.0 kPa and PaO2 8.0 kPa) |
| Garrod 2000 [138] | Stable | Exercise capacity and health status | Yes | 5.9 ± 0.9 | No data | No data | Not excluded | No | Nasal BiPAP, S mode overnight or minimum 8 h/day, settings adjusted individually to obtain the maximal pressure tolerated; EPAP: 4 (4–6) cmH2O; IPAP: 16 (13–24) cmH2O |
| Köhnlein 2014 [93] | Stable | 1-year all-cause mortality | Yes | 7.8 ± 0.8 | No data | 24.8 ± 5.8 | Not excluded | Yes (>20% decrease or PaCO2 < 6.5 kPa) | Pressure support ventilation with high backup rates minimum 6 h/day, preferably during sleep (face or nasal mask). Aim: to reduce ≥20% baseline PaCO2 or PaCO2 < 6.5 kPa |
| Marquez-Martin 2014 [135] | Stable | Exercise capacity | Favors ventilation/training combined group over ventilation alone | NIV group: median 51, NIV-ET group: median 50 | No data | No data | Excluded | No | Nocturnal nasal BiPAP, S/T mode, backup frequency 12/min, 6–8 h/night; EPAP: 4 cmH2O; IPAP: initially 10 cmH2O and increased progressively to a maximum of 20 cmH2O, depending on patient tolerance, clinical response and SpO2 |
| McEvoy 2009 [129] | Stable | Survival | Yes | 7.01 [6.80–7.23] | No data | 25.5 [24.3–26.7] | Excluded | No | BiPAP, VPAP mode, EPAP: lowest possible level (~3 cm H2O); IPAP: gradually increased during daytime and night-time trials to the maximum tolerated with a target PS of ≥10 cm H2O |
| Cheung 2010 [148] | Post AE (>48 h after successful weaning of acute NIV) | Recurrent severe AE with AHRF requiring acute NIV, intubation or resulting in death in the first year | Yes | 7.7 ± 1.0 | Previous acute NIV: 1 [0–3], previous intubation: 0 (0–1), no other data | 19.2 ± 3.6 | Excluded | No | BiPAP, S/T mode, backup frequency: 14/min; EPAP: 5 cmH2O; IPAP: 10–20 cmH2O |
| De Backer 2011 [149] | Post AE (5–12 days after admission) | Arterial blood gas values and functional imaging of the lungs | Yes | 7.39 ± 1.03 | No data | No data | Excluded | Yes (5% decrease) | BiPAP for >5 h a day with a full face mask; modes were adapted until O2 saturation was >90% during 90% of the time, and PaCO2 was decreased 5% in 1 h |
| Funk 2011 [153] | Post AE (before discharge from the ICU or immediately after transfer to regular wards) | Time to clinical worsening Defined as an escalation of mechanical ventilation | Yes | 7.6 ± 1.7 | No data | 24.2 ± 4.3 | Excluded | No | BiPAP EPAP: ~5 cmH2O; IPAP: increasingly raised from 10 to ~20 cmH2O. The inspiratory time was limited to a maximum of 1.3 s |
| Murphy 2017 [150] | Post AE (2–4 weeks after resolution of respiratory acidemia) | Time to readmission or death within 12 months adjusted for the number of previous COPD admissions, previous use of long-term oxygen, age, and BMI | Yes | 7.87 ± 0.93 | ≥3 COPD-related readmissions within past year: NIV-LTOT group: N = 30 (53%) vs. LTOT group: N = 31 (53%) | 21.5 (18.8–24.5) | Excluded | Yes (reduce tcCO2 by at least 4 mmHg) | BiPAP, PS mode, recommended initial titration settings: IPAP 18 cmH2O, EPAP 4 cmH2O, backup rate 14–16/min; target IPAP ≥25 cmH2O. NIV settings and O2 flow rate were titrated to maintain SpO2 >88% and to reduce tcCO2 by ≥4 mmHg |
| Struik 2014 [151] | Post AE (>48 h after termination of ventilatory support) | Time to readmission for respiratory cause or death | No | 7.9 ± 1.2 | Median: 2, min–max: 1–9 | 24.6 ± 5.4 | Excluded | Yes (to achieve normocapnia) | BiPAP, S/T mode starting with a backup frequency of 12/min; IPAP: initial 14 cmH2O and gradually increased to a maximal tolerated level; EPAP: initial 4 cmH2O and increased if auto-PEEP was present or when patients used respiratory muscles to trigger the ventilator. Respiratory rate was set as close as possible to the that of the patient. I:E ratio was 1:3, with a short rise time and then titrated on comfort and effectiveness |
| Category | Recommendation |
|---|---|
| Screening | Patients with severe and very severe COPD and those on long-term oxygen therapy should have regular blood gas assessment. |
| Patients with acute hypercapnic respiratory failure should have a blood gas assessment at 2–4 weeks following discharge. | |
| Assessment | Pharmacological and nonpharmacological COPD treatment and other disorders causing hypercapnia (i.e., obesity, neuromuscular, and chest wall diseases) should be evaluated during assessment. |
| Routine sleep study should be offered to explore the presence of obstructive sleep apnoea and to identify variable (i.e., sleep-phase or positional) episodes of hypoventilation. | |
| Treatment | Pharmacological therapy should be optimised to improve symptoms and reduce the number of exacerbations. |
| Treatable traits contributing to hypercapnia (i.e., obesity and sarcopenia) should be addressed in parallel with NIV. | |
| Long-term NIV should be offered to those with persistent hypercapnic respiratory failure (PaCO2 ≥ 52 mmHg (>6.8 kPa)). | |
| The effect of long-term NIV therapy should be assessed with routine blood gas tests, sleep studies, and COPD-related outcomes (i.e., symptoms, quality of life, and the number of exacerbations). | |
| NIV treatment should be titrated to normalise PaCO2 (PaCO2 < 52 mmHg (<6.8 kPa)). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csoma, B.; Vulpi, M.R.; Dragonieri, S.; Bentley, A.; Felton, T.; Lázár, Z.; Bikov, A. Hypercapnia in COPD: Causes, Consequences, and Therapy. J. Clin. Med. 2022, 11, 3180. https://doi.org/10.3390/jcm11113180
Csoma B, Vulpi MR, Dragonieri S, Bentley A, Felton T, Lázár Z, Bikov A. Hypercapnia in COPD: Causes, Consequences, and Therapy. Journal of Clinical Medicine. 2022; 11(11):3180. https://doi.org/10.3390/jcm11113180
Chicago/Turabian StyleCsoma, Balázs, Maria Rosaria Vulpi, Silvano Dragonieri, Andrew Bentley, Timothy Felton, Zsófia Lázár, and Andras Bikov. 2022. "Hypercapnia in COPD: Causes, Consequences, and Therapy" Journal of Clinical Medicine 11, no. 11: 3180. https://doi.org/10.3390/jcm11113180
APA StyleCsoma, B., Vulpi, M. R., Dragonieri, S., Bentley, A., Felton, T., Lázár, Z., & Bikov, A. (2022). Hypercapnia in COPD: Causes, Consequences, and Therapy. Journal of Clinical Medicine, 11(11), 3180. https://doi.org/10.3390/jcm11113180