
Clonal Hematopoiesis and the Risk of Hematologic Malignancies after Curative Therapies for Sickle Cell Disease




Abstract
1. Introduction
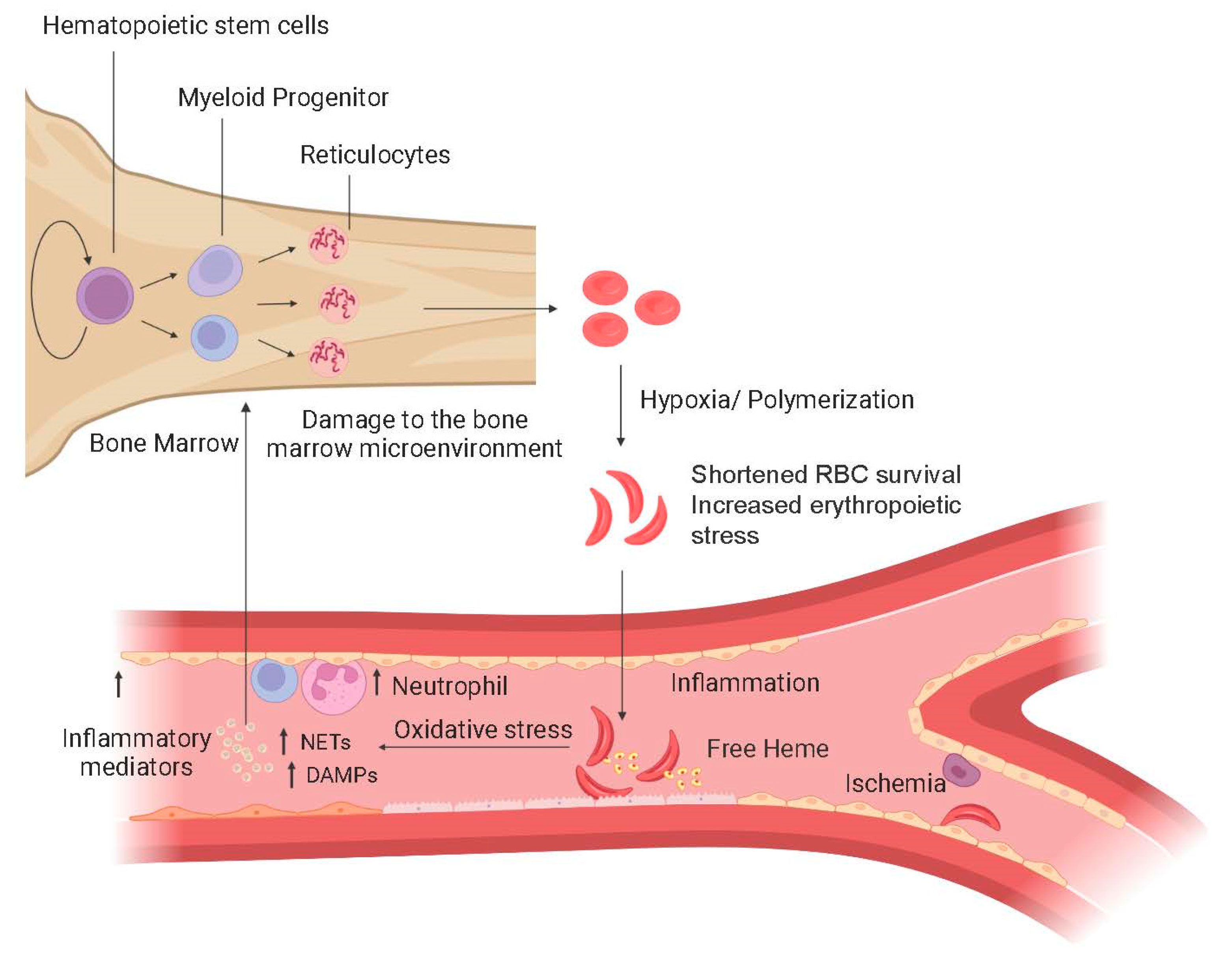
1.1. Pathophysiology of SCD
1.2. Morbidity and Mortality in SCD
1.3. Palliative Therapies in SCD
1.4. Curative Therapies for SCD
2. Clonal Hematopoiesis
2.1. Definition, Prevalence, and Clinical Consequences of CH
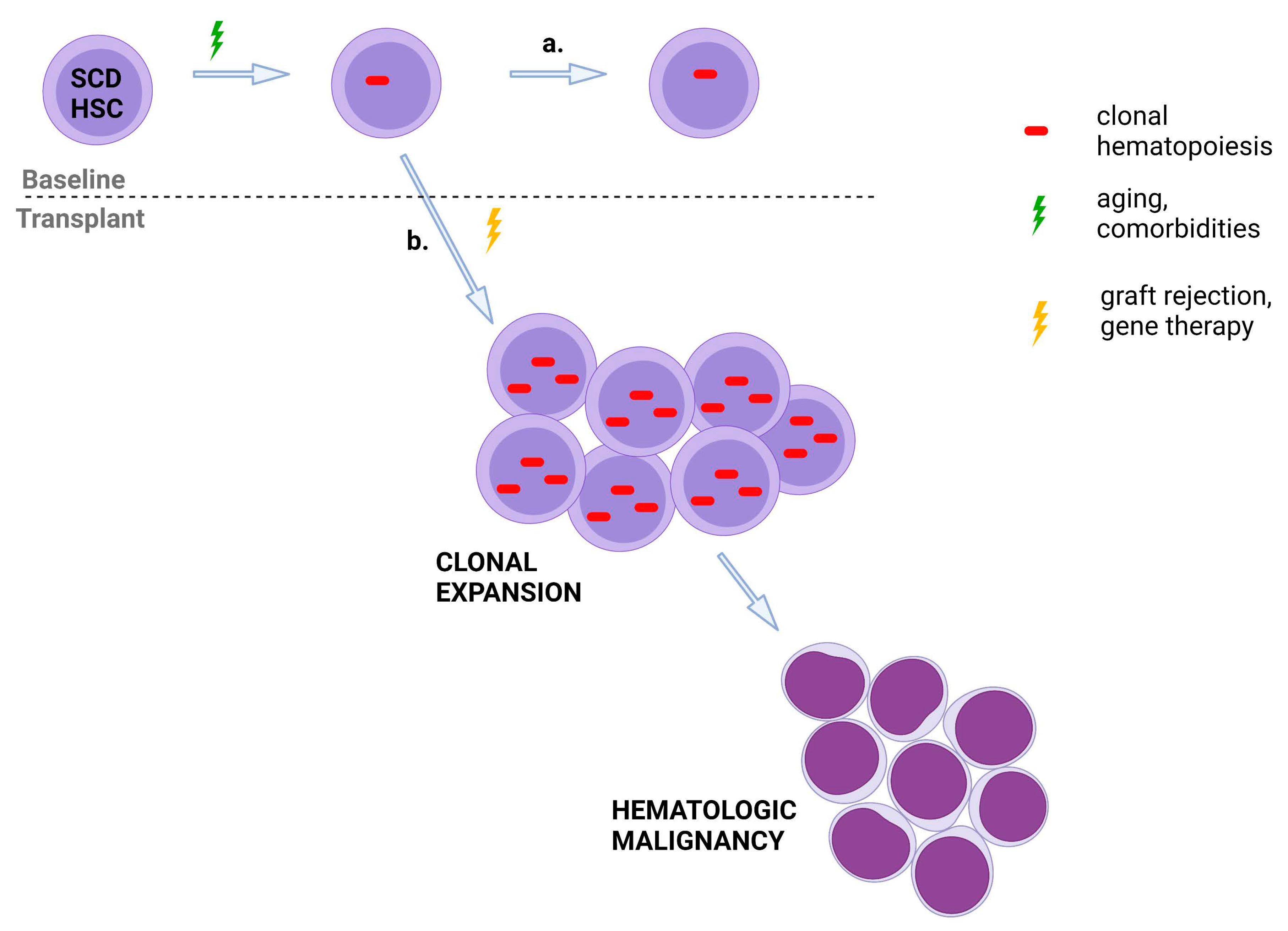
2.2. Risk Factors for Clonal Hematopoiesis
2.3. Clonal Hematopoiesis and Sickle Cell Disease
2.4. Clonal Hematopoiesis and Curative Therapies for Sickle Cell Disease
3. Leukemia and Sickle Cell Disease
3.1. Risk of Leukemia in Sickle Cell Disease
3.2. Risk of Leukemia after Curative Therapies for SCD
4. Why Is the Risk of Leukemia Increased in Patients with SCD after Curative Therapies?
5. How to Counsel Patients about Baseline Risk of Leukemia after Curative Therapies for Sickle Cell Disease—Our Opinion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aygun, B.; Odame, I. A global perspective on sickle cell disease. Pediatr. Blood Cancer 2012, 59, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Pincez, T.; Lee, S.S.K.; Ilboudo, Y.; Preuss, M.; Pham Hung d’Alexandry d’Orengiani, A.L.; Bartolucci, P.; Galactéros, F.; Joly, P.; Bauer, D.E.; Loos, R.J. Clonal hematopoiesis in sickle cell disease. Blood 2021, 138, 2148–2152. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, J.Y.; Xu, X.; Maric, I.; Dillon, L.; Li, Y.; Hsieh, M.M.; Hourigan, C.S.; Fitzhugh, C.D. Baseline TP53 mutations in adults with SCD developing myeloid malignancy following hematopoietic cell transplantation. Blood 2020, 135, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Alexy, T.; Sangkatumvong, S.; Connes, P.; Pais, E.; Tripette, J.; Barthelemy, J.C.; Fisher, T.C.; Meiselman, H.J.; Khoo, M.C.; Coates, T.D. Sickle cell disease: Selected aspects of pathophysiology. Clin. Hemorheol. Microcirc. 2010, 44, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Barabino, G.A.; Platt, M.O.; Kaul, D.K. Sickle cell biomechanics. Annu. Rev. Biomed. Eng. 2010, 12, 345–367. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.; Bonifacino, A.; Dominical, V.M.; Luo, M.; Haro-Mora, J.J.; Demirci, S.; Uchida, N.; Pierciey, F.J., Jr.; Tisdale, J.F. Bone marrow characterization in sickle cell disease: Inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br. J. Haematol. 2019, 186, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Hibino, S.; Kawazoe, T.; Kasahara, H.; Itoh, S.; Ishimoto, T.; Sakata-Yanagimoto, M.; Taniguchi, K. Inflammation-Induced Tumorigenesis and Metastasis. Int. J. Mol. Sci. 2021, 22, 5421. [Google Scholar] [CrossRef]
- Manwani, D.; Frenette, P.S. Vaso-occlusion in sickle cell disease: Pathophysiology and novel targeted therapies. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 362–369. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Conran, N.; Belcher, J.D. Inflammation in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 263–299. [Google Scholar] [CrossRef]
- El Nemer, W.; Godard, A.; El Hoss, S. Ineffective erythropoiesis in sickle cell disease: New insights and future implications. Curr. Opin. Hematol. 2021, 28, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sorensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Grosse, S.D.; Odame, I.; Atrash, H.K.; Amendah, D.D.; Piel, F.B.; Williams, T.N. Sickle cell disease in Africa: A neglected cause of early childhood mortality. Am. J. Prev. Med. 2011, 41 (Suppl. S4), S398–S405. [Google Scholar] [CrossRef] [PubMed]
- McClish, D.K.; Penberthy, L.T.; Bovbjerg, V.E.; Roberts, J.D.; Aisiku, I.P.; Levenson, J.L.; Roseff, S.D.; Smith, W.R. Health related quality of life in sickle cell patients: The PiSCES project. Health Qual. Life Outcomes 2005, 3, 50. [Google Scholar] [CrossRef]
- Verduzco, L.A.; Nathan, D.G. Sickle cell disease and stroke. Blood 2009, 114, 5117–5125. [Google Scholar] [CrossRef]
- Klings, E.S.; Steinberg, M.H. Acute chest syndrome of sickle cell disease: Genetics, risk factors, prognosis, and management. Expert Rev. Hematol. 2022, 15, 117–125. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Barst, R.J.; Gibbs, J.S.; Hildesheim, M.; Sachdev, V.; Nouraie, M.; Hassell, K.L.; Little, J.A.; Schraufnagel, D.E.; Krishnamurti, L.; et al. Risk factors for death in 632 patients with sickle cell disease in the United States and United kingdom. PLoS ONE 2014, 9, e99489. [Google Scholar] [CrossRef] [PubMed]
- Kassim, A.A.; Payne, A.B.; Rodeghier, M.; Macklin, E.A.; Strunk, R.C.; DeBaun, M.R. Low forced expiratory volume is associated with earlier death in sickle cell anemia. Blood 2015, 126, 1544–1550. [Google Scholar] [CrossRef]
- Xu, J.Z.; Garrett, M.E.; Soldano, K.L.; Chen, S.T.; Clish, C.B.; Ashley-Koch, A.E.; Telen, M.J. Clinical and metabolomic risk factors associated with rapid renal function decline in sickle cell disease. Am. J. Hematol. 2018, 93, 1451–1460. [Google Scholar] [CrossRef]
- Telen, M.J.; Afenyi-Annan, A.; Garrett, M.E.; Combs, M.R.; Orringer, E.P.; Ashley-Koch, A.E. Alloimmunization in sickle cell disease: Changing antibody specificities and association with chronic pain and decreased survival. Transfusion 2015, 55, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Lanzkron, S.; Carroll, C.P.; Haywood, C., Jr. Mortality rates and age at death from sickle cell disease: U.S., 1979–2005. Public Health Rep. 2013, 128, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.T.; Rogers, Z.R.; McCavit, T.L.; Buchanan, G.R. Improved survival of children and adolescents with sickle cell disease. Blood 2010, 115, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; McCarthy, W.F.; Castro, O.; Ballas, S.K.; Armstrong, F.D.; Smith, W.; Ataga, K.; Swerdlow, P.; Kutlar, A.; DeCastro, L.; et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am. J. Hematol. 2010, 85, 403–408. [Google Scholar] [CrossRef]
- Ware, R.E.; Dertinger, S.D. Absence of hydroxyurea-induced mutational effects supports higher utilisation for the treatment of sickle cell anaemia. Br. J. Haematol. 2021, 194, 252–266. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef]
- Yawn, B.P.; Buchanan, G.R.; Afenyi-Annan, A.N.; Ballas, S.K.; Hassell, K.L.; James, A.H.; Jordan, L.; Lanzkron, S.M.; Lottenberg, R.; Savage, W.J.; et al. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. Practice Guideline. Research Support, N.I.H., Extramural. Review. JAMA 2014, 312, 1033–1048. [Google Scholar] [CrossRef]
- Walters, M.C.; Patience, M.; Leisenring, W.; Rogers, Z.R.; Aquino, V.M.; Buchanan, G.R.; Roberts, I.A.G.; Yeager, A.M.; Hsu, L.; Adamkiewicz, T.; et al. Bone Marrow Transplantation for Sickle Cell Disease. N. Engl. J. Med. 1996, 335, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Pinto Simões, B.; Ferster, A.; Dupont, S.; De La Fuente, J.; et al. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Fitzhugh, C.D.; Weitzel, R.; Link, M.E.; Coles, W.A.; Zhao, X.; Rodgers, G.P.; Powell, J.D.; Tisdale, J.F. Nonmyeloablative hla-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 2014, 312, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.D.; Cordes, S.; Taylor, T.; Coles, W.; Roskom, K.; Link, M.; Hsieh, M.M.; Tisdale, J.F. At Least 20% Donor Myeloid Chimerism is Necessary to Reverse the Sickle Phenotype after Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2017, 130, 1946–1948. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Hsieh, M.; Eapen, M.; Fitzhugh, C.; Carreras, J.; Keesler, D.; Guilcher, G.; Kamani, N.; Walters, M.C.; Boelens, J.J.; et al. Relationship between Mixed Donor-Recipient Chimerism and Disease Recurrence Following Hematopoietic Stem Cell Transplantation for Sickle Cell Disease. Biol. Blood Marrow Transpl. 2017, 23, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, M.; Damlaj, M.; Jeffries, N.; Alahmari, B.; Singh, A.; Rondelli, D.; Tisdale, J.F.; Saraf, S.L.; Hsieh, M.M. Non-myeloablative human leukocyte antigen-matched related donor transplantation in sickle cell disease: Outcomes from three independent centres. Br. J. Haematol. 2021, 192, 761–768. [Google Scholar] [CrossRef]
- Walters, M.C.; Patience, M.; Leisenring, W.; Eckman, J.R.; Buchanan, G.R.; Rogers, Z.R.; Olivieri, N.E.; Vichinsky, E.; Davies, S.C.; Mentzer, W.C.; et al. Barriers to bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transpl. 1996, 2, 100–104. [Google Scholar]
- Bolanos-Meade, J.; Fuchs, E.J.; Luznik, L.; Lanzkron, S.M.; Gamper, C.J.; Jones, R.J.; Brodsky, R.A. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Clinical Trial, Phase I. Clinical Trial, Phase II. Research Support, N.I.H., Extramural. Blood 2012, 120, 4285–4291. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Hsieh, M.M.; Taylor, T.; Coles, W.; Roskom, K.; Wilson, D.; Wright, E.; Jeffries, N.; Gamper, C.J.; Powell, J.; et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical peripheral blood stem cell transplantation. Blood Adv. 2017, 1, 652–661. [Google Scholar] [CrossRef]
- Dallas, M.H.; Triplett, B.; Shook, D.R.; Hartford, C.; Srinivasan, A.; Laver, J.; Ware, R.; Leung, W. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Research Support, Non-U.S. Gov’t. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2013, 19, 820–830. [Google Scholar] [CrossRef]
- Bolanos-Meade, J.; Cooke, K.R.; Gamper, C.J.; Ali, S.A.; Ambinder, R.F.; Borrello, I.M.; Fuchs, E.J.; Gladstone, D.E.; Gocke, C.B.; Huff, C.A.; et al. Effect of increased dose of total body irradiation on graft failure associated with HLA-haploidentical transplantation in patients with severe haemoglobinopathies: A prospective clinical trial. Lancet Haematol. 2019, 6, e183–e193. [Google Scholar] [CrossRef]
- de la Fuente, J.; Dhedin, N.; Koyama, T.; Bernaudin, F.; Kuentz, M.; Karnik, L.; Socie, G.; Culos, K.A.; Brodsky, R.A.; DeBaun, M.R.; et al. Haploidentical Bone Marrow Transplantation with Post-Transplantation Cyclophosphamide Plus Thiotepa Improves Donor Engraftment in Patients with Sickle Cell Anemia: Results of an International Learning Collaborative. Biol. Blood Marrow Transpl. 2019, 25, 1197–1209. [Google Scholar] [CrossRef]
- Frangoul, H.; Evans, M.; Isbell, J.; Bruce, K.; Domm, J. Haploidentical hematopoietic stem cell transplant for patients with sickle cell disease using thiotepa, fludarabine, thymoglobulin, low dose cyclophosphamide, 200 cGy tbi and post transplant cyclophosphamide. Bone Marrow Transpl. 2018, 53, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.L.; Oh, A.L.; Patel, P.R.; Sweiss, K.; Koshy, M.; Campbell-Lee, S.; Gowhari, M.; Jain, S.; Peace, D.; Quigley, J.G.; et al. Haploidentical Peripheral Blood Stem Cell Transplantation Demonstrates Stable Engraftment in Adults with Sickle Cell Disease. Biol. Blood Marrow Transpl. 2018, 24, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, A.B.; Cheng, J.C.; Karras, N.A.; Sun, W.; Wang, L.D.; Bell, A.D.; Gutierrez, L.; Rosenthal, J. HLA Haploidentical Stem Cell Transplant with Pretransplant Immunosuppression for Patients with Sickle Cell Disease. Biol. Blood Marrow Transpl. 2018, 24, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Kanter, J.; Walters, M.C.; Krishnamurti, L.; Mapara, M.Y.; Kwiatkowski, J.L.; Rifkin-Zenenberg, S.; Aygun, B.; Kasow, K.A.; Pierciey, F.J., Jr.; Bonner, M.; et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N. Engl. J. Med. 2021, 312, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Gibson, C.J.; Kim, H.T.; Zhao, L.; Murdock, H.M.; Hambley, B.; Ogata, A.; Madero-Marroquin, R.; Wang, S.; Green, L.; Fleharty, M.; et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J. Clin. Oncol. 2022, 40, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef] [PubMed]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; Mar, B.G.; Shi, J.; Jaiswal, S.; Bosworth, A.; Francisco, L.; He, J.; Bansal, A.; et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol. 2017, 35, 1598–1605. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Jasek, M.; Gondek, L.P.; Bejanyan, N.; Tiu, R.; Huh, J.; Theil, K.S.; O’Keefe, C.; McDevitt, M.A.; Maciejewski, J.P. TP53 mutations in myeloid malignancies are either homozygous or hemizygous due to copy number-neutral loss of heterozygosity or deletion of 17p. Leukemia 2010, 24, 216–219. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell. 2018, 23, 833–849.e5. [Google Scholar] [CrossRef] [PubMed]
- Hormaechea-Agulla, D.; Matatall, K.A.; Le, D.T.; Kain, B.; Long, X.; Kus, P.; Jaksik, R.; Challen, G.A.; Kimmel, M.; King, K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell. 2021, 28, 1428–1442.e6. [Google Scholar] [CrossRef] [PubMed]
- Heyde, A.; Rohde, D.; McAlpine, C.S.; Zhang, S.; Hoyer, F.F.; Gerold, J.M.; Cheek, D.; Iwamoto, Y.; Schloss, M.J.; Vandoorne, K.; et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 2021, 184, 1348–1361.e22. [Google Scholar] [CrossRef] [PubMed]
- Liggett, L.A.; Cato, L.D.; Weinstock, J.S.; Zhang, Y.; Nouraie, S.M.; Gladwin, M.T.; Garrett, M.E.; Ashley-Koch, A.; Telen, M.; Custer, B.; et al. Clonal hematopoiesis in sickle cell disease. J. Clin. Investig. 2022, 132, e156060. [Google Scholar] [CrossRef]
- Li, Y.; Maule, J.; Neff, J.L.; McCall, C.M.; Rapisardo, S.; Lagoo, A.S.; Yang, L.H.; Crawford, R.D.; Zhao, Y.; Wang, E. Myeloid neoplasms in the setting of sickle cell disease: An intrinsic association with the underlying condition rather than a coincidence; report of 4 cases and review of the literature. Mod. Pathol. 2019, 32, 1712–1726. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Bonner, M.; Pierciey, F.J.; Uchida, N.; Rottman, J.; Demopoulos, L.; Schmidt, M.; Kanter, J.; Walters, M.C.; Thompson, A.A.; et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020, 4, 2058–2063. [Google Scholar] [CrossRef]
- Goyal, S.; Tisdale, J.; Schmidt, M.; Kanter, J.; Jaroscak, J.; Whitney, D.; Bitter, H.; Gregory, P.D.; Parsons, G.; Foos, M.; et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N. Engl. J. Med. 2022, 386, 138–147. [Google Scholar] [CrossRef]
- Brunson, A.; Keegan, T.H.M.; Bang, H.; Mahajan, A.; Paulukonis, S.; Wun, T. Increased risk of leukemia among sickle cell disease patients in California. Blood 2017, 130, 1597–1599. [Google Scholar] [CrossRef]
- Seminog, O.O.; Ogunlaja, O.I.; Yeates, D.; Goldacre, M.J. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J. R. Soc. Med. 2016, 109, 303–309. [Google Scholar] [CrossRef]
- Jones, R.J.; DeBaun, M.R. Leukemia after gene therapy for sickle cell disease: Insertional mutagenesis, busulfan, both, or neither. Blood 2021, 138, 942–947. [Google Scholar] [CrossRef]
- Vermylen, C.; Cornu, G.; Ferster, A.; Brichard, B.; Ninane, J.; Ferrant, A.; Zenebergh, A.; Maes, P.; Dhooge, C.; Benoit, Y.; et al. Haematopoietic stem cell transplantation for sickle cell anaemia: The first 50 patients transplanted in Belgium. Bone Marrow Transpl. 1998, 22, 1–6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bernaudin, F.; Dalle, J.H.; Bories, D.; de Latour, R.P.; Robin, M.; Bertrand, Y.; Pondarre, C.; Vannier, J.P.; Neven, B.; Kuentz, M.; et al. Long-term event-free survival, chimerism and fertility outcomes in 234 patients with sickle-cell anemia younger than 30 years after myeloablative conditioning and matched-sibling transplantation in France. Haematologica 2020, 105, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.; Brazauskas, R.; Walters, M.C.; Bernaudin, F.; Bo-Subait, K.; Fitzhugh, C.D.; Hankins, J.S.; Kanter, J.; Meerpohl, J.J.; Bolanos-Meade, J.; et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: A retrospective multicentre, cohort study. Lancet Haematol. 2019, 6, e585–e596. [Google Scholar] [CrossRef]
- Janakiram, M.; Verma, A.; Wang, Y.; Budhathoki, A.; Suarez Londono, J.; Murakhovskaya, I.; Braunschweig, I.; Minniti, C.P. Accelerated leukemic transformation after haplo-identical transplantation for hydroxyurea-treated sickle cell disease. Leuk. Lymphoma 2018, 59, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Gondek, L.P.; Zheng, G.; Ghiaur, G.; DeZern, A.E.; Matsui, W.; Yegnasubramanian, S.; Lin, M.T.; Levis, M.; Eshleman, J.R.; Varadhan, R.; et al. Donor cell leukemia arising from clonal hematopoiesis after bone marrow transplantation. Leukemia 2016, 30, 1916–1920. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Sample Size | HCT Type | Conditioning Agents | Number (%) with MDS or Leukemia | Time to MDS/Leukemia Diagnosis |
|---|---|---|---|---|---|
| Ghannam et al. [3] | 76 | HLA-matched sibling Haploidentical | Alemtuzumab 300–400 cGy TBI PT-Cy 0–100 mg/kg | 3 (3.9%) | 2–5 years |
| Jones and DeBaun [70] | 47 | Gene therapy | Busulfan | 2 (4.3%) | 3–5.5 years |
| Vermylen et al. [71] | 50 | HLA-matched family member | Busulfan Cy ±r-ATG ±TLI | 1 (2%) | 35 months |
| Bernaudin et al. [72] | 234 | HLA-matched sibling | Busulfan Cy r-ATG | 0 (0%) | N/A |
| Eapen et al. [73] | 910 | Mostly HLA-matched sibling | Mostly Busulfan Cy r-ATG | 5 (0.55%) | 9 to 44 months |
| Seminog et al. [69] | 7512 | N/A | N/A | <35 (<0.47%) | Observed over 12 years |
| Brunson et al. [68] | 6423 | N/A | N/A | 12 (0.19%) | Observed over 23 years |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gondek, L.P.; Sheehan, V.A.; Fitzhugh, C.D. Clonal Hematopoiesis and the Risk of Hematologic Malignancies after Curative Therapies for Sickle Cell Disease. J. Clin. Med. 2022, 11, 3160. https://doi.org/10.3390/jcm11113160
Gondek LP, Sheehan VA, Fitzhugh CD. Clonal Hematopoiesis and the Risk of Hematologic Malignancies after Curative Therapies for Sickle Cell Disease. Journal of Clinical Medicine. 2022; 11(11):3160. https://doi.org/10.3390/jcm11113160
Chicago/Turabian StyleGondek, Lukasz P., Vivien A. Sheehan, and Courtney D. Fitzhugh. 2022. "Clonal Hematopoiesis and the Risk of Hematologic Malignancies after Curative Therapies for Sickle Cell Disease" Journal of Clinical Medicine 11, no. 11: 3160. https://doi.org/10.3390/jcm11113160
APA StyleGondek, L. P., Sheehan, V. A., & Fitzhugh, C. D. (2022). Clonal Hematopoiesis and the Risk of Hematologic Malignancies after Curative Therapies for Sickle Cell Disease. Journal of Clinical Medicine, 11(11), 3160. https://doi.org/10.3390/jcm11113160