
New Treatment Strategies for IgA Nephropathy: Targeting Plasma Cells as the Main Source of Pathogenic Antibodies



Abstract
:1. Introduction
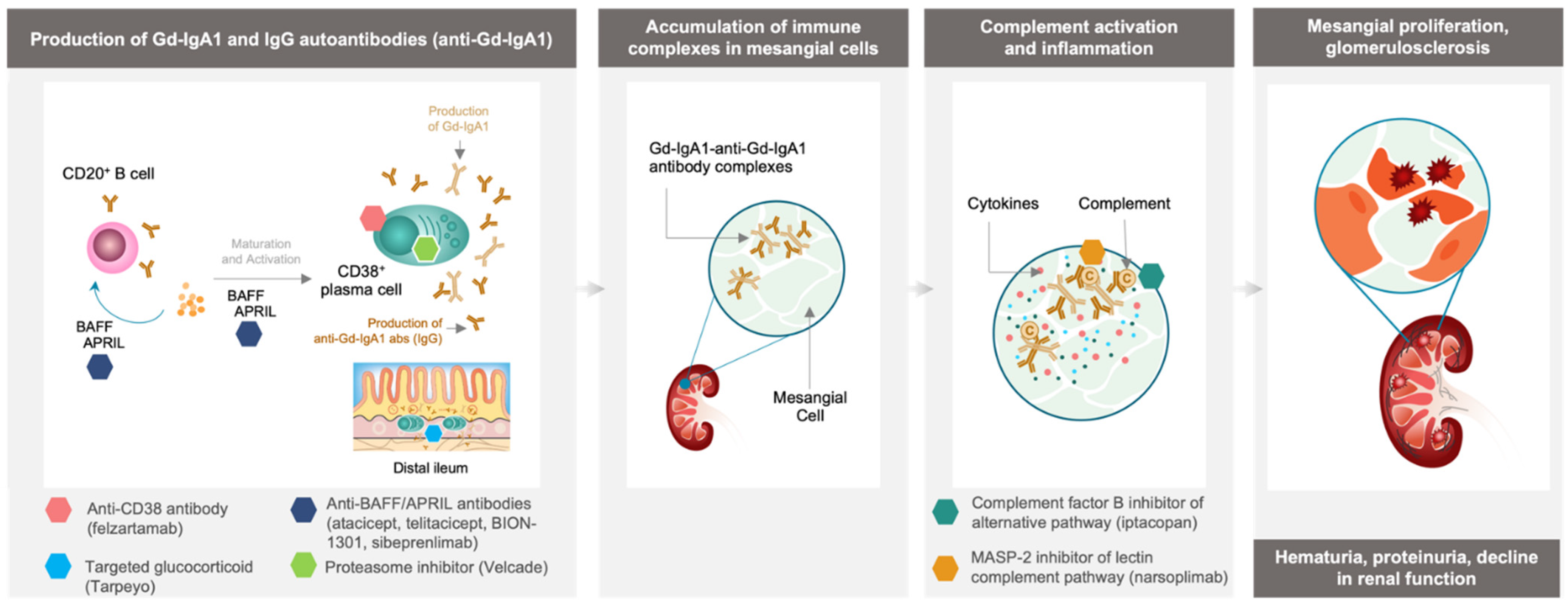
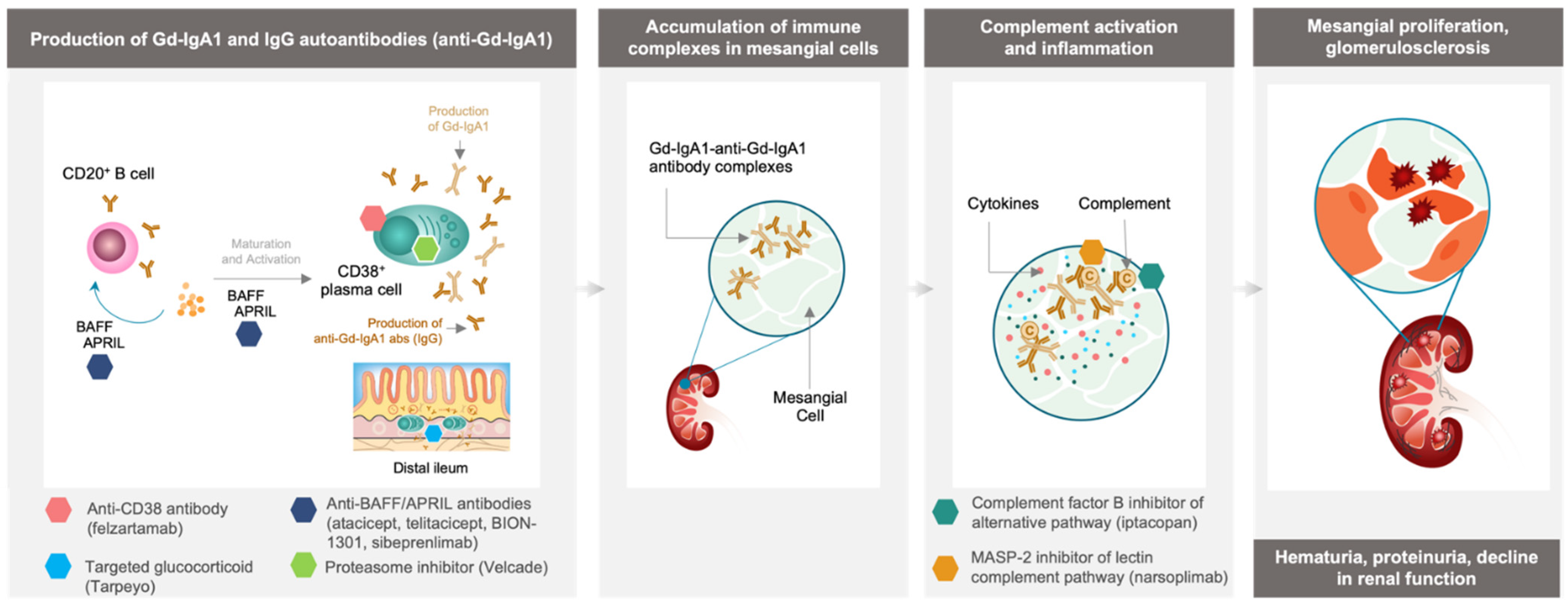
2. New Strategies for the Management of IgAN
3. Inhibition of Immune Complex-Activated Complement Activity
4. Depletion of Gd-IgA1-Producing Immune Cells
4.1. Targeting Cytokines Responsible for B Cell and Plasma Cell Activation and Survival
4.2. Tarpeyo, a Targeted Approach for Immune Cell Depletion in the Small Intestine
4.3. Velcade, Plasma Cell Depletion via Proteasome Inhibition
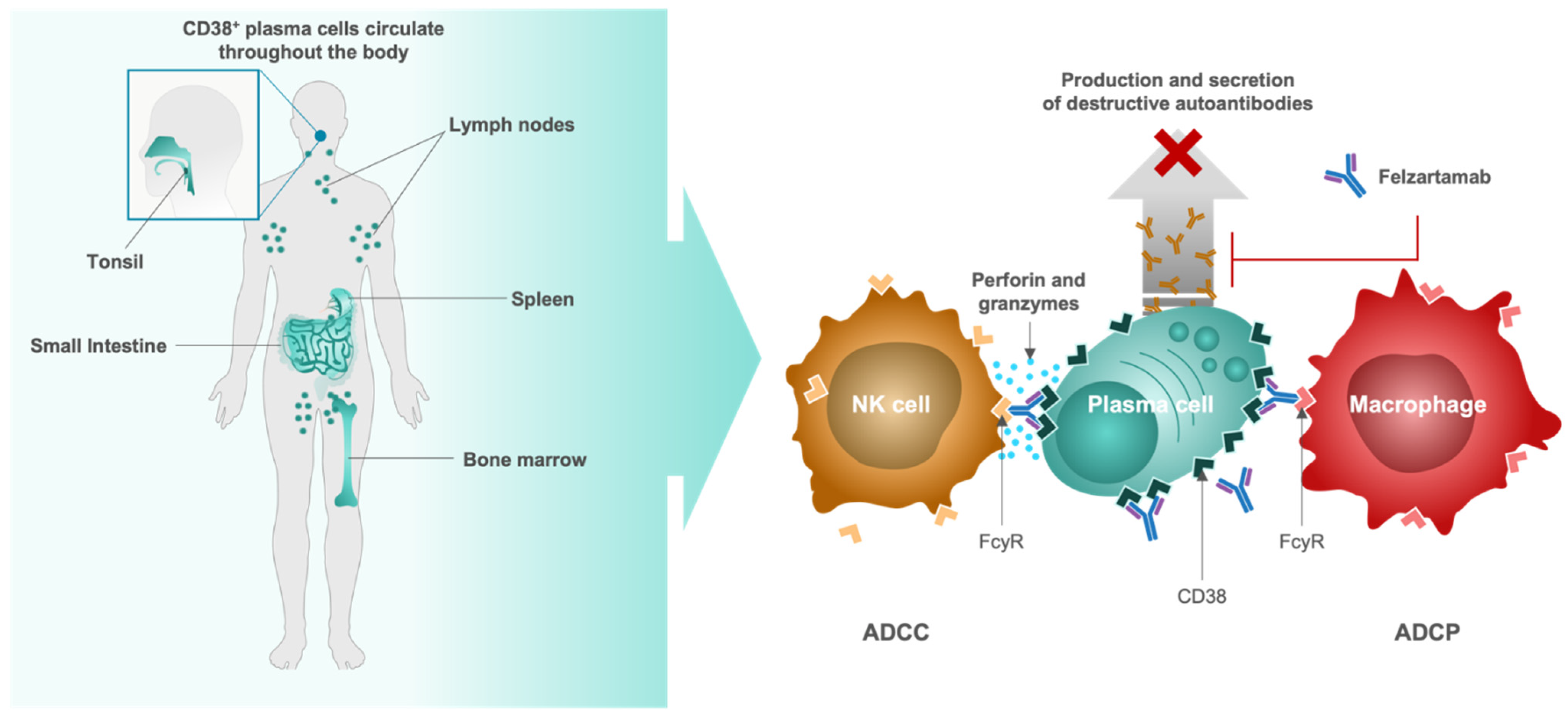
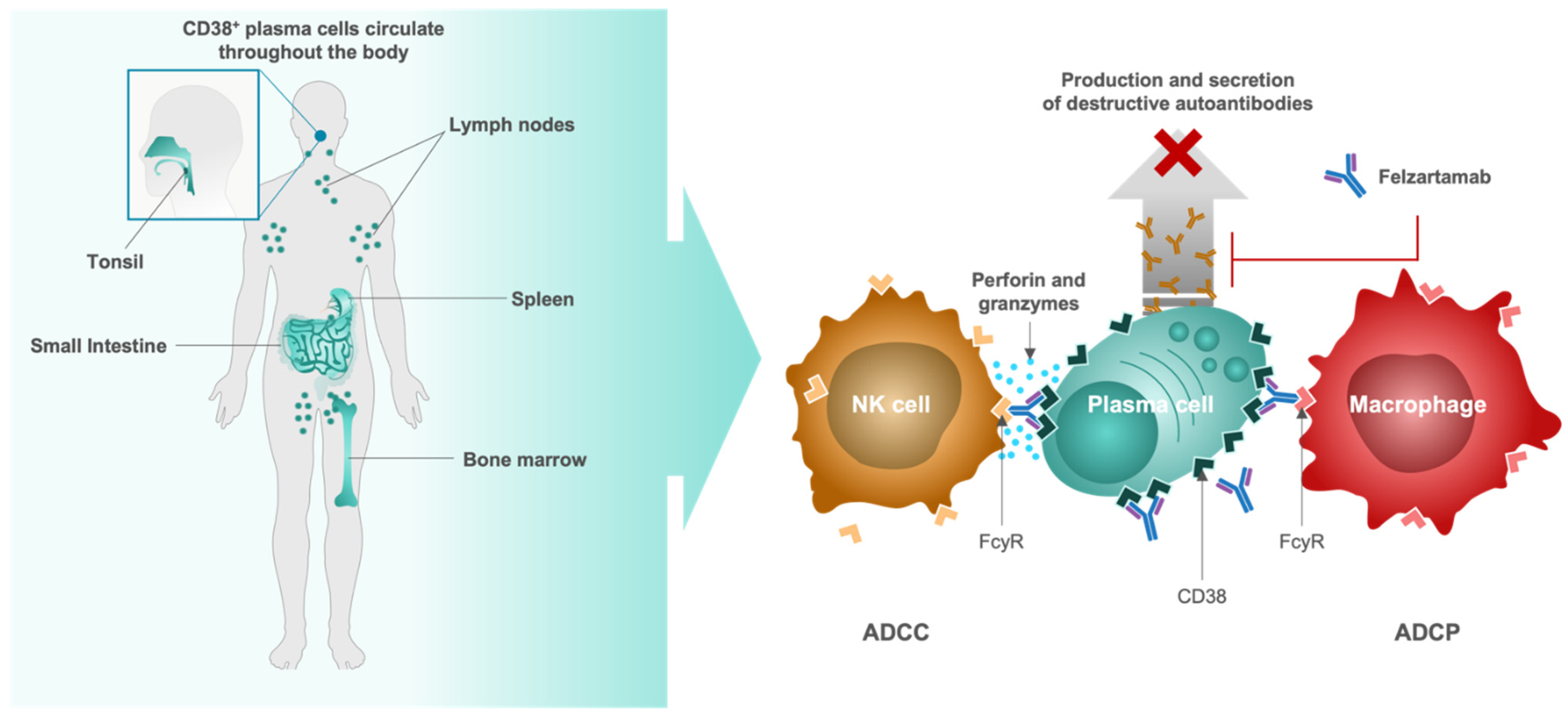
4.4. Felzartamab, Targeted CD38+ Plasma Cell Depletion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rodrigues, J.C.; Haas, M.; Reich, H.N. IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N.; Tang, S.C.W.; Schena, F.P.; Novak, J.; Tomino, Y.; Fogo, A.B.; Glassock, R.J. IgA Nephropathy. Nat. Rev. Dis. Prim. 2016, 2, 16001. [Google Scholar] [CrossRef] [PubMed]
- McGrogan, A.; Franssen, C.F.M.; de Vries, C.S. The Incidence of Primary Glomerulonephritis Worldwide: A Systematic Review of the Literature. Nephrol. Dial. Transplant. 2011, 26, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schena, F.P.; Nistor, I. Epidemiology of IgA Nephropathy: A Global Perspective. Semin. Nephrol. 2018, 38, 435–442. [Google Scholar] [CrossRef]
- Hakim, R.M.; Saha, S. Dialysis Frequency versus Dialysis Time, That Is the Question. Kidney Int. 2014, 85, 1024–1029. [Google Scholar] [CrossRef] [Green Version]
- Tattersall, J.; Martin-Malo, A.; Pedrini, L.; Basci, A.; Canaud, B.; Fouque, D.; Haage, P.; Konner, K.; Kooman, J.; Pizzarelli, F.; et al. EBPG Guideline on Dialysis Strategies. Nephrol. Dial. Transplant. 2007, 22 (Suppl. S2), ii5–ii21. [Google Scholar] [CrossRef] [Green Version]
- Dąbrowska-Bender, M.; Dykowska, G.; Żuk, W.; Milewska, M.; Staniszewska, A. The Impact on Quality of Life of Dialysis Patients with Renal Insufficiency. Patient Prefer. Adherence 2018, 12, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Clinical Epidemiology of Cardiovascular Disease in Chronic Renal Disease. Am. J. Kidney Dis. 1998, 32, S112–S119. [Google Scholar] [CrossRef]
- Komatsu, H.; Kikuchi, M.; Nakagawa, H.; Fukuda, A.; Iwakiri, T.; Toida, T.; Sato, Y.; Kitamura, K.; Fujimoto, S. Long-Term Survival of Patients with IgA Nephropathy after Dialysis Therapy. Kidney Blood Press. Res. 2013, 37, 649–656. [Google Scholar] [CrossRef]
- Jarrick, S.; Lundberg, S.; Welander, A.; Carrero, J.-J.; Höijer, J.; Bottai, M.; Ludvigsson, J.F. Mortality in IgA Nephropathy: A Nationwide Population-Based Cohort Study. J. Am. Soc. Nephrol. 2019, 30, 866–876. [Google Scholar] [CrossRef]
- Wyld, M.L.; Chadban, S.J. Recurrent IgA Nephropathy After Kidney Transplantation. Transplantation 2016, 100, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Moroni, G.; Gallelli, B.; Quaglini, S.; Leoni, A.; Banfi, G.; Passerini, P.; Montagnino, G.; Messa, P. Long-Term Outcome of Renal Transplantation in Patients with Idiopathic Membranous Glomerulonephritis (MN). Nephrol. Dial. Transplant. 2010, 25, 3408–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maixnerova, D.; Hruba, P.; Neprasova, M.; Bednarova, K.; Slatinska, J.; Suchanek, M.; Kollar, M.; Novak, J.; Tesar, V.; Viklicky, O. Outcome of 313 Czech Patients With IgA Nephropathy After Renal Transplantation. Front. Immunol. 2021, 12, 726215. [Google Scholar] [CrossRef] [PubMed]
- Knoppova, B.; Reily, C.; Maillard, N.; Rizk, D.V.; Moldoveanu, Z.; Mestecky, J.; Raska, M.; Renfrow, M.B.; Julian, B.A.; Novak, J. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front. Immunol. 2016, 7, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliley, J.L.; Tipton, C.M.; Liesveld, J.; Rosenberg, A.F.; Darce, J.; Gregoretti, I.V.; Popova, L.; Kaminiski, D.; Fucile, C.F.; Albizua, I.; et al. Long-Lived Plasma Cells Are Contained within the CD19(-)CD38(Hi)CD138(+) Subset in Human Bone Marrow. Immunity 2015, 43, 132–145. [Google Scholar] [CrossRef] [Green Version]
- Khodadadi, L.; Cheng, Q.; Radbruch, A.; Hiepe, F. The Maintenance of Memory Plasma Cells. Front. Immunol. 2019, 10, 721. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H. Biomarkers for IgA Nephropathy on the Basis of Multi-Hit Pathogenesis. Clin. Exp. Nephrol. 2019, 23, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.N. Pathogenesis of IgA Nephropathy. Nat. Rev. Nephrol. 2012, 8, 275–283. [Google Scholar] [CrossRef]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The Pathophysiology of IgA Nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Rizk, D.V.; Maillard, N.; Julian, B.A.; Knoppova, B.; Green, T.J.; Novak, J.; Wyatt, R.J. The Emerging Role of Complement Proteins as a Target for Therapy of IgA Nephropathy. Front. Immunol. 2019, 10, 504. [Google Scholar] [CrossRef]
- Maixnerova, D.; Ling, C.; Hall, S.; Reily, C.; Brown, R.; Neprasova, M.; Suchanek, M.; Honsova, E.; Zima, T.; Novak, J.; et al. Galactose-Deficient IgA1 and the Corresponding IgG Autoantibodies Predict IgA Nephropathy Progression. PLoS ONE 2019, 14, e0212254. [Google Scholar] [CrossRef] [Green Version]
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. Executive Summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, 753–779. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, F.-L.; Zhou, J.; Zhao, Y.-J. IgA Nephropathy Factors That Predict and Accelerate Progression to End-Stage Renal Disease. Cell Biochem. Biophys. 2014, 68, 443–447. [Google Scholar] [CrossRef]
- Maixnerova, D.; Tesar, V. Emerging Modes of Treatment of IgA Nephropathy. Int. J. Mol. Sci. 2020, 21, 9064. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Mani, K.; Swamy, A.; Barwad, A.; Singh, G.; Bhowmik, D.; Agarwal, S.K. Supportive Management of IgA Nephropathy With Renin-Angiotensin Blockade, the AIIMS Primary IgA Nephropathy Cohort (APPROACH) Study. Kidney Int. Rep. 2021, 6, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Travere Therapeutics, Inc. A Randomized, Multicenter, Double-Blind, Parallel-Group, Active-Control Study of the Efficacy and Safety of Sparsentan for the Treatment of Immunoglobulin A Nephropathy; Travere Therapeutics, Inc.: San Diego, CA, USA, 2021. Available online: https://www.clinicaltrials.gov (accessed on 28 March 2022).
- Komers, R.; Plotkin, H. Dual Inhibition of Renin-Angiotensin-Aldosterone System and Endothelin-1 in Treatment of Chronic Kidney Disease. Am. J. Physio.l Regul. Integr. Comp. Physiol. 2016, 310, R877–R884. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.C.; Toto, R.D.; Stefánsson, B.V.; Jongs, N.; Chertow, G.M.; Greene, T.; Hou, F.F.; McMurray, J.J.V.; Pecoits-Filho, R.; Correa-Rotter, R.; et al. A Pre-Specified Analysis of the DAPA-CKD Trial Demonstrates the Effects of Dapagliflozin on Major Adverse Kidney Events in Patients with IgA Nephropathy. Kidney Int. 2021, 100, 215–224. [Google Scholar] [CrossRef]
- Morphosys Farxiga (Dapagliflozin). [Package Insert]. 2021. Available online: https://den8dhaj6zs0e.cloudfront.net/50fd68b9-106b-4550-b5d0-12b045f8b184/0be9cb1b-3b33-41c7-bfc2-04c9f718e442/0be9cb1b-3b33-41c7-bfc2-04c9f718e442_viewable_rendition__v.pdf (accessed on 28 March 2022).
- Rauen, T.; Wied, S.; Fitzner, C.; Eitner, F.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Budde, K.; Benck, U.; et al. After Ten Years of Follow-up, No Difference between Supportive Care plus Immunosuppression and Supportive Care Alone in IgA Nephropathy. Kidney Int. 2020, 98, 1044–1052. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, H.; Wong, M.G.; Jardine, M.J.; Hladunewich, M.; Jha, V.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.; et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2017, 318, 432–442. [Google Scholar] [CrossRef]
- Calliditas Therapeutics Tarpeyo (Budesomide). [Package Insert]. 2021. Available online: https://www.tarpeyo.com/prescribinginformation.pdf (accessed on 28 March 2022).
- FDA. FDA Approves First Drug to Decrease Urine Protein in IgA Nephropathy, a Rare Kidney Disease; FDA: Silver Spring, MD, USA, 2021.
- Schrezenmeier, E.; Jayne, D.; Dörner, T. Targeting B Cells and Plasma Cells in Glomerular Diseases: Translational Perspectives. J. Am. Soc. Nephrol. 2018, 29, 741–758. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Zhang, H. Insights into the Role of Mucosal Immunity in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2018, 13, 1584–1586. [Google Scholar] [CrossRef] [PubMed]
- He, J.-W.; Zhou, X.-J.; Lv, J.-C.; Zhang, H. Perspectives on How Mucosal Immune Responses, Infections and Gut Microbiome Shape IgA Nephropathy and Future Therapies. Theranostics 2020, 10, 11462–11478. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; He, H.; Hu, P.; Xu, X. T Lymphocytes in IgA Nephropathy. Exp. Ther. Med. 2020, 20, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, H.; Ohtake, H.; Ishida, A.; Ohta, N.; Kakehata, S.; Yamakawa, M. IgA Production and Tonsillar Focal Infection in IgA Nephropathy. J. Clin. Exp. Hematop. 2012, 52, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Li, X.-K. The Role of Immune Modulation in Pathogenesis of IgA Nephropathy. Front. Med. 2020, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-Y.; Zhang, L.; Zhao, P.-W.; Ma, L.; Li, C.; Zou, H.-B.; Jiang, Y.-F. Functional Implications of Regulatory B Cells in Human IgA Nephropathy. Scand. J. Immunol. 2014, 79, 51–60. [Google Scholar] [CrossRef]
- Muto, M.; Manfroi, B.; Suzuki, H.; Joh, K.; Nagai, M.; Wakai, S.; Righini, C.; Maiguma, M.; Izui, S.; Tomino, Y.; et al. Toll-Like Receptor 9 Stimulation Induces Aberrant Expression of a Proliferation-Inducing Ligand by Tonsillar Germinal Center B Cells in IgA Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 1227–1238. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Peng, X.; Liu, Y.; Liu, H.; Liu, F.; He, L.; Liu, Y.; Zhang, F.; Guo, C.; Chen, G.; et al. TLR9 and BAFF: Their Expression in Patients with IgA Nephropathy. Mol. Med. Rep. 2014, 10, 1469–1474. [Google Scholar] [CrossRef] [Green Version]
- Selvaskandan, H.; Cheung, C.K.; Muto, M.; Barratt, J. New Strategies and Perspectives on Managing IgA Nephropathy. Clin. Exp. Nephrol. 2019, 23, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Lafayette, R.A.; Canetta, P.A.; Rovin, B.H.; Appel, G.B.; Novak, J.; Nath, K.A.; Sethi, S.; Tumlin, J.A.; Mehta, K.; Hogan, M.; et al. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. J. Am. Soc. Nephrol. 2017, 28, 1306–1313. [Google Scholar] [CrossRef]
- Coppo, R.; Peruzzi, L.; Loiacono, E.; Bergallo, M.; Krutova, A.; Russo, M.L.; Cocchi, E.; Amore, A.; Lundberg, S.; Maixnerova, D.; et al. Defective Gene Expression of the Membrane Complement Inhibitor CD46 in Patients with Progressive Immunoglobulin A Nephropathy. Nephrol. Dial. Transplant. 2019, 34, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Zhai, Y.-L.; Wang, F.-M.; Hou, P.; Lv, J.-C.; Xu, D.-M.; Shi, S.-F.; Liu, L.-J.; Yu, F.; Zhao, M.-H.; et al. Variants in Complement Factor H and Complement Factor H-Related Protein Genes, CFHR3 and CFHR1, Affect Complement Activation in IgA Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1195–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennette, J.C. The Immunohistology of IgA Nephropathy. Am. J. Kidney Dis. 1988, 12, 348–352. [Google Scholar] [CrossRef]
- Barratt, J. Interim Analysis of a Phase 2 Dose Ranging Study to Investigate the Effect and Safety of Iptacopan in Primary IGA Nephropathy. Available online: https://era-edta.conference2web.com/#!resources/interim-analysis-of-a-phase-2-dose-ranging-study-to-investigate-the-efficacy-and-safety-of-iptacopan-in-primary-iga-nephropathy-20ec3f83-fd34-441e-8745-44587bda74da (accessed on 15 March 2022).
- Novartis Announces Iptacopan Met Phase II Study Primary Endpoint in Rare Kidney Disease IgA Nephropathy (IgAN). Available online: https://www.novartis.com/news/media-releases/novartis-announces-iptacopan-met-phase-ii-study-primary-endpoint-rare-kidney-disease-iga-nephropathy-igan (accessed on 15 March 2022).
- Barratt, J.; Rovin, B.; Zhang, H.; Kashihara, N.; Maes, B.; Rizk, D.; Trimarchi, H.; Sprangers, B.; Meier, M.; Kollins, D.; et al. Pos-546 Efficacy and Safety of Iptacopan in Iga Nephropathy: Results of a Randomized Double-Blind Placebo-Controlled Phase 2 Study at 6 Months. Kidney Int. Rep. 2022, 7, S236. [Google Scholar] [CrossRef]
- Lafayette, R.A.; Carroll, K.; Barratt, J. Long-Term Phase 2 Efficacy of the MASP-2 Inhibitor Narsoplimab for Treatment of Severe IgA Nephropathy. In Proceedings of the ASN Kidney Week 2021, San Diego, CA, USA, 4–7 November 2021. [Google Scholar]
- Lafayette, R.A.; Rovin, B.H.; Reich, H.N.; Tumlin, J.A.; Floege, J.; Barratt, J. Safety, Tolerability and Efficacy of Narsoplimab, a Novel MASP-2 Inhibitor for the Treatment of IgA Nephropathy. Kidney Int. Rep. 2020, 5, 2032–2041. [Google Scholar] [CrossRef]
- Wire, B. Omeros Announces Results From Nearly Three-Year Follow-Up of Patients in Phase 2 IgA Nephropathy Trial. Available online: https://www.benzinga.com/node/23920855 (accessed on 15 March 2022).
- Schubart, A.; Anderson, K.; Mainolfi, N.; Sellner, H.; Ehara, T.; Adams, C.M.; Mac Sweeney, A.; Liao, S.-M.; Crowley, M.; Littlewood-Evans, A.; et al. Small-Molecule Factor B Inhibitor for the Treatment of Complement-Mediated Diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 7926–7931. [Google Scholar] [CrossRef] [Green Version]
- Rambaldi, A.; Gritti, G.; Micò, M.C.; Frigeni, M.; Borleri, G.; Salvi, A.; Landi, F.; Pavoni, C.; Sonzogni, A.; Gianatti, A.; et al. Endothelial Injury and Thrombotic Microangiopathy in COVID-19: Treatment with the Lectin-Pathway Inhibitor Narsoplimab. Immunobiology 2020, 225, 152001. [Google Scholar] [CrossRef]
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 597959. [Google Scholar] [CrossRef]
- Samy, E.; Wax, S.; Huard, B.; Hess, H.; Schneider, P. Targeting BAFF and APRIL in Systemic Lupus Erythematosus and Other Antibody-Associated Diseases. Int. Rev. Immunol. 2017, 36, 3–19. [Google Scholar] [CrossRef]
- Zhai, Y.-L.; Zhu, L.; Shi, S.-F.; Liu, L.-J.; Lv, J.-C.; Zhang, H. Increased APRIL Expression Induces IgA1 Aberrant Glycosylation in IgA Nephropathy. Medicine 2016, 95, e3099. [Google Scholar] [CrossRef]
- Struemper, H.; Kurtinecz, M.; Edwards, L.; Freimuth, W.W.; Roth, D.A.; Stohl, W. Reductions in Circulating B Cell Subsets and Immunoglobulin G Levels with Long-Term Belimumab Treatment in Patients with SLE. Lupus Sci. Med. 2022, 9, e000499. [Google Scholar] [CrossRef] [PubMed]
- Couser, W.G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 983–997. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Allergy and Infectious Diseases (NIAID). NCT03949855: Efficacy of Belimumab and Rituximab Compared to Rituximab Alone for the Treatment of Primary Membranous Nephropathy (ITN080AI); National Institute of Allergy and Infectious Diseases (NIAID): Rockville, MD, USA, 2021. Available online: https://www.clinicaltrials.gov (accessed on 28 March 2022).
- Barratt, J.; Hour, B.T.; Schwartz, B.S.; Sorensen, B.; Roy, S.E.; Stromatt, C.L.; MacDonald, M.; Endsley, A.N.; Lo, J.; Glicklich, A.; et al. Pharmacodynamic and Clinical Responses to BION-1301 in Patients with IgA Nephropathy: Initial Results of a Ph1/2 Trial. In Proceedings of the ASN Kidney Week 2021, San Diego, CA, USA, 4–7 November 2021. [Google Scholar]
- Barratt, J.; Tumlin, J.A.; Suzuki, Y.; Kao, A.; Aydemir, A.; Zima, Y.; Appel, G.B. 24-Week Interim Analysis of a Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Atacicept in Patients with IgA Nephropathy and Persistent Proteinuria. In Proceedings of the ASN Kidney Week 2020, Denver, CO, USA, 20–25 October 2020. [Google Scholar]
- Vera Therapeutics, Inc. NCT04716231: A Phase IIb Randomized, Double-Blinded, Placebo-Controlled, Dose-Ranging Study to Evaluate the Efficacy and Safety of Atacicept in Subjects With IgA Nephropathy (IGAN); Vera Therapeutics, Inc.: South San Francisco, CA, USA, 2022. Available online: https://www.clinicaltrials.gov (accessed on 28 March 2022).
- Lv, J.; Liu, L.-J.; Hao, C.-M.; Li, G.; Fu, P.; Xing, G.; Zheng, H.; Chen, N.; Caili, W.; Luo, P.; et al. A Phase 2, Randomized, Double-Blind, Placebo-Controlled Trial of Telitacicept in Patients with IgA Nephropathy and Persistent Proteinuria. In Proceedings of the ASN Kidney Week 2021, San Diego, CA, USA, 4–7 November 2021. [Google Scholar]
- RemeGen Co. Ltd. NCT04905212: A Phase 2, Randomized, Double-Blind, Multicenter Study of Telitacicept for Injection (RC18) in Subjects With IgA Nephropathy; RemeGen Co., Ltd.: Yantai, China, 2022. Available online: https://www.clinicaltrials.gov (accessed on 28 March 2022).
- Mathur, M.; Barratt, J.; Suzuki, Y.; Engler, F.; Pasetti, M.F.; Yarbrough, J.; Sloan, S.; Oldach, D. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of VIS649 (Sibeprenlimab), an APRIL-Neutralizing IgG2 Monoclonal Antibody, in Healthy Volunteers. Kidney Int. Rep. 2022, 7, 993–1003. [Google Scholar] [CrossRef]
- Visterra NCT04287985: Safety and Efficacy Study of VIS649 for IgA Nephropathy—Full Text View—ClinicalTrials.Gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04287985?term=nct04287985&draw=2&rank=1 (accessed on 22 March 2022).
- Barratt, J.; Rovin, B.H.; Cattran, D.; Floege, J.; Lafayette, R.; Tesar, V.; Trimarchi, H.; Zhang, H.; NefIgArd Study Steering Committee. Why Target the Gut to Treat IgA Nephropathy? Kidney Int. Rep. 2020, 5, 1620–1624. [Google Scholar] [CrossRef]
- Macpherson, A.J.; McCoy, K.D.; Johansen, F.-E.; Brandtzaeg, P. The Immune Geography of IgA Induction and Function. Mucosal Immunol. 2008, 1, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Kano, T.; Suzuki, H.; Makita, Y.; Fukao, Y.; Suzuki, Y. Nasal-Associated Lymphoid Tissue Is the Major Induction Site for Nephritogenic IgA in Murine IgA Nephropathy. Kidney Int. 2021, 100, 364–376. [Google Scholar] [CrossRef]
- Nakata, J.; Suzuki, Y.; Suzuki, H.; Sato, D.; Kano, T.; Yanagawa, H.; Matsuzaki, K.; Horikoshi, S.; Novak, J.; Tomino, Y. Changes in Nephritogenic Serum Galactose-Deficient IgA1 in IgA Nephropathy Following Tonsillectomy and Steroid Therapy. PLoS ONE 2014, 9, e89707. [Google Scholar] [CrossRef]
- Lanzillotta, M.; Della-Torre, E.; Milani, R.; Bozzolo, E.; Bozzalla-Cassione, E.; Rovati, L.; Arcidiacono, P.G.; Partelli, S.; Falconi, M.; Ciceri, F.; et al. Increase of Circulating Memory B Cells after Glucocorticoid-Induced Remission Identifies Patients at Risk of IgG4-Related Disease Relapse. Arthritis Res. Ther. 2018, 20, 222. [Google Scholar] [CrossRef] [Green Version]
- Floege, J. Mucosal Corticosteroid Therapy of IgA Nephropathy. Kidney Int. 2017, 92, 278–280. [Google Scholar] [CrossRef]
- Coppo, R.; Mariat, C. Systemic Corticosteroids and Mucosal-Associated Lymphoid Tissue-Targeted Therapy in Immunoglobulin A Nephropathy: Insight from the NEFIGAN Study. Nephrol. Dial. Transplant. 2020, 35, 1291–1294. [Google Scholar] [CrossRef] [Green Version]
- Fellström, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-Release Budesonide versus Placebo in Patients with IgA Nephropathy (NEFIGAN): A Double-Blind, Randomised, Placebo-Controlled Phase 2b Trial. Lancet 2017, 389, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Barratt, J.; Stone, A.; Kristensen, J. POS-830 NEFECON for the Treatment of IgA Nephropathy in Patients at Risk of Progressing to End-Stage Renal Disease: The NEFIgArd Phase 3 Trial Results. Kidney Int. Rep. 2021, 6, S361. [Google Scholar] [CrossRef]
- Calliditas Therapeutics AB. NCT03643965: A Randomized, Double-Blind, Placebo Controlled Study to Evaluate Efficacy and Safety of Nefecon in Patients With Primary IgA (Immunoglobulin A) Nephropathy at Risk of Progressing to End-Stage Renal Disease (NefIgArd); Calliditas Therapeutics AB: Stockholm, Sweden, 2021. Available online: https://www.clinicaltrials.gov (accessed on 28 March 2022).
- Hartono, C.; Chung, M.; Perlman, A.S.; Chevalier, J.M.; Serur, D.; Seshan, S.V.; Muthukumar, T. Bortezomib for Reduction of Proteinuria in IgA Nephropathy. Kidney Int. Rep. 2018, 3, 861–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MorphoSys AG. NCT05065970: A Double Blind, Randomized, Placebo-Controlled, Multicenter Phase IIa, Clinical Trial to Assess Efficacy and Safety of the Human Anti-CD38 Antibody Felzartamab in IgA Nephropathy; MorphoSys AG: Planegg, Germany, 2021. Available online: https://www.clinicaltrials.gov (accessed on 28 March 2022).
- Boxhammer, R.; Weirather, J.; Steidl, S.; Endell, J. MOR202, a Human Anti-CD38 Monoclonal Antibody, Mediates Potent Tumoricidal Activity In Vivo and Shows Synergistic Efficacy in Combination with Different Antineoplastic Compounds. Blood 2015, 126, 3015. [Google Scholar] [CrossRef]
- Raab, M.S.; Engelhardt, M.; Blank, A.; Goldschmidt, H.; Agis, H.; Blau, I.W.; Einsele, H.; Ferstl, B.; Schub, N.; Röllig, C.; et al. MOR202, a Novel Anti-CD38 Monoclonal Antibody, in Patients with Relapsed or Refractory Multiple Myeloma: A First-in-Human, Multicentre, Phase 1-2a Trial. Lancet Haematol. 2020, 7, e381–e394. [Google Scholar] [CrossRef]
- Endell, J.; Boxhammer, R.; Wurzenberger, C.; Ness, D.; Steidl, S. The Activity of MOR202, a Fully Human Anti-CD38 Antibody, Is Complemented by ADCP and Is Synergistically Enhanced by Lenalidomide in Vitro and in Vivo. Blood 2012, 120, 4018. [Google Scholar] [CrossRef]
- Tawara, T.; Hasegawa, K.; Sugiura, Y.; Harada, K.; Miura, T.; Hayashi, S.; Tahara, T.; Ishikawa, M.; Yoshida, H.; Kubo, K.; et al. Complement Activation Plays a Key Role in Antibody-Induced Infusion Toxicity in Monkeys and Rats. J. Immunol. 2008, 180, 2294–2298. [Google Scholar] [CrossRef] [Green Version]
- Rovin, B.H.; Adler, S.G.; Hoxha, E.; Sprangers, B.; Stahl, R.; Wetzels, J.F.; Schwamb, B.; Boxhammer, R.; Nguyen, Q.; Haertle, S.; et al. Felzartamab in Patients with Anti-Phospholipase A2 Receptor Autoantibody Positive (Anti-PLA2R+) Membranous Nephropathy (MN): Interim Results from the M-PLACE Study. In Proceedings of the ASN Kidney Week 2021, San Diego, CA, USA, 4–7 November 2021. [Google Scholar]
- Rovin, B.; Adler, S.G.; Hoxha, E.; Sprangers, B.; Stahl, R.; Wetzels, J.F.; Jauch-Lembach, J.; Griese, J.; Boxhammer, R.; Xu, L.; et al. Felzartamab in Patients with Anti-Phospholipase A2 Receptor Autoantibody-Positive (Anti-PLA2R Ab+) Membranous Nephropathy (MN): Preliminary Results from the M-PLACE Study. In Proceedings of the National Kidney Foundation Spring Clinical Meetings, Boston, MA, USA, 6–10 April 2022. [Google Scholar]
- Liyasova, M.; McDonald, Z.; Taylor, P.; Gorospe, K.; Xu, X.; Yao, C.; Liu, Q.; Yang, L.; Atenafu, E.G.; Piza, G.; et al. A Personalized Mass Spectrometry-Based Assay to Monitor M-Protein in Patients with Multiple Myeloma (EasyM). Clin. Cancer Res. 2021, 27, 5028–5037. [Google Scholar] [CrossRef]
- Raab, M.S.; Chatterjee, M.; Goldschmidt, H.; Agis, H.; Blau, I.; Einsele, H.; Engelhardt, M.; Ferstl, B.; Gramatzki, M.; Röllig, C.; et al. A Phase I/IIa Study of the CD38 Antibody MOR202 Alone and in Combination with Pomalidomide or Lenalidomide in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2016, 128, 1152. [Google Scholar] [CrossRef]
- Heesterbeek, D.A.C.; Angelier, M.L.; Harrison, R.A.; Rooijakkers, S.H.M. Complement and Bacterial Infections: From Molecular Mechanisms to Therapeutic Applications. J. Innate Immun. 2018, 10, 455–464. [Google Scholar] [CrossRef]
- Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Alberts, B., Ed.; Garland Science: New York, NY, USA, 2002; ISBN 978-0-8153-3218-3. [Google Scholar]

{kind=link}
{kind=link}
| Agent | Target | Modality | Mechanism of Action |
|---|---|---|---|
| Atacicept | BAFF and APRIL | Fusion protein/antibody | Inhibits maturation and activation of B cells |
| BION-1301 | APRIL | Monoclonal antibody | Inhibits maturation and activation of B cells |
| Felzartamab (MOR202/TJ202) | CD38 | Monoclonal antibody | Depletes CD38+ plasma cells |
| Iptacopan | Factor B | small molecule | Inhibits complement alternative pathway activation |
| Narsoplimab | MASP-2 | Monoclonal antibody | Inhibits complement lectin pathway activation |
| Sibeprenlimab | APRIL | Monoclonal antibody | Inhibits maturation and activation of B cells |
| Tarpeyo (targeted-release budesonide) | Glucocorticoid receptors | Corticosteroid | Depletes B cells and plasma cells in the small intestine |
| Telitacicept | BAFF and APRIL | Fusion protein/antibody | Inhibits maturation and activation of B cells |
| Velcade (bortezomib) | Proteasome | Peptide | Inhibits proteasome activity in plasma cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maixnerova, D.; El Mehdi, D.; Rizk, D.V.; Zhang, H.; Tesar, V. New Treatment Strategies for IgA Nephropathy: Targeting Plasma Cells as the Main Source of Pathogenic Antibodies. J. Clin. Med. 2022, 11, 2810. https://doi.org/10.3390/jcm11102810
Maixnerova D, El Mehdi D, Rizk DV, Zhang H, Tesar V. New Treatment Strategies for IgA Nephropathy: Targeting Plasma Cells as the Main Source of Pathogenic Antibodies. Journal of Clinical Medicine. 2022; 11(10):2810. https://doi.org/10.3390/jcm11102810
Chicago/Turabian StyleMaixnerova, Dita, Delphine El Mehdi, Dana V. Rizk, Hong Zhang, and Vladimir Tesar. 2022. "New Treatment Strategies for IgA Nephropathy: Targeting Plasma Cells as the Main Source of Pathogenic Antibodies" Journal of Clinical Medicine 11, no. 10: 2810. https://doi.org/10.3390/jcm11102810
APA StyleMaixnerova, D., El Mehdi, D., Rizk, D. V., Zhang, H., & Tesar, V. (2022). New Treatment Strategies for IgA Nephropathy: Targeting Plasma Cells as the Main Source of Pathogenic Antibodies. Journal of Clinical Medicine, 11(10), 2810. https://doi.org/10.3390/jcm11102810