
Sarcoidosis-Like Cancer-Associated Granulomatosis: Characteristics and a Case-Control Comparison with Sarcoidosis

 ,
,
Abstract
1. Introduction
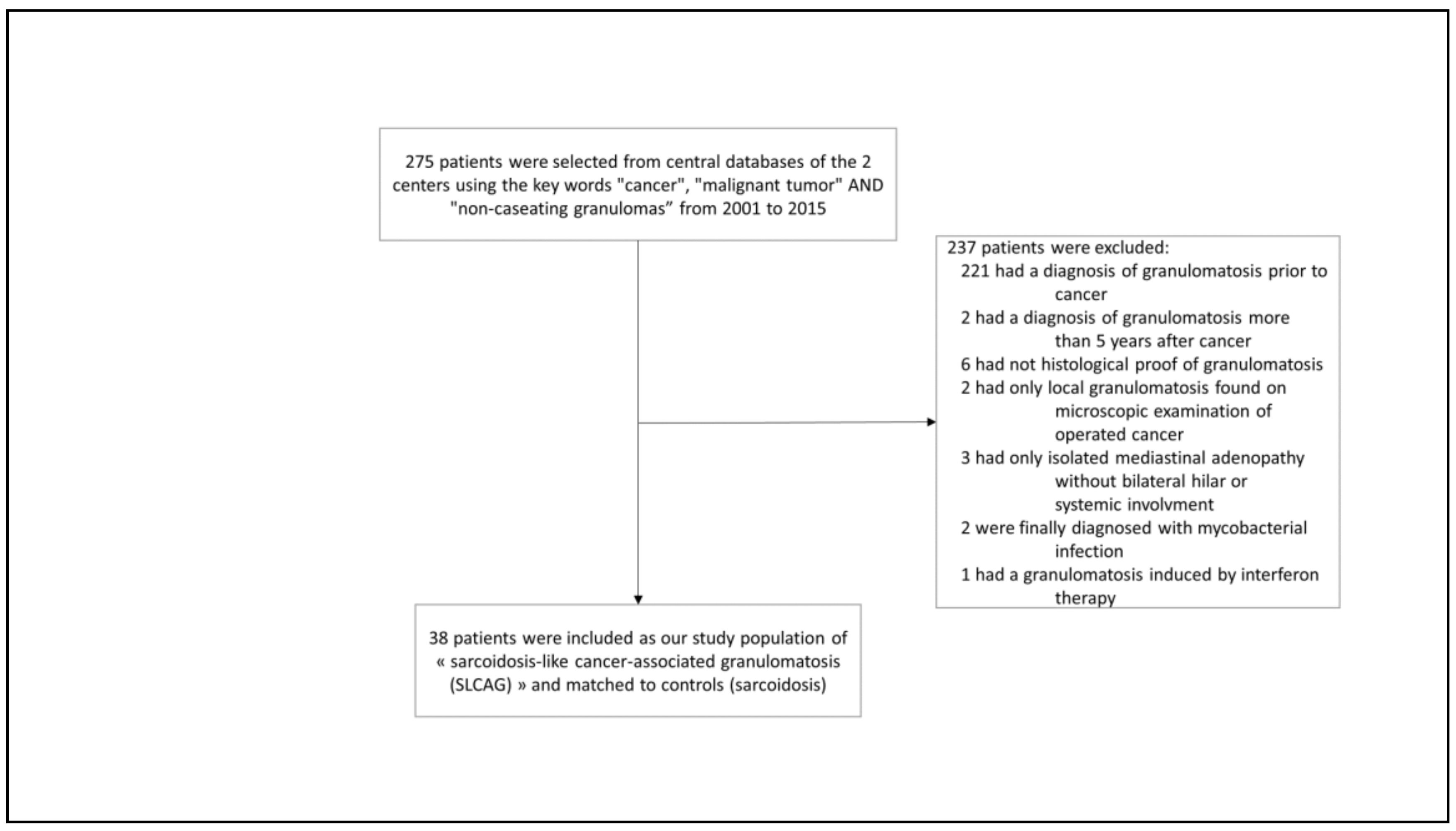
2. Patients and Methods
Statistical Analysis
3. Results
3.1. Clinical and Laboratory Findings
3.2. Outcome and Treatment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brincker, H. Sarcoid reactions in malignant tumours. Cancer Treat. Rev. 1986, 13, 147–156. [Google Scholar] [CrossRef]
- Shigemitsu, H. Is sarcoidosis frequent in patients with cancer? Curr. Opin. Pulm. Med. 2008, 14, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, F.U.; Sheerin, F.; Bradley, K.M.; Gleeson, F.V. Sarcoid-like reaction to malignancy on whole-body integrated 18F-FDG PET/CT: Prevalence and disease pattern. Clin. Radiol. 2009, 64, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Ravaglia, C.; Gurioli, C.; Casoni, G.L.; Romagnoli, M.; Tomassetti, S.; Gurioli, C.; Dubini, A.; Poletti, V. Sarcoid-like lesion is a frequent benign cause of lymphadenopathy in neoplastic patients. Eur. Respir. J. 2013, 41, 754–755. [Google Scholar] [CrossRef]
- Hunt, B.M.; Vallières, E.; Buduhan, G.; Aye, R.; Louie, B. Sarcoidosis as a benign cause of lymphadenopathy in cancer patients. Am. J. Surg. 2009, 197, 629–632. [Google Scholar] [CrossRef]
- Grados, A.; Ebbo, M.; Bernit, E.; Veit, V.; Mazodier, K.; Jean, R.; Coso, D.; Aurran-Schleinitz, T.; Broussais, F.; Bouabdallah, R.; et al. Sarcoidosis occurring after solid cancer: A nonfortuitous association: Report of 12 cases and review of the literature. Medicine 2015, 94, e928. [Google Scholar] [CrossRef]
- Maayan, H.; Ashkenazi, Y.; Nagler, A.; Izbicki, G. Sarcoidosis and lymphoma: Case series and literature review. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2011, 28, 146–152. [Google Scholar]
- London, J.; Grados, A.; Fermé, C.; Charmillon, A.; Maurier, F.; Deau, B.; Crickx, E.; Brice, P.; Chapelon-Abric, C.; Haioun, C.; et al. Sarcoidosis occurring after lymphoma: Report of 14 patients and review of the literature. Medicine 2014, 93, e121. [Google Scholar] [CrossRef]
- Cerri, S.; Fontana, M.; Balduzzi, S.; Potenza, L.; Faverio, P.; Luppi, M.; D’Amico, R.; Spagnolo, P.; Clini, E.; Luppi, F. Clinical differences in sarcoidosis patients with and without lymphoma: A single-centre retrospective cohort analysis. Eur. Respir. J. 2019, 54, 1802470. [Google Scholar] [CrossRef]
- Dagaonkar, R.S.; Choong, C.V.; Asmat, A.B.; Ahmed, D.B.; Chopra, A.; Lim, A.Y.; Tai, D.Y.; Kor, A.C.; Goh, S.K.; Abisheganaden, J.; et al. Significance of coexistent granulomatous inflammation and lung cancer. J. Clin. Pathol. 2017, 70, 337–341. [Google Scholar] [CrossRef]
- Reich, J.M. Neoplasia in the Etiology of Sarcoidosis. Eur. J. Intern. Med. 2006, 17, 81–87. [Google Scholar] [CrossRef]
- O’Connell, M.J.; Schimpff, S.C.; Kirschner, R.H.; Abt, A.B.; Wiernik, P.H. Epithelioid granulomas in Hodgkin disease: A favorable prognostic sign? JAMA 1975, 233, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Sacks, E.L.; Donaldson, S.S.; Gordon, J.; Dorfman, R.F. Epithelioid granulomas associated with Hodgkin’s disease. Clinical correlations in 55 previously untreated patients. Cancer 1978, 41, 562–567. [Google Scholar] [CrossRef]
- Steinfort, D.P.; Tsui, A.; Grieve, J.; Hibbs, M.L.; Anderson, G.P.; Irving, L.B. Sarcoidal reactions in regional lymph nodes of patients with early stage non–small cell lung cancer predict improved disease-free survival: A pilot case-control study. Hum. Pathol. 2012, 43, 333–338. [Google Scholar] [CrossRef]
- Murthi, M.; Yoshioka, K.; Cho, J.H.; Arias, S.; Danna, E.; Zaw, M.; Holt, G.; Tatsumi, K.; Kawasaki, T.; Mirsaeidi, M. Presence of concurrent sarcoid-like granulomas indicates better survival in cancer patients: A retrospective cohort study. ERJ Open Res. 2020, 6, 00061–02020. [Google Scholar] [CrossRef] [PubMed]
- Statement on Sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am. J. Respir. Crit. Care Med. 1999, 160, 736–755.
- Judson, M.A.; Costabel, U.; Drent, M.; Wells, A.; Maier, L.; Koth, L.; Shigemitsu, H.; Culver, D.A.; Gelfand, J.; Valeyre, D.; et al. The WASOG Sarcoidosis Organ Assessment Instrument: An update of a previous clinical tool. Sarcoidosis Vasc. Diffus. Lung Dis. 2014, 31, 19–27. [Google Scholar]
- Crouser, E.D.; Maier, L.A.; Wilson, K.C.; Bonham, C.A.; Morgenthau, A.S.; Patterson, K.C.; Abston, E.; Bernstein, R.C.; Blankstein, R.; Chen, E.S.; et al. Diagnosis and detection of sarcoidosis. An official American Thoracic Society clinical practice guideline. Am. J. Respir. Crit. Care Med. 2020, 201, e26–e51. [Google Scholar] [CrossRef] [PubMed]
- Prasse, A.; Katic, C.; Germann, M.; Buchwald, A.; Zissel, G.; Muller-Quernheim, J. Phenotyping sarcoidosis from a pulmonary perspective. Am. J. Respir. Crit. Care Med. 2008, 177, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Nagai, S.; Balter, M.; Costabel, U.; Drent, M.; Du Bois, R.; Grutters, J.C.; Judson, M.A.; Lambiri, I.; Lower, E.E.; et al. Defining the clinical outcome status (COS) in sarcoidosis: Results of WASOG Task Force. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2011, 28, 56–64. [Google Scholar]
- Pacheco, Y.; Calender, A.; Israël-Biet, D.; Roy, P.; Lebecque, S.; Cottin, V.; Bouvry, D.; Nunes, H.; Sève, P.; Pérard, L.; et al. Familial vs. sporadic sarcoidosis: BTNL2 polymorphisms, clinical presentations, and outcomes in a French cohort. Orphanet. J. Rare Dis. 2016, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.; Alzebdeh, R.; Kable, T.D.; Soubani, A.O. Non-caseating granulomas in patients after the diagnosis of cancer: Clinical characteristics and outcome. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2011, 28, 44–49. [Google Scholar]
- Kennedy, M.P.; Jimenez, C.A.; Mhatre, A.D.; Morice, R.C.; Eapen, G.A. Clinical implications of granulomatous inflammation detected by endobronchial ultrasound transbronchial needle aspiration in patients with suspected cancer recurrence in the mediastinum. J. Cardiothorac. Surg. 2008, 3, 8. [Google Scholar] [CrossRef]
- Lashari, B.H.; Asai, M.; Randleman, G.; Sack, M.; Patel, R. Sarcoid-like mediastinal lymphadenopathy in gynecologic malignancy. Pulm. Med. 2018, 2018, 5141575. [Google Scholar] [CrossRef]
- Tamada, T.; Nara, M.; Murakami, K.; Gamo, S.; Aritake, H.; Shimizu, M.; Kazama, I.; Ichinose, M.; Sugiura, H. The Clinical Features of Patients with Sarcoidosis and Malignant Diseases in Japan. Intern. Med. Tokyo Jpn. 2021, 60, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Ergin, A.B.; Nasr, C.E. Thyroid cancer & sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2014, 31, 239–243. [Google Scholar]
- Shima, T.; Tanaka, Y.; Katsuragi, K.; Fujio, N.; Nakatani, S.; Kobayashi, Y.; Hida, T. Sarcoid reaction in the spleen after sigmoid colon cancer resection: A case report. Surg. Case Rep. 2016, 2, 115. [Google Scholar] [CrossRef]
- Majori, M.; Anghinolfi, M.; Gnetti, L.; Casalini, A.G. Pulmonary metastases from low grade sarcoma in a patient with pulmonary sarcoidosis. Sarcoidosis or sarcoid-like reaction? Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2016, 33, 171–174. [Google Scholar]
- Valeyre, D.; Prasse, A.; Nunes, H.; Uzunhan, Y.; Brillet, P.-Y.; Müller-Quernheim, J. Sarcoidosis. Lancet Lond. Engl. 2014, 383, 1155–1167. [Google Scholar] [CrossRef]
- Iannuzzi, M.C.; Rybicki, B.A.; Teirstein, A.S. Sarcoidosis. N. Engl. J. Med. 2007, 357, 2153–2165. [Google Scholar] [CrossRef]
- Spagnolo, P.; Rossi, G.; Trisolini, R.; Sverzellati, N.; Baughman, R.P.; Wells, A.U. Pulmonarysarcoidosis. Lancet Respir. Med. 2018, 6, 389–402. [Google Scholar] [CrossRef]
- Duchemann, B.; Annesi-Maesano, I.; de Naurois, C.J.; Sanyal, S.; Brillet, P.Y.; Brauner, M.; Kambouchner, M.; Huynh, S.; Naccache, J.M.; Borie, R.; et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur. Respir. J. 2017, 50, 1602419. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Ishii, H.; Eishi, Y.; Uchida, K.; Yoshimura, M.; Iwasaki, A.; Fujita, M.; Nabeshima, K.; Watanabe, K. Histological differences between sarcoidosis and lung cancer-related sarcoid reaction. Respir. Investig. 2020, 58, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Paydas, S.; Yavuz, S.; Disel, U.; Zeren, H.; Hastürk, S.; Hanta, I.; Ergin, M.; Sahin, B. Granulomatous reaction after chemotherapy for Hodgkin’s disease. Leuk. Res. 2002, 26, 967–970. [Google Scholar] [CrossRef]


{kind=link}
| SLCAG Patients | ||
|---|---|---|
| N | 38 | |
| Age, years (mean ± SD) | 51 ± 14 | |
| Females, n (%) | 25 (66%) | |
| Caucasian/black ethnicity, n (%) | 32 (84%)/4 (11%) | |
| Smokers, n (%, mean pack-years) | 12 (33%, 30) | |
| Site of malignancy n (%) | Breast | 7 (17%) |
| Gynecologic (endometrial or ovarian) | 5 (13%) | |
| Lung | 4 (11%) | |
| Colorectal | 4 (11%) | |
| Head and neck | 4 (11%) | |
| Renal | 3 (8%) | |
| Prostatic | 2 (5%) | |
| Skin (melanoma) | 2 (5%) | |
| Others | 7 (19%) | |
| Cancer stage at diagnosis n (%) | Limited | 21 (55%) |
| Regional lymph nodes extension | 5 (13%) | |
| Metastatic extension | 0 | |
| Not available or not applicable | 12 (32%) | |
| 5 years remission rate, n (%) | 29 (76%) | |
| SLCAG | Sarcoidosis Controls | p Value | ||
|---|---|---|---|---|
| Symptomatic, N (%) | 13 (34%) | 32 (86%) | <0.0001 | |
| Symptoms n (%) | General | 4 (11%) | 12 (33%) | 0.04 |
| Respiratory | 11 (29%) | 21 (58%) | 0.04 | |
| Cutaneous | 1 (3%) | 9 (25%) | 0.01 | |
| Ophthalmic | 0 | 4 (11%) | 0.11 | |
| Löfgren syndrome | 0 | 3 (8%) | 0.24 | |
| Organs involved n (%) | Thorax | 38 (100%) | 38 (100%) | |
| Bilateral lymph nodes with or without mediastinal ones | 36 (95%) | 38 (100%) | 0.49 | |
| Thorax alone | 26 (68%) | 27 (71%) | 1.0 | |
| Pulmonary infiltrates | 22 (58%) | 23 (60%) | 1.0 | |
| Extrathoracic | 12 (32%) | 11 (29%) | 1.0 | |
| Extrathoracic lymphadenopathy | 9 (24%) | 3 (8%) | 0.11 | |
| Liver | 4 (10%) | 2 (5%) | 0.67 | |
| Heart | 3 (8%) | 2 (5%) | 1.0 | |
| SLCAG | Sarcoidosis Controls | p Value | ||
|---|---|---|---|---|
| X-ray staging n (%) | 1 | 18 (48%) | 17 (45%) | |
| 2 | 14 (37%) | 15 (39%) | ||
| 3 | 4 (10%) | 2 (5%) | ||
| 4 | 2 (5%) | 3 (8%) | ||
| PFT n (%) | n | 33 | 38 | |
| TLC < 80% pred | 4 (12%) | 9 (24%) | 0.24 | |
| FVC < 80% pred | 6 (18%) | 9 (24%) | 0.77 | |
| FEV1/FVC < 70% | 5 (15%) | 5 (13%) | 1.0 | |
| TLCO < 80% pred | 22 (66%) | 31 (82%) | 0.18 | |
| BAL | n | 18 | 30 | |
| Lymphocyte count, mean ± SD, % | 21 ± 17 | 27 ± 20 | 0.08 | |
| Lymphocyte count ≥20%, n (%) | 8 (44%) | 17 (57%) | 0.55 | |
| CD4/CD8 T-cell ratio >3.5, n (%) | 6/10 (60%) | 10/16 (62%) | 1.0 | |
| Increased SACE, n (%) | 16/32 (50%) | 17/37 (46%) | 0.81 | |
| SLCAG | Sarcoidosis Controls | p Value | ||
|---|---|---|---|---|
| Follow-up, Median (Range), Months | 32 (4–239) | 73 (0–370) | 0.12 | |
| PFT change from baseline at last FU, n/available (%) | FVC improvement * | 10/27 (37%) | 8/28 (29%) | 0.57 |
| FVC deterioration † | 6/27 (22%) | 5/28 (18%) | 0.75 | |
| TLCO improvement * | 8/27 (30%) | 11/33 (33%) | 0.79 | |
| TLCO deterioration † | 4/27 (15%) | 5/33 (15%) | 1.0 | |
| Treatment, n (%) | 12 (32%) | 22 (58%) | 0.04 | |
| Corticosteroids, n (%) | 9 (24%) | 19 (50%) | 0.03 | |
| Others, n (%) | 9 (24%) | 13 (34%) | 0.45 | |
| SCAC n (%) | n | 38 | 36 | |
| 1—asymptomatic—no treatment | 20 (53%) | 4 (11%) | 0.0002 | |
| 2—asymptomatic—treatment ≤12 months | 0 | 1 (3%) | 0.49 | |
| 3—asymptomatic—treatment >12months | 4 (10%) | 0 | 0.11 | |
| 4—symptomatic—no treatment | 6 (16%) | 13 (36%) | 0.06 | |
| 5—symptomatic—treatment ≤12 months | 2 (5%) | 1 (3%) | 1.0 | |
| 6—symptomatic—treatment >12 months | 6 (16%) | 17 (53%) | 0.005 | |
| Classification of outcome n/available (%) | (1) Recovery within 3 years | 12/29 (41%) | 13/33 (39%) | 1.0 |
| (2) Recovery between 3 and 5 years | 0 | 0 | ||
| (3) Persistent GD activity signs at 5 years | 2/13 (15%) | 11/19 (58%) | 0.03 | |
| (4) Death | 0 | 0 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastré, J.; Bouvry, D.; Juvin, K.; Benattia, A.; Annesi-Maesano, I.; Valeyre, D.; Nunes, H.; Israël-Biet, D. Sarcoidosis-Like Cancer-Associated Granulomatosis: Characteristics and a Case-Control Comparison with Sarcoidosis. J. Clin. Med. 2021, 10, 1988. https://doi.org/10.3390/jcm10091988
Pastré J, Bouvry D, Juvin K, Benattia A, Annesi-Maesano I, Valeyre D, Nunes H, Israël-Biet D. Sarcoidosis-Like Cancer-Associated Granulomatosis: Characteristics and a Case-Control Comparison with Sarcoidosis. Journal of Clinical Medicine. 2021; 10(9):1988. https://doi.org/10.3390/jcm10091988
Chicago/Turabian StylePastré, Jean, Diane Bouvry, Karine Juvin, Amira Benattia, Isabella Annesi-Maesano, Dominique Valeyre, Hilario Nunes, and Dominique Israël-Biet. 2021. "Sarcoidosis-Like Cancer-Associated Granulomatosis: Characteristics and a Case-Control Comparison with Sarcoidosis" Journal of Clinical Medicine 10, no. 9: 1988. https://doi.org/10.3390/jcm10091988
APA StylePastré, J., Bouvry, D., Juvin, K., Benattia, A., Annesi-Maesano, I., Valeyre, D., Nunes, H., & Israël-Biet, D. (2021). Sarcoidosis-Like Cancer-Associated Granulomatosis: Characteristics and a Case-Control Comparison with Sarcoidosis. Journal of Clinical Medicine, 10(9), 1988. https://doi.org/10.3390/jcm10091988