
Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Novel Aspects of Early Topographic Modeling of Human OA
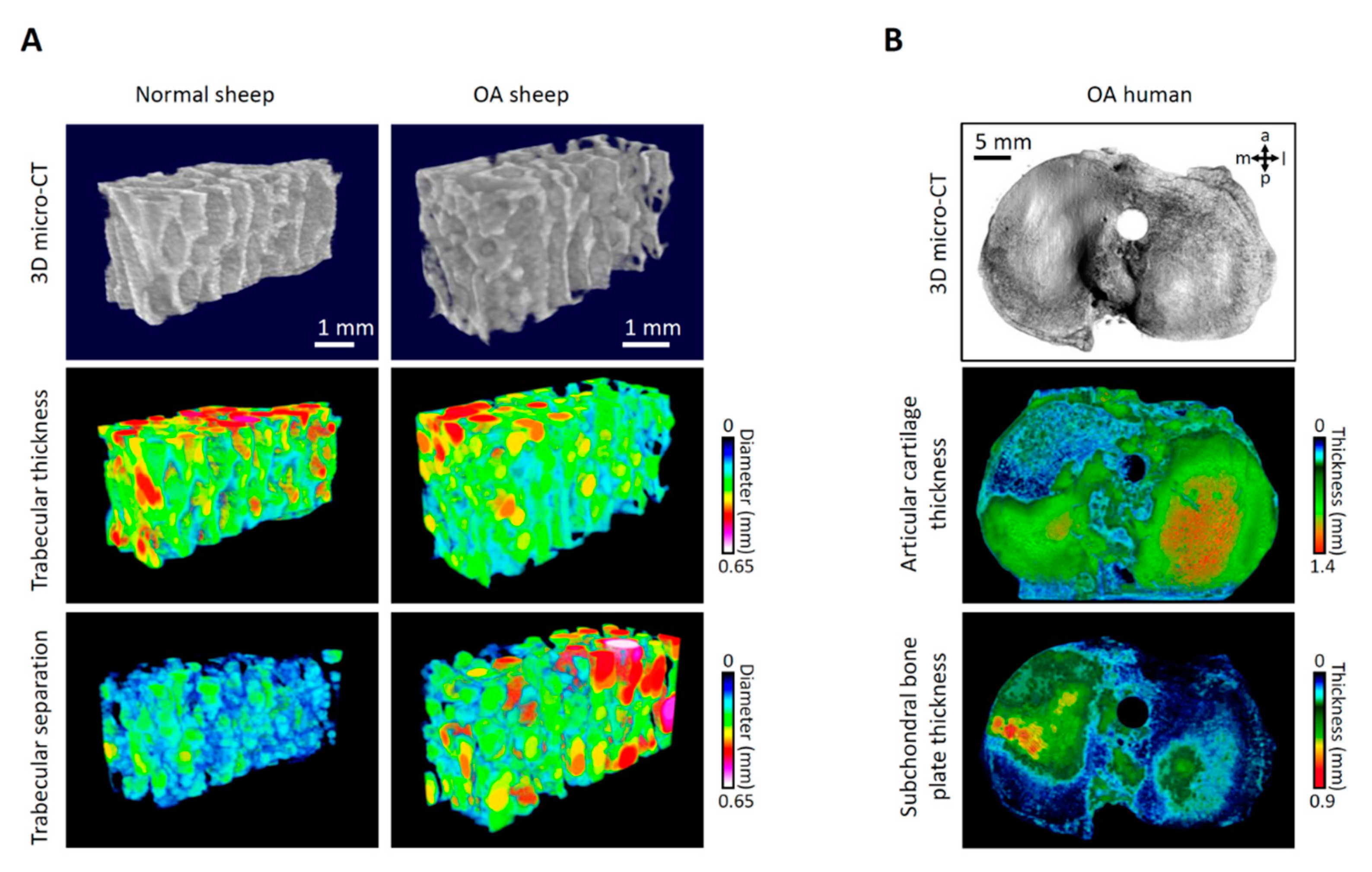
2.1. Structural Changes in Human Early OA
2.2. What Initiates the Breakdown of the Osteochondral Unit?
2.3. Meniscal Tears Lead to OA
2.4. Topographic Modeling of Human OA
3. The Role of Proteases and Cytokines in OA
3.1. Proteases in OA
3.2. Cartilage ECM Protein Fragments
3.3. Cytokines in OA
4. Impact of the Peripheral Nervous System and Its Neuropeptides on OA
4.1. Sensory Nerve Fibers in OA Joints
4.2. Sensory Neuropeptides in OA Joints
4.2.1. Substance P Effects
4.2.2. αCGRP Effects
4.3. Neuropeptides as Biomarkers
5. Conclusions and Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Litwic, A.; Edwards, M.H.; Dennison, E.M.; Cooper, C. Epidemiology and burden of osteoarthritis. Br. Med. Bull. 2013, 105. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.-P. Osteoarthritis. Nat. Rev. Dis. Primers 2016, 2, 16072. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage-bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Ratneswaran, A.; Kapoor, M. Osteoarthritis year in review: Genetics, genomics, epigenetics. Osteoarthr. Cartil. 2021, 29, 151–160. [Google Scholar] [CrossRef]
- Richard, D.; Liu, Z.; Cao, J.; Kiapour, A.M.; Willen, J.; Yarlagadda, S.; Jagoda, E.; Kolachalama, V.B.; Sieker, J.T.; Chang, G.H.; et al. Evolutionary Selection and Constraint on Human Knee Chondrocyte Regulation Impacts Osteoarthritis Risk. Cell 2020, 181, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Kania, K.; Colella, F.; Riemen, A.H.K.; Wang, H.; Howard, K.A.; Aigner, T.; Dell’Accio, F.; Capellini, T.D.; Roelofs, A.J.; De Bari, C. Regulation of Gdf5 expression in joint remodelling, repair and osteoarthritis. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Olah, T.; Reinhard, J.; Gao, L.; Haberkamp, S.; Goebel, L.K.H.; Cucchiarini, M.; Madry, H. Topographic modeling of early human osteoarthritis in sheep. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Jenei-Lanzl, Z.; Meurer, A.; Zaucke, F. Interleukin-1beta signaling in osteoarthritis—Chondrocytes in focus. Cell Signal. 2019, 53, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Grassel, S.; Muschter, D. Peripheral Nerve Fibers and Their Neurotransmitters in Osteoarthritis Pathology. Int. J. Mol. Sci. 2017, 18, 931. [Google Scholar] [CrossRef]
- Madry, H.; van Dijk, C.N.; Mueller-Gerbl, M. The basic science of the subchondral bone. Knee Surg. Sports Traumatol. Arthrosc. 2010, 18, 419–433. [Google Scholar] [CrossRef]
- Luyten, F.P.; Denti, M.; Filardo, G.; Kon, E.; Engebretsen, L. Definition and classification of early osteoarthritis of the knee. Knee Surg. Sports Traumatol. Arthrosc. 2012, 20, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Angele, P.; Madry, H.; Kon, E.; Early, O.A. Point of no return or a chance for regenerative approaches. Knee Surg. Sports Traumatol. Arthrosc. 2016, 24, 1741–1742. [Google Scholar] [CrossRef] [PubMed]
- Emery, C.A.; Whittaker, J.L.; Mahmoudian, A.; Lohmander, L.S.; Roos, E.M.; Bennell, K.L.; Toomey, C.M.; Reimer, R.A.; Thompson, D.; Ronsky, J.L.; et al. Establishing outcome measures in early knee osteoarthritis. Nat. Rev. Rheumatol. 2019, 15, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T.; Hodgson, R. Identifying and treating preclinical and early osteoarthritis. Rheum. Dis. Clin. N. Am. 2014, 40, 699–710. [Google Scholar] [CrossRef]
- Madry, H.; Luyten, F.P.; Facchini, A. Biological aspects of early osteoarthritis. Knee Surg. Sports Traumatol. Arthrosc. 2012, 20, 407–422. [Google Scholar] [CrossRef]
- Rao, A.J.; Johnston, T.R.; Harris, A.H.; Smith, R.L.; Costouros, J.G. Inhibition of chondrocyte and synovial cell death after exposure to commonly used anesthetics: Chondrocyte apoptosis after anesthetics. Am. J. Sports Med. 2014, 42, 50–58. [Google Scholar] [CrossRef]
- Klose-Jensen, R.; Nielsen, A.W.; Hartlev, L.B.; Thomsen, J.S.; Boel, L.W.T.; Laursen, M.; Keller, K.K.; Hauge, E.-M. Histomorphometric case-control study of subarticular osteophytes in patients with osteoarthritis of the hip. BMC Musculoskelet. Disord. 2020, 21, 653. [Google Scholar] [CrossRef] [PubMed]
- Pritzker, K.P.; Aigner, T. Terminology of osteoarthritis cartilage and bone histopathology—A proposal for a consensus. Osteoarthr. Cartil. 2010, 18 (Suppl. 3), S7–S9. [Google Scholar] [CrossRef]
- Pritzker, K.P.; Gay, S.; Jimenez, S.A.; Ostergaard, K.; Pelletier, J.P.; Revell, P.A.; Salter, D.; Berg, W.V.D. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 2006, 14, 13–29. [Google Scholar] [CrossRef]
- Rolauffs, B.; Williams, J.M.; Aurich, M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Proliferative remodeling of the spatial organization of human superficial chondrocytes distant from focal early osteoarthritis. Arthritis Rheum. 2010, 62, 489–498. [Google Scholar] [PubMed]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef]
- Madry, H.; Kon, E.; Condello, V.; Peretti, G.M.; Steinwachs, M.; Seil, R.; Berruto, M.; Engebretsen, L.; Filardo, G.; Angele, P. Early osteoarthritis of the knee. Knee Surg. Sports Traumatol. Arthrosc. 2016, 24, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Berenbaum, F.; Walker, C. Osteoarthritis and inflammation: A serious disease with overlapping phenotypic patterns. Postgrad. Med. 2020, 132, 377–384. [Google Scholar] [CrossRef]
- Weinans, H.; Siebelt, M.; Agricola, R.; Botter, S.M.; Piscaer, T.M.; Waarsing, J.H. Pathophysiology of peri-articular bone changes in osteoarthritis. Bone 2012, 51, 190–196. [Google Scholar] [CrossRef]
- Klose-Jensen, R.; Hartlev, L.B.; Boel, L.W.T.; Laursen, M.B.; Stengaard-Pedersen, K.; Keller, K.K.; Hauge, E.-M. Subchondral bone turnover, but not bone volume, is increased in early stage osteoarthritic lesions in the human hip joint. Osteoarthr. Cartil. 2015, 23, 2167–2173. [Google Scholar] [CrossRef]
- Nagira, K.; Ikuta, Y.; Shinohara, M.; Sanada, Y.; Omoto, T.; Kanaya, H.; Nakasa, T.; Ishikawa, M.; Adachi, N.; Miyaki, S.; et al. Histological scoring system for subchondral bone changes in murine models of joint aging and osteoarthritis. Sci. Rep. 2020, 10, 10077. [Google Scholar] [CrossRef] [PubMed]
- Hada, S.; Ishijima, M.; Kaneko, H.; Kinoshita, M.; Liu, L.; Sadatsuki, R.; Futami, I.; Yusup, A.; Takamura, T.; Arita, H.; et al. Association of medial meniscal extrusion with medial tibial osteophyte distance detected by T2 mapping MRI in patients with early-stage knee osteoarthritis. Arthritis Res. Ther. 2017, 19, 201. [Google Scholar] [CrossRef]
- Sandell, L.J. Etiology of osteoarthritis: Genetics and synovial joint development. Nat. Rev. Rheumatol. 2012, 8, 77–89. [Google Scholar] [CrossRef]
- Graf, R. Developmental hip disorders in infants. Diagnosis and therapy. Orthopade 1997, 26, 1. [Google Scholar] [PubMed]
- Englund, M.; Roos, E.M.; Lohmander, L.S. Impact of type of meniscal tear on radiographic and symptomatic knee osteoarthritis: A sixteen-year followup of meniscectomy with matched controls. Arthritis Rheum. 2003, 48, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Frobell, R.B.; Roos, E.M.; Roos, H.P.; Ranstam, J.; Lohmander, L.S. A randomized trial of treatment for acute anterior cruciate ligament tears. N. Engl. J. Med. 2010, 363, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Sanders, T.L.; Pareek, A.; Obey, M.R.; Johnson, N.R.; Carey, J.L.; Stuart, M.J.; Krych, A.J. High Rate of Osteoarthritis After Osteochondritis Dissecans Fragment Excision Compared with Surgical Restoration at a Mean 16-Year Follow-up. Am. J. Sports Med. 2017, 45, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Brenner, R.E. Evidence of necroptosis in osteoarthritic disease: Investigation of blunt mechanical impact as possible trigger in regulated necrosis. Cell Death Dis. 2019, 10, 683. [Google Scholar] [CrossRef] [PubMed]
- Berenbaum, F.; Wallace, I.J.; Lieberman, D.E.; Felson, D.T. Modern-day environmental factors in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2018, 14, 674–681. [Google Scholar] [CrossRef]
- Courties, A.; Sellam, J.; Berenbaum, F. Metabolic syndrome-associated osteoarthritis. Curr. Opin. Rheumatol. 2017, 29, 214–222. [Google Scholar] [CrossRef]
- Haberkamp, S.; Olah, T.; Orth, P.; Cucchiarini, M.; Madry, H. Analysis of spatial osteochondral heterogeneity in advanced knee osteoarthritis exposes influence of joint alignment. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Sharma, L.; Chmiel, J.S.; Almagor, O.; Felson, D.; Guermazi, A.; Roemer, F.; Lewis, C.E.; Segal, N.; Torner, J.; Cooke, T.D.V.; et al. The role of varus and valgus alignment in the initial development of knee cartilage damage by MRI: The MOST study. Ann. Rheum. Dis. 2013, 72, 235–240. [Google Scholar] [CrossRef]
- Sharma, L.; Song, J.; Dunlop, D.; Felson, D.; Lewis, C.E.; Segal, N.; Torner, J.; Cooke, T.D.V.; Hietpas, J.; Lynch, J.; et al. Varus and valgus alignment and incident and progressive knee osteoarthritis. Ann. Rheum. Dis. 2010, 69, 1940–1945. [Google Scholar] [CrossRef]
- Bastick, A.N.; Belo, J.N.; Runhaar, J.; Bierma-Zeinstra, S.M. What Are the Prognostic Factors for Radiographic Progression of Knee Osteoarthritis? A Meta-analysis. Clin. Orthop. Relat. Res. 2015, 473, 2969–2989. [Google Scholar] [CrossRef]
- Moyer, R.F.; Birmingham, T.B.; Chesworth, B.M.; Kean, C.O.; Giffin, J.R. Alignment, body mass and their interaction on dynamic knee joint load in patients with knee osteoarthritis. Osteoarthr. Cartil. 2010, 18, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Niu, J.; Felson, D.T.; Harvey, W.F.; Gross, K.D.; McCree, P.; Aliabadi, P.; Sack, B.; Zhang, Y. Knee alignment does not predict incident osteoarthritis: The Framingham osteoarthritis study. Arthritis Rheum. 2007, 56, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T.; Niu, J.; Gross, K.D.; Englund, M.; Sharma, L.; Cooke, T.D.; Guermazi, A.; Roemer, F.W.; Segal, N.; Goggins, J.M.; et al. Valgus malalignment is a risk factor for lateral knee osteoarthritis incidence and progression: Findings from the Multicenter Osteoarthritis Study and the Osteoarthritis Initiative. Arthritis Rheum. 2013, 65, 355–362. [Google Scholar] [CrossRef]
- Losina, E.; Ranstam, J.; Collins, J.E.; Schnitzer, T.J.; Katz, J.N. OARSI Clinical Trials Recommendations: Key analytic considerations in design, analysis, and reporting of randomized controlled trials in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 677–685. [Google Scholar] [CrossRef]
- Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of human knee menisci: Structure, composition, and function. Sports Health 2012, 4, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Radin, E.L.; de Lamotte, F.; Maquet, P. Role of the menisci in the distribution of stress in the knee. Clin. Orthop. Relat. Res. 1984, 185, 290–294. [Google Scholar] [CrossRef]
- Ahmed, A.M.; Burke, D.L. In-vitro measurement of static pressure distribution in synovial joints--Part I: Tibial surface of the knee. J. Biomech. Eng. 1983, 105, 216–225. [Google Scholar] [CrossRef]
- Crema, M.D.; Guermazi, A.; Li, L.; Nogueira-Barbosa, M.H.; Marra, M.D.; Roemer, F.W.; Eckstein, F.; Le Graverand, M.H.; Wyman, B.; Hunter, D. The association of prevalent medial meniscal pathology with cartilage loss in the medial tibiofemoral compartment over a 2-year period. Osteoarthr. Cartil. 2010, 18, 336–343. [Google Scholar] [CrossRef]
- Bloecker, K.; Wirth, W.; Guermazi, A.; Hunter, D.J.; Resch, H.; Hochreiter, J.; Eckstein, F. Relationship Between Medial Meniscal Extrusion and Cartilage Loss in Specific Femorotibial Subregions: Data from the Osteoarthritis Initiative. Arthritis Care Res. 2015, 67, 1545–1552. [Google Scholar] [CrossRef]
- Eckstein, F.; Buck, R.; Wirth, W. Location-independent analysis of structural progression of osteoarthritis-Taking it all apart, and putting the puzzle back together makes the difference. Semin. Arthritis Rheum. 2017, 46, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F.; Le Graverand, M.P. Plain radiography or magnetic resonance imaging (MRI): Which is better in assessing outcome in clinical trials of disease-modifying osteoarthritis drugs? Summary of a debate held at the World Congress of Osteoarthritis 2014. Semin. Arthritis Rheum. 2015, 45, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, P. Suprastructures of extracellular matrices: Paradigms of functions controlled by aggregates rather than molecules. Cell Tissue Res. 2010, 339, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, C.; Klatt, A.R.; Wagener, R.; Paulsson, M.; Bateman, J.F.; Heinegard, D.; Mörgelin, M. Complexes of matrilin-1 and biglycan or decorin connect collagen VI microfibrils to both collagen II and aggrecan. J. Biol. Chem. 2003, 278, 37698–37704. [Google Scholar] [CrossRef] [PubMed]
- Zaucke, F.; Grässel, S. Genetic mouse models for the functional analysis of the perifibrillar components collagen IX, COMP and matrilin-3: Implications for growth cartilage differentiation and endochondral ossification. Histol. Histopathol. 2009, 24, 1067–1079. [Google Scholar] [PubMed]
- Danfelter, M.; Onnerfjord, P.; Heinegard, D. Fragmentation of proteins in cartilage treated with interleukin-1: Specific cleavage of type IX collagen by matrix metalloproteinase 13 releases the NC4 domain. J. Biol. Chem. 2007, 282, 36933–36941. [Google Scholar] [CrossRef]
- Heinegard, D.; Saxne, T. The role of the cartilage matrix in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 50–56. [Google Scholar] [CrossRef]
- Mehana, E.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef]
- Durigova, M.; Nagase, H.; Mort, J.S.; Roughley, P.J. MMPs are less efficient than ADAMTS5 in cleaving aggrecan core protein. Matrix Biol. 2011, 30, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Tonge, D.P.; Pearson, M.J.; Jones, S.W. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthr. Cartil. 2014, 22, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Khalil, R.A. Matrix Metalloproteinase Inhibitors as Investigational and Therapeutic Tools in Unrestrained Tissue Remodeling and Pathological Disorders. Prog. Mol. Biol. Transl. Sci. 2017, 148, 355–420. [Google Scholar] [PubMed]
- Wan, Y.; Li, W.; Liao, Z.; Yan, M.; Chen, X.; Tang, Z. Selective MMP-13 Inhibitors: Promising Agents for the Therapy of Osteoarthritis. Curr. Med. Chem. 2020, 27, 3753–3769. [Google Scholar] [CrossRef] [PubMed]
- Stanton, H.; Rogerson, F.M.; East, C.J.; Golub, S.B.; Lawlor, K.E.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.K.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Vo, P.; Kanakis, I.; Liu, K.; Bou-Gharios, G. Aggrecanase-selective tissue inhibitor of metalloproteinase-3 (TIMP3) protects articular cartilage in a surgical mouse model of osteoarthritis. Sci. Rep. 2020, 10, 9288. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Inhibition of MMPs and ADAM/ADAMTS. Biochem. Pharmacol. 2019, 165, 33–40. [Google Scholar] [CrossRef]
- Hao, H.Q.; Zhang, J.F.; He, Q.Q.; Wang, Z. Cartilage oligomeric matrix protein, C-terminal cross-linking telopeptide of type II collagen, and matrix metalloproteinase-3 as biomarkers for knee and hip osteoarthritis (OA) diagnosis: A systematic review and meta-analysis. Osteoarthr. Cartil. 2019, 27, 726–736. [Google Scholar] [CrossRef]
- Schulz, J.N.; Nuchel, J.; Niehoff, A.; Bloch, W.; Schonborn, K.; Hayashi, S.; Kamper, M.; Brinckmann, J.; Plomann, M.; Paulsson, M.; et al. COMP-assisted collagen secretion--a novel intracellular function required for fibrosis. J. Cell Sci. 2016, 129, 706–716. [Google Scholar] [CrossRef]
- Halasz, K.; Kassner, A.; Morgelin, M.; Heinegard, D. COMP acts as a catalyst in collagen fibrillogenesis. J. Biol. Chem. 2007, 282, 31166–31173. [Google Scholar] [CrossRef]
- Clark, A.G.; Jordan, J.M.; Vilim, V.; Renner, J.B.; Dragomir, A.D.; Luta, G.; Kraus, V.B. Serum cartilage oligomeric matrix protein reflects osteoarthritis presence and severity: The Johnston County Osteoarthritis Project. Arthritis Rheum. 1999, 42, 2356–2364. [Google Scholar] [CrossRef]
- Posey, K.L.; Coustry, F.; Hecht, J.T. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol. 2018, 71–72, 161–173. [Google Scholar] [CrossRef]
- Ahrman, E.; Lorenzo, P.; Holmgren, K.; Grodzinsky, A.J.; Dahlberg, L.E.; Saxne, T.; Heinegård, D.; Önnerfjord, P. Novel cartilage oligomeric matrix protein (COMP) neoepitopes identified in synovial fluids from patients with joint diseases using affinity chromatography and mass spectrometry. J. Biol. Chem. 2014, 289, 20908–20916. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, P.; Aspberg, A.; Saxne, T.; Onnerfjord, P. Quantification of cartilage oligomeric matrix protein (COMP) and a COMP neoepitope in synovial fluid of patients with different joint disorders by novel automated assays. Osteoarthr. Cartil. 2017, 25, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Thomsson, K.A.; Jin, C.; Alweddi, S.; Struglics, A.; Rolfson, O.; Björkman, L.I.; Kalamajski, S.; Schmidt, T.A.; Jay, G.D.; et al. Cathepsin g Degrades Both Glycosylated and Unglycosylated Regions of Lubricin, a Synovial Mucin. Sci. Rep. 2020, 10, 4215. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.Y.; Yang, M.; Guo, J.; Zhang, C.; Lin, L.L.; Liu, Y.; Wei, R.-X. Identification of the Biomarkers and Pathological Process of Osteoarthritis: Weighted Gene Co-expression Network Analysis. Front. Physiol. 2019, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Guo, H.; Yang, X.; Li, Z.; Zhang, D.; Li, B.; Zhang, D.; Li, Q.; Xiong, Y. Potential candidate biomarkers associated with osteoarthritis: Evidence from a comprehensive network and pathway analysis. J. Cell. Physiol. 2019, 234, 17433–17443. [Google Scholar] [CrossRef]
- Ruthard, J.; Hermes, G.; Hartmann, U.; Sengle, G.; Pongratz, G.; Ostendorf, B.; Schneider, M.; Höllriegl, S.; Zaucke, F.; Wagener, R.; et al. Identification of antibodies against extracellular matrix proteins in human osteoarthritis. Biochem. Biophys. Res. Commun. 2018, 503, 1273–1277. [Google Scholar] [CrossRef]
- Bersellini Farinotti, A.; Wigerblad, G.; Nascimento, D.; Bas, D.B.; Morado Urbina, C.; Nandakumar, K.S.; Sandor, K.; Xu, B.; Abdelmoaty, S.; Hunt, M.A.; et al. Cartilage-binding antibodies induce pain through immune complex-mediated activation of neurons. J. Exp. Med. 2019, 216, 1904–1924. [Google Scholar] [CrossRef]
- Lees, S.; Golub, S.B.; Last, K.; Zeng, W.; Jackson, D.C.; Sutton, P.; Fosang, A.J. Bioactivity in an Aggrecan 32-mer Fragment Is Mediated via Toll-like Receptor 2. Arthritis Rheumatol. 2015, 67, 1240–1249. [Google Scholar] [CrossRef]
- Miller, R.E.; Ishihara, S.; Tran, P.B.; Golub, S.B.; Last, K.; Miller, R.J.; Fosang, A.J.; Malfait, A.-M. An aggrecan fragment drives osteoarthritis pain through Toll-like receptor 2. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Klatt, A.R.; Klinger, G.; Paul-Klausch, B.; Kuhn, G.; Renno, J.H.; Wagener, R.; Paulsson, M.; Schmidt, J.; Malchau, G.; Wielckens, K. Matrilin-3 activates the expression of osteoarthritis-associated genes in primary human chondrocytes. FEBS Lett. 2009, 583, 3611–3617. [Google Scholar] [CrossRef]
- Ruthard, J.; Kamper, M.; Renno, J.H.; Kuhn, G.; Hillebrand, U.; Hollriegl, S.; Johannis, W.; Zaucke, F.; Klatt, A.R. COMP does not directly modify the expression of genes involved in cartilage homeostasis in contrast to several other cartilage matrix proteins. Connect. Tissue Res. 2014, 55, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Andres Sastre, E.; Zaucke, F.; Witte-Bouma, J.; van Osch, G.; Farrell, E. Cartilage Oligomeric Matrix Protein-Derived Peptides Secreted by Cartilage Do Not Induce Responses Commonly Observed during Osteoarthritis. Cartilage 2020. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.L.; Mienaltowski, M.J.; MacLeod, J.N.; Schittny, J.; Kasper, S.; Fluck, M. Tenascin-C expression controls the maturation of articular cartilage in mice. BMC Res. Notes 2020, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Hasegawa, M.; Iino, T.; Imanaka-Yoshida, K.; Yoshida, T.; Sudo, A. Tenascin-C Prevents Articular Cartilage Degeneration in Murine Osteoarthritis Models. Cartilage 2018, 9, 80–88. [Google Scholar] [CrossRef]
- Hattori, T.; Hasegawa, M.; Unno, H.; Iino, T.; Fukai, F.; Yoshida, T.; Sudo, A. TNIIIA2, The Peptide of Tenascin-C, as a Candidate for Preventing Articular Cartilage Degeneration. Cartilage 2020. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Cain, S.A.; Tian, P.; Baldwin, A.K.; Uppanan, P.; Kielty, C.M.; Kimber, S.J. Recombinant Extracellular Matrix Protein Fragments Support Human Embryonic Stem Cell Chondrogenesis. Tissue Eng. Part A 2018, 24, 968–978. [Google Scholar] [CrossRef]
- Yeh, C.H.; Chen, D.; Aghdasi, B.; Xiao, L.; Ding, M.; Jin, L.; Li, X. Link protein N-terminal peptide and fullerol promote matrix production and decrease degradation enzymes in rabbit annulus cells. Connect. Tissue Res. 2018, 59, 191–200. [Google Scholar] [CrossRef]
- Alaqeel, M.; Grant, M.P.; Epure, L.M.; Salem, O.; AlShaer, A.; Huk, O.L.; Bergeron, S.G.; Zukor, D.J.; Kc, R.; Im, H.J.; et al. Link N suppresses interleukin-1beta-induced biological effects on human osteoarthritic cartilage. Eur. Cell Mater. 2020, 39, 65–76. [Google Scholar] [CrossRef]
- Melrose, J.; Fuller, E.S.; Roughley, P.J.; Smith, M.M.; Kerr, B.; Hughes, C.E.; Caterson, B.; Little, C.B. Fragmentation of decorin, biglycan, lumican and keratocan is elevated in degenerate human meniscus, knee and hip articular cartilages compared with age-matched macroscopically normal and control tissues. Arthritis Res. Ther. 2008, 10. [Google Scholar] [CrossRef]
- Adair-Kirk, T.L.; Senior, R.M. Fragments of extracellular matrix as mediators of inflammation. Int. J. Biochem. Cell Biol. 2008, 40, 1101–1110. [Google Scholar] [CrossRef]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, M.H.J.; van Lent, P.; van der Kraan, P.M. Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthr. Cartil. 2020, 28, 532–543. [Google Scholar] [CrossRef]
- Melchiorri, C.; Meliconi, R.; Frizziero, L.; Silvestri, T.; Pulsatelli, L.; Mazzetti, I.; Borzì, R.M.; Uguccioni, M.; Facchini, A. Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis Rheum. 1998, 41, 2165–2174. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; McCollum, R.; DiBattista, J.; Faure, M.P.; Chin, J.A.; Fournier, S.; Sarfati, M.; Pelletier, J.-P. The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis Rheum. 1992, 35, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Sadouk, M.B.; Pelletier, J.P.; Tardif, G.; Kiansa, K.; Cloutier, J.M.; Martel-Pelletier, J. Human synovial fibroblasts coexpress IL-1 receptor type I and type II mRNA. The increased level of the IL-1 receptor in osteoarthritic cells is related to an increased level of the type I receptor. Lab. Investig. J. Tech. Methods Pathol. 1995, 73, 347–355. [Google Scholar]
- Malemud, C.J. Anticytokine therapy for osteoarthritis: Evidence to date. Drugs Aging 2010, 27, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Grassel, S.; Muschter, D. Recent advances in the treatment of osteoarthritis. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Vincent, T.L. IL-1 in osteoarthritis: Time for a critical review of the literature. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Saklatvala, J. Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature 1986, 322, 547–549. [Google Scholar] [CrossRef]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Huang, W.; Dehnade, F.; Ahmad, M.; Zafarullah, M. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by MAP kinases, AP-1, and NF-kappaB transcription factors in articular chondrocytes. Exp. Cell Res. 2003, 288, 208–217. [Google Scholar] [CrossRef]
- Vincent, T.L. Of mice and men: Converging on a common molecular understanding of osteoarthritis. Lancet Rheumatol. 2020, 2, e633–e645. [Google Scholar] [CrossRef]
- Chow, Y.Y.; Chin, K.Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 8293921. [Google Scholar] [CrossRef] [PubMed]
- Stannus, O.; Jones, G.; Cicuttini, F.; Parameswaran, V.; Quinn, S.; Burgess, J.; Ding, C. Circulating levels of IL-6 and TNF-alpha are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr. Cartil. 2010, 18, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 2017, 76, 748–755. [Google Scholar] [CrossRef]
- Richette, P.; Latourte, A.; Sellam, J.; Wendling, D.; Piperno, M.; Goupille, P.; Pers, Y.-M.; Eymard, F.; Ottaviani, S.; Ornetti, P.; et al. Efficacy of tocilizumab in patients with hand osteoarthritis: Double blind, randomised, placebo-controlled, multicentre trial. Ann. Rheum. Dis. 2020, 80. [Google Scholar] [CrossRef]
- Raghu, H.; Lepus, C.M.; Wang, Q.; Wong, H.H.; Lingampalli, N.; Oliviero, F.; Punzi, L.; Giori, N.J.; Goodman, S.B.; Chu, C.R.; et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann. Rheum. Dis. 2017, 76, 914–922. [Google Scholar] [CrossRef]
- Xu, Z.; Li, J.; Yang, H.; Jiang, L.; Zhou, X.; Huang, Y.; Xu, N. Association of CCL2 Gene Variants with Osteoarthritis. Arch. Med. Res. 2019, 50, 86–90. [Google Scholar] [CrossRef]
- Yuan, G.H.; Masuko-Hongo, K.; Sakata, M.; Tsuruha, J.; Onuma, H.; Nakamura, H.; Aoki, H.; Kato, T.; Nishioka, K. The role of C-C chemokines and their receptors in osteoarthritis. Arthritis Rheum. 2001, 44, 1056–1070. [Google Scholar] [CrossRef]
- Sherwood, J.; Bertrand, J.; Nalesso, G.; Poulet, B.; Pitsillides, A.; Brandolini, L.; Karystinou, A.; De Bari, C.; Luyten, F.P.; Pitzalis, C.; et al. A homeostatic function of CXCR2 signalling in articular cartilage. Ann. Rheum. Dis. 2015, 74, 2207–2215. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef]
- Komatsu, M.; Nakamura, Y.; Maruyama, M.; Abe, K.; Watanapokasin, R.; Kato, H. Expression profiles of human CCN genes in patients with osteoarthritis or rheumatoid arthritis. J. Orthop. Sci. 2015, 20, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Peng, H.; Yao, R.; Zhang, Z.; Mao, G.; Yu, H.; Qiu, Y. Inhibition of cellular communication network factor 1 (CCN1)-driven senescence slows down cartilage inflammaging and osteoarthritis. Bone 2020, 139, 115522. [Google Scholar] [CrossRef] [PubMed]
- Chijiiwa, M.; Mochizuki, S.; Kimura, T.; Abe, H.; Tanaka, Y.; Fujii, Y.; Shimizu, H.; Enomoto, H.; Toyama, Y.; Okada, Y. CCN1 (Cyr61) Is Overexpressed in Human Osteoarthritic Cartilage and Inhibits ADAMTS-4 (Aggrecanase 1) Activity. Arthritis Rheumatol. 2015, 67, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, M.H.; Blom, A.B.; Kram, V.; Maeda, A.; Sikka, S.; Gabet, Y.; Kilts, T.M.; van Den Berg, W.B.; van Lent, P.L.; van der Kraan, P.M.; et al. WISP1/CCN4 aggravates cartilage degeneration in experimental osteoarthritis. Osteoarthr. Cartil. 2017, 25, 1900–1911. [Google Scholar] [CrossRef]
- Seymour, J.F.; Keating, M.J. Parathyroid hormone related protein in hypercalcaemia in CLL. Br. J. Haematol. 1995, 89, 685–686. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Kubota, S.; Takigawa, M. In Vivo Evaluation of Cartilage Regenerative Effects of CCN2 Protein. Methods Mol. Biol. 2017, 1489, 273–282. [Google Scholar] [PubMed]
- Hayashi, M.; Muneta, T.; Takahashi, T.; Ju, Y.J.; Tsuji, K.; Sekiya, I. Intra-articular injections of bone morphogenetic protein-7 retard progression of existing cartilage degeneration. J. Orthop. Res. 2010, 28, 1502–1506. [Google Scholar] [CrossRef]
- Cucchiarini, M.; Madry, H. Overexpression of human IGF-I via direct rAAV-mediated gene transfer improves the early repair of articular cartilage defects in vivo. Gene Ther. 2014, 21, 811–919. [Google Scholar] [CrossRef] [PubMed]
- Gigout, A.; Guehring, H.; Froemel, D.; Meurer, A.; Ladel, C.; Reker, D.; Bay-Jensen, A.; Karsdal, M.; Lindemann, S. Sprifermin (rhFGF18) enables proliferation of chondrocytes producing a hyaline cartilage matrix. Osteoarthr. Cartil. 2017, 25, 1858–1867. [Google Scholar] [CrossRef]
- Muller, S.; Lindemann, S.; Gigout, A. Effects of Sprifermin, IGF1, IGF2, BMP7, or CNP on Bovine Chondrocytes in Monolayer and 3D Culture. J. Orthop. Res. 2020, 38, 653–662. [Google Scholar] [CrossRef]
- Eckstein, F.; Kraines, J.L.; Aydemir, A.; Wirth, W.; Maschek, S.; Hochberg, M.C. Intra-articular sprifermin reduces cartilage loss in addition to increasing cartilage gain independent of location in the femorotibial joint: Post-hoc analysis of a randomised, placebo-controlled phase II clinical trial. Ann. Rheum. Dis. 2020, 79, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Zeng, N.; Chen, X.-Y.; Yan, Z.-P.; Li, J.-T.; Liao, T.; Ni, G.-X. Efficacy and safety of sprifermin injection for knee osteoarthritis treatment: A meta-analysis. Arthritis Res. 2021, 23, 1–10. [Google Scholar] [CrossRef]
- Zhang, W.; Robertson, W.B.; Zhao, J.; Chen, W.; Xu, J. Emerging Trend in the Pharmacotherapy of Osteoarthritis. Front. Endocrinol. 2019, 10, 431. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.S.; Mithoefer, K. Current Applications of Growth Factors for Knee Cartilage Repair and Osteoarthritis Treatment. Curr. Rev. Musculoskelet. Med. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, X.; Xing, L.; Tian, F. Wnt signaling: A promising target for osteoarthritis therapy. Cell Commun. Signal. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Deshmukh, V.; O’Green, A.; Bossard, C.; Seo, T.; Lamangan, L.; Ibanez, M.; Ghias, A.; Lai, C.; Do, L.; Cho, S.; et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr. Cartil. 2019, 27, 1347–1360. [Google Scholar] [CrossRef]
- Deshmukh, V.; Hu, H.; Barroga, C.; Bossard, C.; Kc, S.; Dellamary, L.; Stewart, J.; Chiu, K.; Ibanez, M.; Pedraza, M.; et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 2018, 26, 18–27. [Google Scholar] [CrossRef]
- Yazici, Y.; McAlindon, T.; Fleischmann, R.; Gibofsky, A.; Lane, N.; Kivitz, A.; Skrepnik, N.; Armas, E.; Swearingen, C.; DiFrancesco, A.; et al. A novel Wnt pathway inhibitor, SM04690, for the treatment of moderate to severe osteoarthritis of the knee: Results of a 24-week, randomized, controlled, phase 1 study. Osteoarthr. Cartil. 2017, 25, 1598–1606. [Google Scholar] [CrossRef]
- Yazici, Y.; McAlindon, T.E.; Gibofsky, A.; Lane, N.E.; Clauw, D.; Jones, M.; Bergfeld, J.; Swearingen, C.J.; DiFrancesco, A.; Simsek, I.; et al. Lorecivivint, a Novel Intra-articular CLK/DYRK1A Inhibitor and Wnt Pathway Modulator for Treatment of Knee Osteoarthritis: A Phase 2 Randomized Trial. Arthritis Rheumatol 2020, 72, 1694–1706. [Google Scholar] [CrossRef]
- Sabha, M.; Siaton, B.C.; Hochberg, M.C. Lorecivivint, an intra-articular potential disease-modifying osteoarthritis drug. Expert Opin. Investig. Drugs 2020, 29, 1339–1346. [Google Scholar] [CrossRef]
- Hukkanen, M.; Konttinen, Y.T.; Rees, R.G.; Santavirta, S.; Terenghi, G.; Polak, J.M. Distribution of nerve endings and sensory neuropeptides in rat synovium, meniscus and bone. Int. J. Tissue React. 1992, 14, 1–10. [Google Scholar]
- Suri, S.; Gill, S.E.; Massena, D.C.; Wilson, D.; McWilliams, D.F.; Walsh, D.A. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann. Rheum. Dis. 2007, 66, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Aso, K.; Shahtaheri, S.M.; Hill, R.; Wilson, D.; McWilliams, D.F.; Nwosu, L.N.; Chapman, V.; Walsh, D. Contribution of nerves within osteochondral channels to osteoarthritis knee pain in humans and rats. Osteoarthr. Cartil. 2020, 28, 1245–1254. [Google Scholar] [CrossRef]
- Koeck, F.X.; Schmitt, M.; Baier, C.; Stangl, H.; Beckmann, J.; Grifka, J.; Straub, R.H. Predominance of synovial sensory nerve fibers in arthrofibrosis following total knee arthroplasty compared to osteoarthritis of the knee. J. Orthop. Surg. Res. 2016, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Kawarai, Y.; Orita, S.; Nakamura, J.; Miyamoto, S.; Suzuki, M.; Inage, K.; Hagiwara, S.; Suzuki, T.; Nakajima, T.; Akazawa, T.; et al. Changes in proinflammatory cytokines, neuropeptides, and microglia in an animal model of monosodium iodoacetate-induced hip osteoarthritis. J. Orthop. Res. 2018, 36, 2978–2986. [Google Scholar] [CrossRef] [PubMed]
- Omae, T.; Nakamura, J.; Ohtori, S.; Orita, S.; Yamauchi, K.; Miyamoto, S.; Hagiwara, S.; Kishida, S.; Takahashi, K. A novel rat model of hip pain by intra-articular injection of nerve growth factor-characteristics of sensory innervation and inflammatory arthritis. Mod. Rheumatol. 2015, 25, 931–936. [Google Scholar] [CrossRef]
- Miyamoto, S.; Nakamura, J.; Ohtori, S.; Orita, S.; Nakajima, T.; Omae, T.; Hagiwara, S.; Takazawa, M.; Suzuki, M.; Suzuki, T.; et al. Pain-related behavior and the characteristics of dorsal-root ganglia in a rat model of hip osteoarthritis induced by mono-iodoacetate. J. Orthop. Res. 2017, 35, 1424–1430. [Google Scholar] [CrossRef]
- Miller, R.; Tran, P.; Ishihara, S.; Syx, D.; Ren, D.; Valdes, A.; Malfait, A. Microarray analyses of the dorsal root ganglia support a role for innate neuro-immune pathways in persistent pain in experimental osteoarthritis. Osteoarthr. Cartil. 2020, 28, 581–592. [Google Scholar] [CrossRef]
- Grassel, S.G. The role of peripheral nerve fibers and their neurotransmitters in cartilage and bone physiology and pathophysiology. Arthritis Res. Ther. 2014, 16, 485. [Google Scholar] [CrossRef]
- Murakami, K.; Nakagawa, H.; Nishimura, K.; Matsuo, S. Changes in peptidergic fiber density in the synovium of mice with collagenase-induced acute arthritis. Can. J. Physiol. Pharmacol. 2015, 93, 435–441. [Google Scholar] [CrossRef]
- Buma, P.; Verschuren, C.; Versleyen, D.; Van der, K.P.; Oestreicher, A.B. Calcitonin gene-related peptide, substance P and GAP-43/B-50 immunoreactivity in the normal and arthrotic knee joint of the mouse. Histochemistry 1992, 98, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Koshino, T. Distribution of neuropeptides in synovium of the knee with osteoarthritis. Clin. Orthop. Relat. Res. 2000, 376, 172–182. [Google Scholar] [CrossRef]
- Saxler, G.; Loer, F.; Skumavc, M.; Pfortner, J.; Hanesch, U. Localization of SP- and CGRP-immunopositive nerve fibers in the hip joint of patients with painful osteoarthritis and of patients with painless failed total hip arthroplasties. Eur. J. Pain 2007, 11, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Gronblad, M.; Konttinen, Y.T.; Korkala, O.; Liesi, P.; Hukkanen, M.; Polak, J.M. Neuropeptides in synovium of patients with rheumatoid arthritis and osteoarthritis. J. Rheumatol. 1988, 15, 1807–1810. [Google Scholar]
- Li, F.X.; Xu, F.; Lin, X.; Wu, F.; Zhong, J.Y.; Wang, Y.; Guo, B.; Zheng, M.-H.; Shan, S.-K.; Yuan, L.-Q. The Role of Substance P in the Regulation of Bone and Cartilage Metabolic Activity. Front. Endocrinol. 2020, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Muschter, D.; Fleischhauer, L.; Taheri, S.; Schilling, A.F.; Clausen-Schaumann, H.; Grassel, S. Sensory neuropeptides are required for bone and cartilage homeostasis in a murine destabilization-induced osteoarthritis model. Bone 2020, 133, 115181. [Google Scholar] [CrossRef]
- Warner, S.C.; Walsh, D.A.; Laslett, L.L.; Maciewicz, R.A.; Soni, A.; Hart, D.J.; Zhang, W.; Muir, K.R.; Dennison, E.M.; Leaverton, P.; et al. Pain in knee osteoarthritis is associated with variation in the neurokinin 1/substance P receptor (TACR1) gene. Eur. J. Pain 2017, 21, 1277–1284. [Google Scholar] [CrossRef]
- Millward-Sadler, S.J.; Mackenzie, A.; Wright, M.O.; Lee, H.S.; Elliot, K.; Gerrard, L.; Fiskerstrand, C.E.; Salter, D.M.; Quinn, J.P. Tachykinin expression in cartilage and function in human articular chondrocyte mechanotransduction. Arthritis Rheum. 2003, 48, 146–156. [Google Scholar] [CrossRef]
- Howard, M.R.; Millward-Sadler, S.J.; Vasilliou, A.S.; Salter, D.M.; Quinn, J.P. Mechanical stimulation induces preprotachykinin gene expression in osteoarthritic chondrocytes which is correlated with modulation of the transcription factor neuron restrictive silence factor. Neuropeptides 2008, 42, 681–686. [Google Scholar] [CrossRef]
- Xiao, J.; Yu, W.; Wang, X.; Wang, B.; Chen, J.; Liu, Y.; Li, Z. Correlation between neuropeptide distribution, cancellous bone microstructure and joint pain in postmenopausal women with osteoarthritis and osteoporosis. Neuropeptides 2016, 56, 97–104. [Google Scholar] [CrossRef]
- He, L.; He, T.; Xing, J.; Zhou, Q.; Fan, L.; Liu, C.; Chen, Y.; Wu, D.; Tian, Z.; Liu, B.; et al. Bone marrow mesenchymal stem cell-derived exosomes protect cartilage damage and relieve knee osteoarthritis pain in a rat model of osteoarthritis. Stem Cell Res. Ther. 2020, 11, 276. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Miyazaki, T.; Watanabe, S.; Takahashi, A.; Honjoh, K.; Nakajima, H.; Oki, H.; Kokubo, Y.; Matsumine, A. Intraarticular injection of processed lipoaspirate cells has anti-inflammatory and analgesic effects but does not improve degenerative changes in murine monoiodoacetate-induced osteoarthritis. BMC Musculoskelet. Disord. 2019, 20, 335. [Google Scholar] [CrossRef] [PubMed]
- Araya, N.; Miyatake, K.; Tsuji, K.; Katagiri, H.; Nakagawa, Y.; Hoshino, T.; Onuma, H.; An, S.; Nishio, H.; Saita, Y.; et al. Intra-articular Injection of Pure Platelet-Rich Plasma Is the Most Effective Treatment for Joint Pain by Modulating Synovial Inflammation and Calcitonin Gene-Related Peptide Expression in a Rat Arthritis Model. Am. J. Sports Med. 2020, 48, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Tsuji, K.; Onuma, H.; Udo, M.; Ueki, H.; Akiyama, M.; Abula, K.; Katagiri, H.; Miyatake, K.; Watanabe, T.; et al. Persistent synovial inflammation plays important roles in persistent pain development in the rat knee before cartilage degradation reaches the subchondral bone. BMC Musculoskelet. Disord. 2018, 19, 291. [Google Scholar] [CrossRef]
- Nakasa, T.; Ishikawa, M.; Takada, T.; Miyaki, S.; Ochi, M. Attenuation of cartilage degeneration by calcitonin gene-related paptide receptor antagonist via inhibition of subchondral bone sclerosis in osteoarthritis mice. J. Orthop. Res. 2016, 34. [Google Scholar] [CrossRef]
- Aso, K.; Izumi, M.; Sugimura, N.; Okanoue, Y.; Ushida, T.; Ikeuchi, M. Nociceptive phenotype alterations of dorsal root ganglia neurons innervating the subchondral bone in osteoarthritic rat knee joints. Osteoarthr. Cartil. 2016, 24, 1596–1603. [Google Scholar] [CrossRef]
- Zhu, S.; Zhu, J.; Zhen, G.; Hu, Y.; An, S.; Li, Y.; Zheng, Q.; Chen, Z.; Yang, Y.; Wan, M.; et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J. Clin. Investig. 2019, 129. [Google Scholar] [CrossRef]
- Allen, P.I.; Conzemius, M.G.; Evans, R.B.; Kiefer, K. Correlation between synovial fluid cytokine concentrations and limb function in normal dogs and in dogs with lameness from spontaneous osteoarthritis. Vet. Surg. 2019, 48, 770–779. [Google Scholar] [CrossRef]
- Inoue, H.; Shimoyama, Y.; Hirabayashi, K.; Kajigaya, H.; Yamamoto, S.; Oda, H.; Koshihara, Y. Production of neuropeptide substance P by synovial fibroblasts from patients with rheumatoid arthritis and osteoarthritis. Neurosci. Lett. 2001, 303, 149–152. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; He, J.Y.; Zheng, X.F.; Li, D.; Li, Z.; Zhu, J.-F.; Shen, C.; Cai, G.-Q.; Chen, X.-D. Increasing expression of substance P and calcitonin gene-related peptide in synovial tissue and fluid contribute to the progress of arthritis in developmental dysplasia of the hip. Arthritis Res. Ther. 2015, 17, 4. [Google Scholar] [CrossRef]
- Dong, T.; Chang, H.; Zhang, F.; Chen, W.; Zhu, Y.; Wu, T.; Zhang, Y. Calcitonin gene-related peptide can be selected as a predictive biomarker on progression and prognosis of knee osteoarthritis. Int. Orthop. 2015, 39, 1237–1243. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grässel, S.; Zaucke, F.; Madry, H. Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology. J. Clin. Med. 2021, 10, 1938. https://doi.org/10.3390/jcm10091938
Grässel S, Zaucke F, Madry H. Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology. Journal of Clinical Medicine. 2021; 10(9):1938. https://doi.org/10.3390/jcm10091938
Chicago/Turabian StyleGrässel, Susanne, Frank Zaucke, and Henning Madry. 2021. "Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology" Journal of Clinical Medicine 10, no. 9: 1938. https://doi.org/10.3390/jcm10091938
APA StyleGrässel, S., Zaucke, F., & Madry, H. (2021). Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology. Journal of Clinical Medicine, 10(9), 1938. https://doi.org/10.3390/jcm10091938