
Exercise Testing, Physical Training and Fatigue in Patients with Mitochondrial Myopathy Related to mtDNA Mutations


Abstract
1. Introduction
2. Oxygen Delivery during Exercise in Healthy Skeletal Muscle
2.1. Oxygen Delivery: From Air to Contracting Muscle
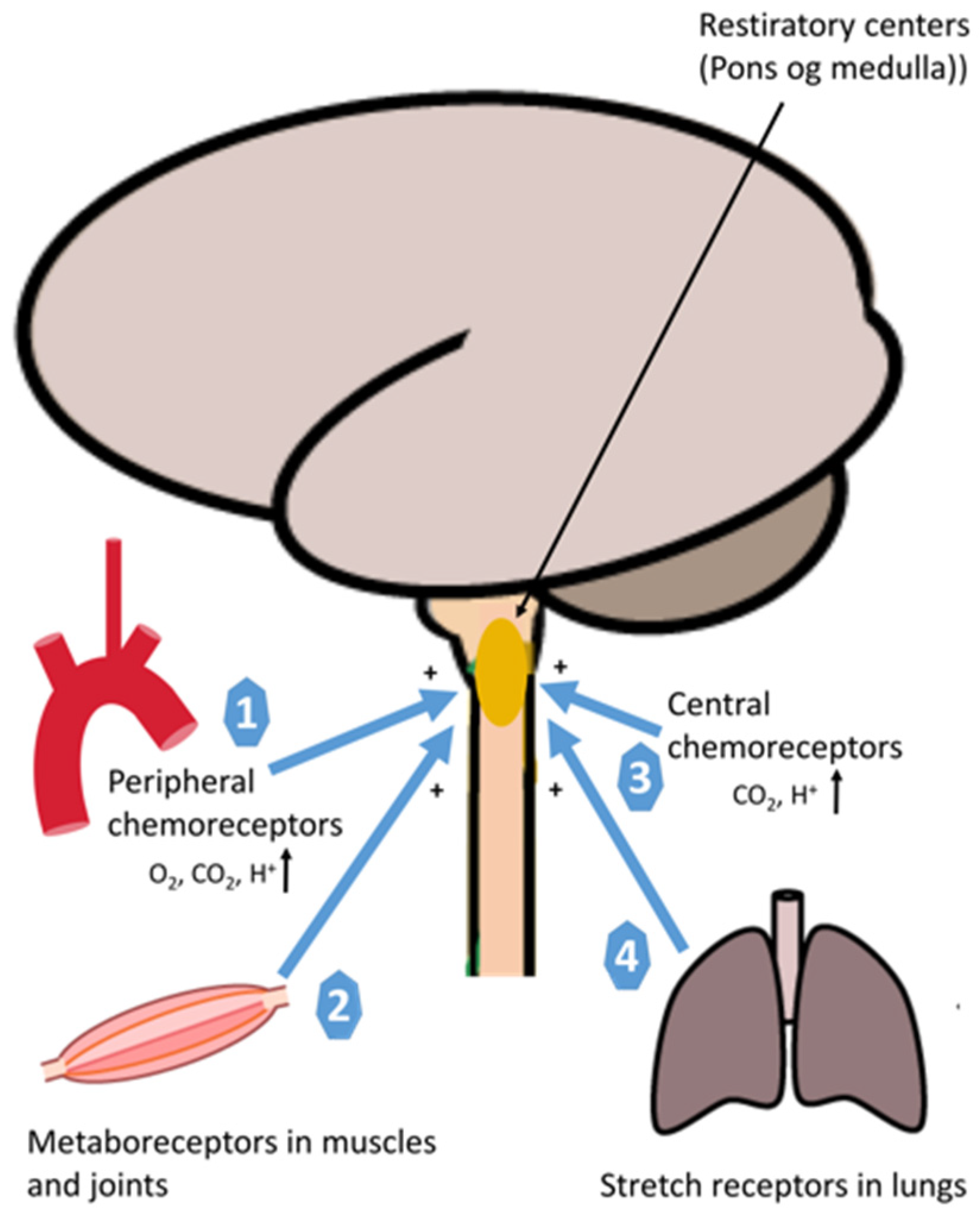
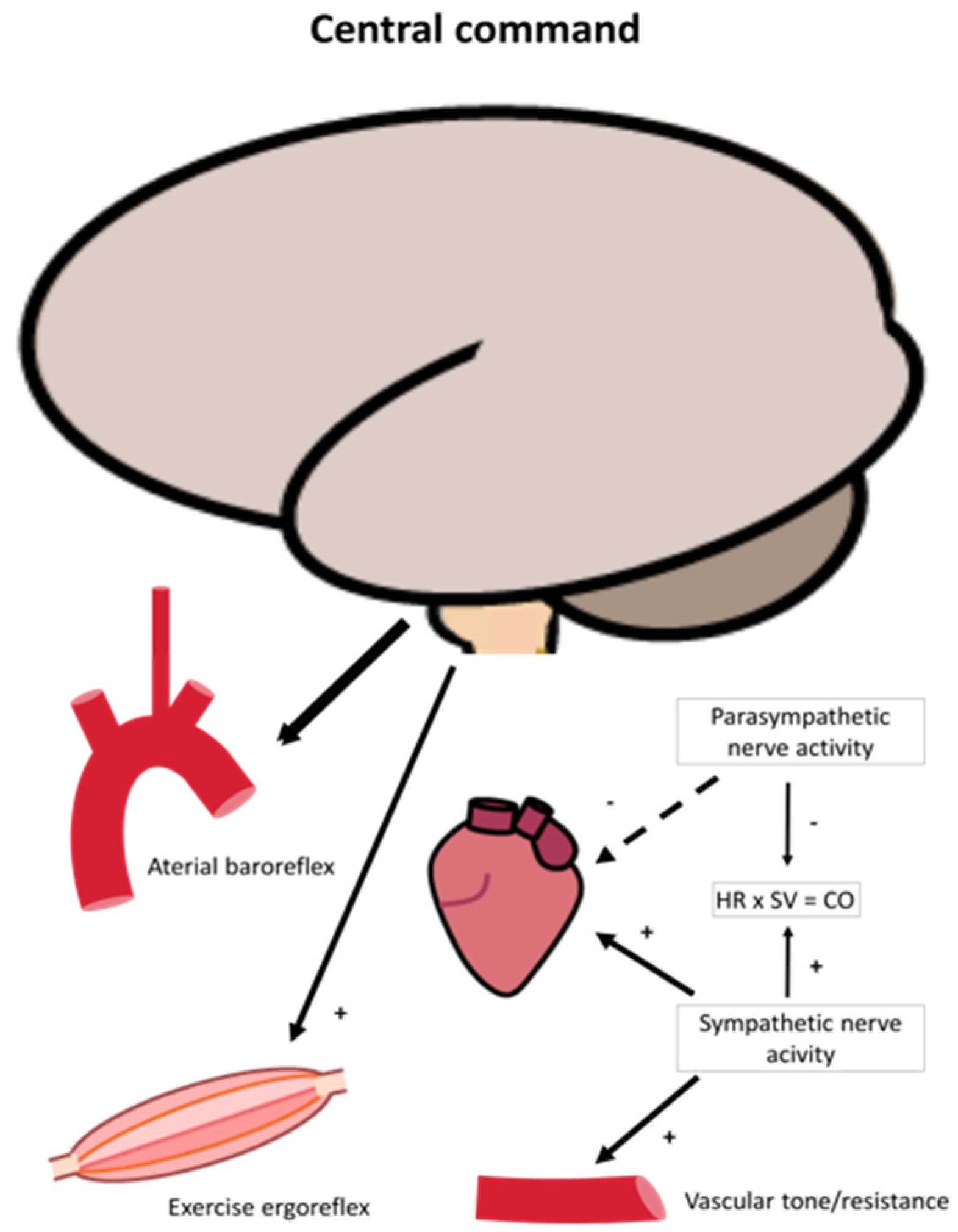
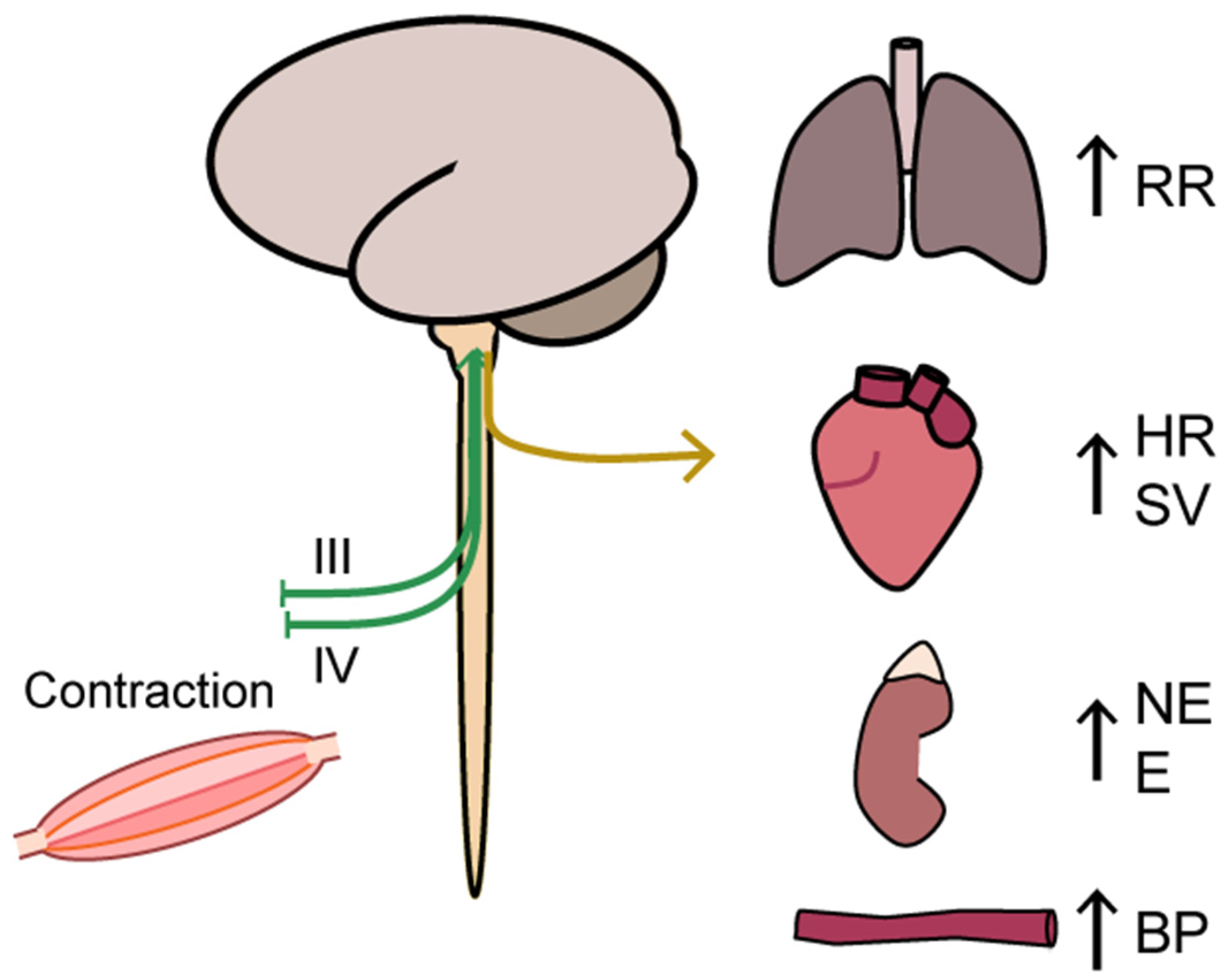
2.2. Oxygen Delivery: Neural Regulation
2.3. Oxygen Delivery: Rate Limiting Step
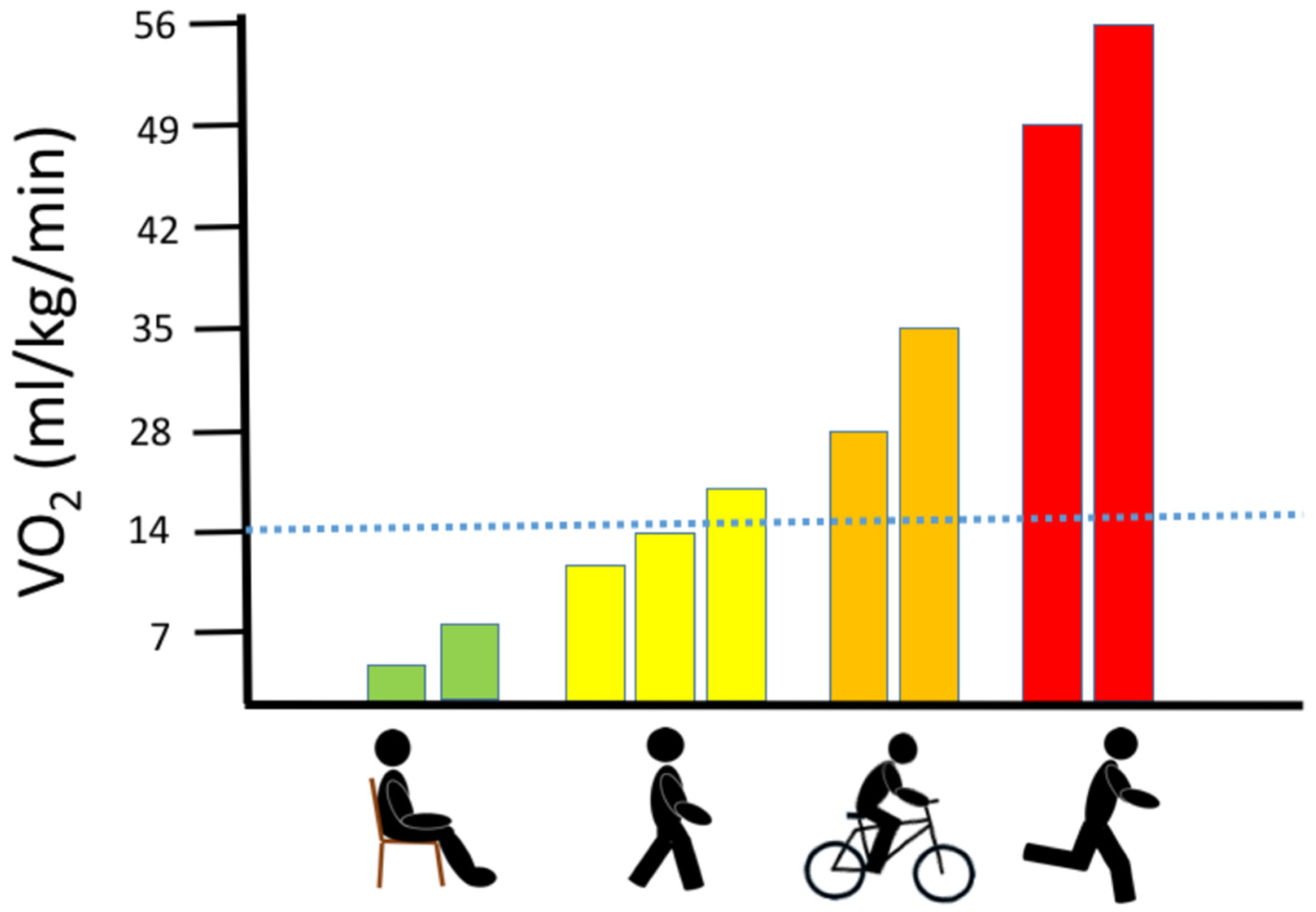
3. Oxygen Consumption during Exercise in Healthy Muscle
4. Exercise Testing
4.1. Whole-Body Exercise
4.1.1. Whole-Body Exercise: Maximal Oxygen Uptake (VO2max)
4.1.2. Whole-Body Exercise: Ventilation Rate
4.1.3. Whole-Body Exercise: Oxygen Delivery and Extraction
4.1.4. Whole-Body Exercise: Autonomic Nervous System Regulation
4.1.5. Whole-Body Exercise: Lactate Turnover
4.1.6. Whole-Body Exercise: Diagnostic Yield
4.1.7. Whole-Body Exercise: GDF-15 as a Diagnostic Biomarker
4.2. One-Extremity Exercise
4.2.1. One-Extremity Exercise: Oxygen Delivery-Extraction and Oxidative Capacity
4.2.2. One-Extremity Exercise: Lactate
5. Outcome Measures
5.1. Maximal Exercise Testing
5.2. Submaximal Exercise Testing
6. Physical Fatigue
7. Exercise Training
8. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Chinnery, P.F. Mitochondrial Disorders Overview. In GeneReviews®; Pagon, R.A., Adam, M.P., Bird, T.D., Dolan, C.R., Fong, C.-T., Smith, R.J., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Jeppesen, T.D.; Schwartz, M.; Olsen, D.B.; Vissing, J. Oxidative capacity correlates with muscle mutation load in mitochondrial myopathy. Ann. Neurol. 2003, 54, 86–92. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Schwartz, M.; Frederiksen, A.L.; Wibrand, F.; Olsen, D.B.; Vissing, J. Muscle Phenotype and Mutation Load in 51 Persons with the 3243A>G Mitochondrial DNA Mutation. Arch. Neurol. 2006, 63, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S. Mitochondrial myopathies. Curr. Opin. Rheumatol. 2006, 18, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.; Minetti, C.; Moggio, M.; Mongini, T.; Servidei, S.; Tonin, P.; et al. Fatigue and exercise intolerance in mitochondrial diseases. Literature revision and experience of the Italian Network of mitochondrial diseases. Neuromuscul. Disord. 2012, 22, S226–S229. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.; Saltin, B. Maximal perfusion of skeletal muscle in man. J. Physiol. 1985, 366, 233–249. [Google Scholar] [CrossRef]
- Taivassalo, T.; Jensen, T.D.; Kennaway, N.; DiMauro, S.; Vissing, J.; Haller, R.G. The spectrum of exercise tolerance in mitochondrial myopathies: A study of 40 patients. Brain 2003, 126 Pt 2, 413–423. [Google Scholar] [CrossRef]
- Frederiksen, A.L.; Andersen, P.H.; Kyvik, K.O.; Jeppesen, T.D.; Vissing, J.; Schwartz, M. Tissue specific distribution of the 3243A->G mtDNA mutation. J. Med. Genet. 2006, 43, 671–677. [Google Scholar] [CrossRef]
- Bassett, D.R., Jr.; Howley, E.T. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med. Sci. Sports Exerc. 2000, 32, 70–84. [Google Scholar] [CrossRef]
- Taivassalo, T.; Abbott, A.; Wyrick, P.; Haller, R.G. Venous oxygen levels during aerobic forearm exercise: An index of impaired oxidative metabolism in mitochondrial myopathy. Ann. Neurol. 2002, 51, 38–44. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Vissing, J.; González-Alonso, J. Influence of erythrocyte oxygenation and intravascular ATP on resting and exer-cising skeletal muscle blood flow in humans with mitochondrial myopathy. Mitochondrion 2012, 12, 414–422. [Google Scholar] [CrossRef]
- Hammarén, E.; Rafsten, L.; Kreuter, M.; Lindberg, C. Modified Exercise Test in Screening for Mitochondrial Myopathies—Adjustment of Workload in Relation to Muscle Strength. Eur. Neurol. 2004, 51, 38–41. [Google Scholar] [CrossRef]
- Vissing, J.; Galbo, H.; Haller, R.G. Exercise fuel mobilization in mitochondrial myopathy: A metabolic dilemma. Ann. Neurol. 1996, 40, 655–662. [Google Scholar] [CrossRef]
- Vissing, J.; Gansted, U.; Quistorff, B. Exercise intolerance in mitochondrial myopathy is not related to lactic acidosis. Ann. Neurol. 2001, 49, 672–676. [Google Scholar] [CrossRef]
- Delaney, N.F.; Sharma, R.; Tadvalkar, L.; Clish, C.B.; Haller, R.G.; Mootha, V.K. Metabolic profiles of exercise in patients with McArdle disease or mitochondrial myopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 8402–8407. [Google Scholar] [CrossRef]
- Ng, K.; Winter, S.; Sue, C.; Burke, D. Preserved motor axonal membrane potential in mitochondrial disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 844–846. [Google Scholar] [CrossRef] [PubMed]
- Steensberg, A.; Vissing, J.; Pedersen, B.K. Lack of IL-6 production during exercise in patients with mitochondrial myopathy. Eur. J. Appl. Physiol. 2001, 84, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, K.; Taivassalo, T.; Wyrick, P.; Wood, H.; Babb, T.G.; Haller, R.G. Exertional dyspnea in mitochondrial myopathy: Clinical features and physiological mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R873–R884. [Google Scholar] [CrossRef] [PubMed]
- Dandurand, R.J.; Matthews, P.M.; Arnold, D.L.; Eidelman, D.H. Mitochondrial disease. Pulmonary function, exercise performance, and blood lactate levels. Chest 1995, 108, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Reproducibility of the lactate stress test. Metab. Brain Dis. 2003, 18, 155–160. [Google Scholar] [CrossRef]
- Brooke, M.H.; Carroll, J.E.; Davis, J.E.; Hagberg, J.M. The prolonged exercise test. Neurology 1979, 29, 636. [Google Scholar] [CrossRef]
- Tarnopolsky, M. Exercise testing as a diagnostic entity in mitochondrial myopathies. Mitochondrion 2004, 4, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Haller, R.G.; Mukherjee, A.; A Gaffney, F.; Blomquist, C.G. Mitochondrial myopathy presenting as exercise intolerance. Trans. Am. Neurol. Assoc. 1978, 103, 6–10. [Google Scholar] [PubMed]
- Trenell, M.I.; Sue, C.M.; Thompson, C.H.; Kemp, G.J. Supplemental oxygen and muscle metabolism in mitochondrial myopathy patients. Eur. J. Appl. Physiol. 2007, 99, 541–547. [Google Scholar] [CrossRef]
- Gimenes, A.C.; Neder, J.A.; Dal Corso, S.; Nogueira, C.R.; Nápolis, L.; Mello, M.T.; Bulle, A.S.; Nery, L.E. Relationship between work rate and oxygen uptake in mitochondrial myopathy during ramp-incremental exercise. Braz. J. Med. Biol. Res. 2011, 44, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, H.; Löfberg, M.; Somer, H.; Näveri, H.; Sovijärvi, A. Abnormal blood lactate accumulation after exercise in patients with multiple mitochondrial DNA deletions and minor muscular symptoms. Clin. Physiol. Funct. Imaging 2004, 24, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Grassi, B.; Porcelli, S.; Marzorati, M. Translational Medicine: Exercise Physiology Applied to Metabolic Myopathies. Med. Sci. Sports Exerc. 2019, 51, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Bravo, D.M.; Gimenes, A.C.; Nascimento, R.B.; Ferreira, E.V.M.; Siqueira, A.C.B.; Meda, E.D.S.; Neder, J.A.; Nery, L.E. Skeletal muscle reoxygenation after high-intensity exercise in mitochondrial myopathy. Eur. J. Appl. Physiol. 2011, 112, 1763–1771. [Google Scholar] [CrossRef]
- Galán, F.; De Lavera, I.; Cotán, D.; Sánchez-Alcázar, J.A. Mitochondrial Myopathy in Follow-up of a Patient With Chronic Fatigue Syndrome. J. Investig. Med. High Impact Case Rep. 2015, 3. [Google Scholar] [CrossRef]
- Parikh, S.; Galioto, R.; Lapin, B.; Haas, R.; Hirano, M.; Koenig, M.K.; Saneto, R.P.; Zolkipli-Cunningham, Z.; Goldstein, A.; Karaa, A. Fatigue in primary genetic mitochondrial disease: No rest for the weary. Neuromuscul. Disord. 2019, 29, 895–902. [Google Scholar] [CrossRef]
- Barcia, G.; Khirani, S.; Amaddeo, A.; Assouline, Z.; Pennisi, A.; Boddaert, N.; Romero, N.; Desguerre, I.; Schiff, M.; Rötig, A.; et al. Evidence of diaphragmatic dysfunction with severe alveolar hypoventilation syndrome in mitochondrial respiratory chain deficiency. Neuromuscul. Disord. 2020, 30, 593–598. [Google Scholar] [CrossRef]
- Uchida, K.; Murata, K.; Kobayashi, S.; Nakamura, H.; Wada, Y.; Okuda, S.; Oshita, C.; Susa, T.; Murakami, W.; Matsuzaki, M. A case of mitochondrial disease with severe left ventricular hypertrophy. J. Med. Ultrason. 2011, 38, 157–159. [Google Scholar] [CrossRef]
- Xu, S.; Xu, X.; Zhang, J.; Ying, K.; Shao, Y.; Zhang, R. Pulmonary hypertension as a manifestation of mitochondrial disease: A case report and review of the literature. Medicine 2017, 96, e8716. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.H.; Sproule, B.J.; Chapman, C.B. The physiological meaning of the maximal oxygen intake test. Clin. Investig. 1958, 37, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, J.A.; Vidruk, E.H.; Mitchell, G.S. Pulmonary control systems in exercise: Update. Fed. Proc. 1985, 44, 2260–2270. [Google Scholar] [PubMed]
- Ward, S.A. Peripheral and central chemoreceptor control of ventilation during exercise in humans. Can. J. Appl. Physiol. 1994, 19, 305–333. [Google Scholar] [CrossRef] [PubMed]
- Saltin, B. Exercise hyperaemia: Magnitude and aspects on regulation in humans. J. Physiol. 2007, 583 Pt 3, 819–823. [Google Scholar] [CrossRef]
- Saltin, B.; Strange, S. Maximal oxygen uptake: “old” and “new” arguments for a cardiovascular limitation. Med. Sci. Sports Exerc. 1992, 24, 30–37. [Google Scholar] [CrossRef] [PubMed]
- A Davis, J.; Whipp, B.J.; Lamarra, N.; Huntsman, D.J.; Frank, M.H.; Wasserman, K. Effect of ramp slope on determination of aerobic parameters from the ramp exercise test. Med. Sci. Sports Exerc. 1982, 14, 339–343. [Google Scholar] [CrossRef]
- Martin, B.J.; Weil, J.V. CO2 and exercise tidal volume. J. Appl. Physiol. 1979, 46, 322–325. [Google Scholar] [CrossRef]
- Goldstein, I.; Goldstein, S.; Urbanetti, J.A.; Anthonisen, N.R. Effects of expiratory threshold loading during steady-state exercise. J. Appl. Physiol. 1975, 39, 697–701. [Google Scholar] [CrossRef]
- Forster, H.V.; Forster, H.V.; Pan, L.G.; Pan, L.G. Breathing During Exercise: Demands, Regulation, Limitations. Adv. Exp. Med. Biol. 1988, 227, 257–276. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.A.; Bassett, J.; Hughes, P.; Gass, G.C. Anaerobic threshold and lactate turnpoint. Eur. J. Appl. Physiol. 1983, 50, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Bruce, R.M.; Jolley, C.; White, M.J. Control of exercise hyperpnoea: Contributions from thin-fibre skeletal muscle afferents. Exp. Physiol. 2019, 104, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Forster, H.V.; Pan, L.G. The role of the carotid chemoreceptors in the control of breathing during exercise. Med. Sci. Sports Exerc. 1994, 26, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-J.; Zucker, I.H.; Wang, W. Muscle reflex in heart failure: The role of exercise training. Front. Physiol. 2012, 3, 398. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; Ferrari, A.; Gregorini, L.; Leonetti, G.; Parati, G.; Picotti, G.B.; Ravazzani, C.; Zanchetti, A. Plasma catecholamines do not invariably reflect sym-pathetically induced changes in blood pressure in man. Clin. Sci. 1983, 65, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Parati, G.; Pomidossi, G.; Grassi, G.; Gavazzi, C.; Ramirez, A.; Gregorini, L.; Mancia, G. Mechanisms of antihypertensive action of beta-adrenergic blocking drugs: Evidence against potentiation of baroreflexes. Eur. Heart J. 1983, 4, D19–D25. [Google Scholar] [CrossRef] [PubMed]
- Potts, J.T.; Shi, X.R.; Raven, P.B. Carotid baroreflex responsiveness during dynamic exercise in humans. Am. J. Physiol. 1993, 265 Pt 2, H1928–H1938. [Google Scholar] [CrossRef]
- Goodwin, G.M.; McCloskey, D.I.; Mitchell, J.H. Cardiovascular and respiratory responses to changes in central command during isometric exercise at constant muscle tension. J. Physiol. 1972, 226, 173–190. [Google Scholar] [CrossRef]
- McCord, J.L.; Kaufman, M.P. Reflex Autonomic Responses Evoked by Group III and IV Muscle Afferents. In Translational Pain Research: From Mouse to Man; Kruger, L., Light, A.R., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2010. [Google Scholar]
- Ellsworth, M.L. The red blood cell as an oxygen sensor: What is the evidence? Acta Physiol. Scand. 2000, 168, 551–559. [Google Scholar] [CrossRef]
- González-Alonso, J.; Mortensen, S.P.; Dawson, E.A.; Secher, N.H.; Damsgaard, R. Erythrocytes and the regulation of human skeletal muscle blood flow and oxygen delivery: Role of erythrocyte count and oxygenation state of haemoglobin. J. Physiol. 2006, 572 Pt 1, 295–305. [Google Scholar] [CrossRef]
- González-Alonso, J.; Olsen, D.B.; Saltin, B. Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: Role of circulating ATP. Circ. Res. 2002, 91, 1046–1055. [Google Scholar] [CrossRef]
- Burnstock, G. Release of vasoactive substances from endothelial cells by shear stress and purinergic mechanosensory trans-duction. J. Anat. 1999, 194 Pt 3, 335–342. [Google Scholar] [CrossRef]
- Sprague, R.S.; Stephenson, A.H.; Ellsworth, M.L. Red not dead: Signaling in and from erythrocytes. Trends Endocrinol. Metab. 2007, 18, 350–355. [Google Scholar] [CrossRef]
- Kinfig, A.E.; Hayes, S.G.; Kaufman, M.P. Purinergic 2 receptor blockade prevents the responses of group IV afferents to post-contraction circulatory occlusion. J. Physiol. 2007, 578, 301–308. [Google Scholar]
- Kindig, A.E.; Hayes, S.G.; Kaufman, M.P. Blockade of purinergic 2 receptors attenuates the mechanoreceptor component of the exercise pressor reflex. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2995–H3000. [Google Scholar] [CrossRef]
- Kim, J.K.; Hayes, S.G.; Kindig, A.E.; Kaufman, M.P. Thin-fiber mechanoreceptors reflexly increase renal sympathetic nerve activity during static contraction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H866–H873. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.D.; Stray-Gundersen, J. “Living high-training low”: Effect of moderate-altitude acclimatization with low-altitude training on performance. J. Appl. Physiol. 1997, 83, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Hoppeler, H.; Weibel, E.R. Limits for oxygen and substrate transport in mammals. J. Exp. Biol. 1998, 201 Pt 8, 1051–1064. [Google Scholar]
- di Prampero, P.E. Factors limiting maximal performance in humans. Eur. J. Appl. Physiol. 2003, 90, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Berglund, B.; Ekblom, B. Effect of recombinant human erythropoietin treatment on blood pressure and some haematological parameters in healthy men. J. Intern. Med. 1991, 229, 125–130. [Google Scholar] [CrossRef]
- Saltin, B.; O Astrand, P. Free fatty acids and exercise. Am. J. Clin. Nutr. 1993, 57, 752S–758S. [Google Scholar] [CrossRef]
- Rasmussen, U.F.; Rasmussen, H.N.; Krustrup, P.; Quistorff, B.; Saltin, B.; Bangsbo, J. Aerobic metabolism of human quadriceps muscle: In vivo data parallel measurements on isolated mitochondria. Am. J. Physiol. Endocrino. Metab. 2001, 280, E301–E307. [Google Scholar] [CrossRef]
- Van Hall, G.; Jensen-Urstad, M.; Rosdahl, H.; Holmberg, H.-C.; Saltin, B.; Calbet, J.A.L. Leg and arm lactate and substrate kinetics during exercise. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E193–E205. [Google Scholar] [CrossRef] [PubMed]
- Consoli, A.; Nurjhan, N.; Reilly, J.J., Jr.; Bier, D.M.; Gerich, J.E. Contribution of liver and skeletal muscle to alanine and lactate metab-olism in humans. Am. J. Physiol. 1990, 259 Pt 1, E677–E684. [Google Scholar]
- Åstrand, P.-O. Quantification of exercise capability and evaluation of physical capacity in man. Prog. Cardiovasc. Dis. 1976, 19, 51–67. [Google Scholar] [CrossRef]
- Linderholm, H.; Müllert, R.; Ringqvist, T.; Sörnäs, R. Hereditary abnormal muscle metabolism with hyperkinetic circulation during exercise. Acta Medica Scand. 1969, 185, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Koo, P.; Sethi, J.M. Metabolic Myopathies and the Respiratory System. Clin. Chest Med. 2018, 39, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, J.M.; Busch, H.F.M.; Arts, W.F.M.; Heijsteeg, M.; Stam, H. Metabolic and Ventilatory Responses to Exercise in Patients with a Deficient O2 Utilization by a Mitochondrial Myopathy. Adv. Exp. Med. Biol. 1985, 191, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, P.; Brandner, S.; Kleinle, S.; Frauchiger, B. Hyperventilation due to mitochondrial myopathy. J. R. Soc. Med. 2000, 93, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.J. Potassium and ventilation in exercise. J. Appl. Physiol. 1992, 72, 811–820. [Google Scholar] [CrossRef]
- Giannoni, A.; Aimo, A.; Mancuso, M.; Piepoli, M.F.; Orsucci, D.; Aquaro, G.D.; Barison, A.; De Marchi, D.; Taddei, C.; Cameli, M.; et al. Autonomic, functional, skeletal muscle, and cardiac abnormalities are associated with increased ergoreflex sensitivity in mitochondrial disease. Eur. J. Heart Fail. 2017, 19, 1701–1709. [Google Scholar] [CrossRef]
- Piepoli, M.; Clark, A.L.; Volterrani, M.; Adamopoulos, S.; Sleight, P.; Coats, A.J. Contribution of muscle afferents to the hemodynamic, autonomic, and ventilatory responses to exercise in patients with chronic heart failure: Effects of physical training. Circulation 1996, 93, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Haller, R.G.; Lewis, S.F.; Estabrook, R.W.; DiMauro, S.; Servidei, S.; Foster, D.W. Exercise intolerance, lactic acidosis, and abnormal cardiopulmonary regulation in exercise associated with adult skeletal muscle cytochrome c oxidase deficiency. J. Clin. Investig. 1989, 84, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Boushel, R.; Gnaiger, E.; Calbet, J.A.; Gonzalez-Alonso, J.; Wright-Paradis, C.; Sondergaard, H.; Ara, I.; Helge, J.W.; Saltin, B. Muscle mitochondrial capacity exceeds maximal oxygen delivery in humans. Mitochondrion 2011, 11, 303–307. [Google Scholar] [CrossRef] [PubMed]
- McCoy, J.; Bates, M.; Eggett, C.; Siervo, M.; Cassidy, S.; Newman, J.; Moore, S.A.; Gorman, G.; Trenell, M.I.; Velicki, L.; et al. Pathophysiology of exercise intolerance in chronic diseases: The role of diminished cardiac performance in mitochondrial and heart failure patients. Open Heart 2017, 4, e000632. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Ørngreen, M.C.; Van Hall, G.; Haller, R.G.; Vissing, J. Fat Metabolism During Exercise in Patients With Mitochondrial Disease. Arch. Neurol. 2009, 66, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, T.D.; Orngreen, M.C.; Van Hall, G.; Vissing, J. Lactate metabolism during exercise in patients with mitochondrial myopathy. Neuromuscul. Disord. 2013, 23, 629–636. [Google Scholar] [CrossRef]
- Siciliano, G.; Renna, M.; Manca, M.L.; Prontera, C.; Zucchelli, G.; Ferrannini, E.; Murri, L. The relationship of plasma catecholamine and lactate during anaerobic threshold exercise in mitochondrial myopathies. Neuromuscul. Disord. 1999, 9, 411–416. [Google Scholar] [CrossRef]
- Siciliano, G.; Manca, M.L.; Renna, M.; Prontera, C.; Mercuri, A.; Murri, L. Effects of aerobic training on lactate and catecholaminergic exercise responses in mitochondrial myopathies. Neuromuscul. Disord. 2000, 10, 40–45. [Google Scholar] [CrossRef]
- Reinöhl, J.; Hoheisel, U.; Unger, T.; Mense, S. Adenosine triphosphate as a stimulant for nociceptive and non-nociceptive muscle group IV receptors in the rat. Neurosci. Lett. 2003, 338, 25–28. [Google Scholar] [CrossRef]
- Finsterer, J.; Shorny, S.; Capek, J.; Cerny-Zacharias, C.; Pelzl, B.; Messner, R.; E Bittner, R.; Mamoli, B. Lactate stress test in the diagnosis of mitochondrial myopathy. J. Neurol. Sci. 1998, 159, 176–180. [Google Scholar] [CrossRef]
- Löfberg, M.; Lindholm, H.; Näveri, H.; Majander, A.; Suomalainen, A.; Paetau, A.; Sovijärvi, A.; Härkönen, M.; Somer, H. ATP, phosphocreatine and lactate in exercising muscle in mitochondrial disease and McArdle’s disease. Neuromuscul. Disord. 2001, 11, 370–375. [Google Scholar] [CrossRef]
- Chan, A.; Reichmann, H.; Kögel, A.; Beck, A.; Gold, R. Metabolic changes in patients with mitochondrial myopathies and effects of coenzyme Q 10 therapy. J. Neurol. 1998, 245, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Dengler, R.; Wohlfarth, K.; Zierz, S.; Jobges, M.; Schubert, M. Muscle fatigue, lactate, and pyruvate in mitochondrial myopathy with progressive external ophthalmoplegia. Muscle Nerve 1996, 19, 456–462. [Google Scholar] [CrossRef]
- Nashef, L.; Lane, R.J. Screening for mitochondrial cytopathies: The sub-anaerobic threshold exercise test (SATET). J. Neurol. Neurosurg. Psychiatry 1989, 52, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, E.; Osari, S.; Yamanouchi, H.; Matsuda, H.; Goto, Y.; Nonaka, I. Long-term therapy with cytochrome c, flavin mono-nucleotide and thiamine diphosphate for a patient with Kearns-Sayre syndrome. Brain Dev. 1996, 18, 68–70. [Google Scholar] [CrossRef]
- Siciliano, G.; Rossi, B.; Manca, L.; Angelini, C.; Tessa, A.; Vergani, L.; Martinuzzi, A.; Muratoyio, A. Residual muscle cytochrome c oxidase activity accounts for submaximal exercise lactate threshold in chronic progressive external ophthalmoplegia. Muscle Nerve 1996, 19, 342–349. [Google Scholar] [CrossRef]
- Hanisch, F.; Müller, T.; Muser, A.; Deschauer, M.; Zierz, S. Lactate increase and oxygen desaturation in mitochondrial disor-ders--evaluation of two diagnostic screening protocols. J. Neurol. 2006, 253, 417–423. [Google Scholar] [CrossRef]
- Weber, K.T.; Janicki, J.S. Lactate production during maximal and submaximal exercise in patients with chronic heart failure. J. Am. Coll. Cardiol. 1985, 6, 717–724. [Google Scholar] [CrossRef][Green Version]
- Ichiwata, T.; Sasao, G.; Abe, T.; Kikuchi, K.; Koyama, K.; Fujiwara, H.; Nagai, A.; Kuwahira, I.; Nagao, K. Oxidative capacity of the skeletal muscle and lactic acid kinetics during exercise in healthy subjects and patients with COPD. Adv. Exp. Med. Biol. 2010, 662, 537–543. [Google Scholar] [PubMed]
- Gregg, S.G.; Mazzeo, R.S.; Budinger, T.F.; Brooks, G.A. Acute anemia increases lactate production and decreases clearance during exercise. J. Appl. Physiol. 1989, 67, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, T.D.; Olsen, D.; Vissing, J. Cycle ergometry is not a sensitive diagnostic test for mitochondrial myopathy. J. Neurol. 2003, 250, 293–299. [Google Scholar] [CrossRef]
- Finsterer, J.; Milvay, E. Stress lactate in mitochondrial myopathy under constant, unadjusted workload. Eur. J. Neurol. 2004, 11, 811–816. [Google Scholar] [CrossRef]
- Finsterer, J.; Milvay, E. Diagnostic yield of the lactate stress test in respiratory chain disorders under absolute and relative workload. J. Neurosci. Methods 2001, 108, 65–70. [Google Scholar] [CrossRef]
- Volpi, L.; Ricci, G.; Orsucci, D.; Alessi, R.; Bertolucci, F.; Piazza, S.; Simoncini, C.; Mancuso, M.; Siciliano, G. Metabolic myopathies: Functional evaluation by different exercise testing approaches. Musculoskelet. Surg. 2011, 95, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Elliot, D.L.; Buist, N.R.; Goldberg, L.; Kennaway, N.G.; Powell, B.R.; Kuehl, K.S. Metabolic myopathies: Evaluation by graded exercise testing. Medicine 1989, 68, 163–172. [Google Scholar] [CrossRef]
- Charlton, G.A.; Crawford, M.H. Physiologic consequences of training. Cardiol. Clin. 1997, 15, 345–354. [Google Scholar] [CrossRef]
- Jones, A.M.; Carter, H. The Effect of Endurance Training on Parameters of Aerobic Fitness. Sports Med. 2000, 29, 373–386. [Google Scholar] [CrossRef]
- Haller, R.G.; Vissing, J. Spontaneous “second wind” and glucose-induced second “second wind” in McArdle disease: Oxidative mechanisms. Arch. Neurol. 2002, 59, 1395–1402. [Google Scholar] [CrossRef]
- Lewis, S.F.; Haller, R.G. The pathophysiology of McArdle’s disease: Clues to regulation in exercise and fatigue. J. Appl. Physiol. 1986, 61, 391–401. [Google Scholar] [CrossRef]
- Haller, R.G.; Lewis, S.F.; Cook, J.D.; Blomqvist, C.G. Myophosphorylase deficiency impairs muscle oxidative metabolism. Ann. Neurol. 1985, 17, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Sheel, A.W.; Foster, G.E.; Romer, L.M. Exercise and its impact on dyspnea. Curr. Opin. Pharmacol. 2011, 11, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Haller, R.G.; Lewis, S.F. Pathophysiology of exercise performance in muscle disease. Med. Sci. Sports Exerc. 1984, 16, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, K.C.; Bourassa, M.W.; Adamolekun, B.; Bergeron, G.; Bettendorff, L.; Brown, K.H.; Cox, L.; Fattal-Valevski, A.; Fischer, P.R.; Frank, E.L.; et al. Thiamine deficiency disorders: Diagnosis, prevalence, and a roadmap for global control programs. Ann. N. Y. Acad. Sci. 2018, 1430, 3–43. [Google Scholar] [CrossRef] [PubMed]
- Hooper, R.G.; Thomas, A.R.; Kearl, R.A. Mitochondrial Enzyme Deficiency Causing Exercise Limitation in Normal-Appearing Adults. Chest 1995, 107, 317–322. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wald, J.; Weisman, I.M.; Zeballos, R.J.; Schork, M.A.; Blaivas, M.; Rubenfire, M.; Martinez, F.J. Unexplained exertional limitation: Characteri-zation of patients with a mitochondrial myopathy. Am. J. Respir. Crit. Care Med. 2001, 164, 425–432. [Google Scholar] [CrossRef]
- Koene, S.; De Laat, P.; Van Tienoven, D.H.; Weijers, G.; Vriens, D.; Sweep, F.C.G.J.; Timmermans, J.; Kapusta, L.; Janssen, M.C.H.; Smeitink, J.A.M.; et al. Serum GDF15 Levels Correlate to Mitochondrial Disease Severity and Myocardial Strain, but Not to Disease Progression in Adult m.3243A>G Carriers. JIMD Rep. 2015, 24, 69–81. [Google Scholar] [CrossRef]
- Montero, R.; Yubero, D.; Villarroya, J.; Henares, D.; Jou, C.; Rodríguez, M.A.; Ramos, F.; Nascimento, A.; Ortez, C.I.; Campistol, J.; et al. GDF-15 Is Elevated in Children with Mitochondrial Diseases and Is Induced by Mitochondrial Dysfunction. PLoS ONE 2016, 11, e0148709. [Google Scholar] [CrossRef]
- Yatsuga, S.; Fujita, Y.; Ishii, A.; Fukumoto, Y.; Arahata, H.; Kakuma, T.; Kojima, T.; Ito, M.; Tanaka, M.; Saiki, R.; et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 2015, 78, 814–823. [Google Scholar] [CrossRef]
- Fujita, Y.; Ito, M.; Kojima, T.; Yatsuga, S.; Koga, Y.; Tanaka, M. GDF15 is a novel biomarker to evaluate efficacy of pyruvate therapy for mitochondrial diseases. Mitochondrion 2015, 20, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhao, L.; Ji, K.; Zhao, Y.; Li, W.; Zhang, R.; Hou, Y.; Lu, J.; Yan, C. Growth Differentiation Factor 15 Is a Novel Diagnostic Biomarker of Mitochondrial Diseases. Mol. Neurobiol. 2017, 54, 8110–8116. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, N.S.; Madsen, K.L.; Hornsyld, T.M.; Eisum, A.-S.V.; Fornander, F.; Buch, A.E.; Stemmerik, M.G.; Ruiz-Ruiz, C.; Krag, T.O.; Vissing, J. Growth and differentiation factor 15 as a biomarker for mitochondrial myopathy. Mitochondrion 2020, 50, 35–41. [Google Scholar] [CrossRef]
- Kalko, S.G.; Paco, S.; Jou, C.; Rodríguez, M.A.; Meznaric, M.; Rogac, M.; Jekovec-Vrhovsek, M.; Sciacco, M.; Moggio, M.; Fagiolari, G.; et al. Transcriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genom. 2014, 15, 91. [Google Scholar] [CrossRef]
- Davis, R.L.; Liang, C.; Sue, C.M. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology 2016, 86, 2010–2015. [Google Scholar] [CrossRef]
- Kleinert, M.; Clemmensen, C.; Sjøberg, K.A.; Carl, C.S.; Jeppesen, J.F.; Wojtaszewski, J.F.; Kiens, B.; Richter, E.A. Exercise increases circulating GDF15 in humans. Mol. Metab. 2018, 9, 187–191. [Google Scholar] [CrossRef]
- Boone, J.; Celie, B.; Dumortier, J.; Barstow, T.J.; De Bleecker, J.; Smet, J.; Van Lander, A.; Van Coster, R.; Bourgois, J. Forearm muscle oxygenation responses during and following arterial occlusion in patients with mitochondrial myopathy. Respir. Physiol. Neurobiol. 2014, 190, 70–75. [Google Scholar] [CrossRef]
- Jensen, T.D.; Kazemi-Esfarjani, P.; Skomorowska, E.; Vissing, J. A forearm exercise screening test for mitochondrial myopathy. Neurology 2002, 58, 1533–1538. [Google Scholar] [CrossRef]
- Bank, W.; Park, J.; Lech, G.; Chance, B. Near-infrared spectroscopy in the diagnosis of mitochondrial disorders. BioFactors 1998, 7, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Celie, B.M.; Boone, J.; Smet, J.E.; Vanlander, A.V.; De Bleecker, J.L.; Van Coster, R.N.; Bourgois, J.G. Forearm deoxyhemoglobin and deoxymyo-globin (deoxy[Hb + Mb]) measured by near-infrared spectroscopy (NIRS) using a handgrip test in mitochondrial myopathy. Appl. Spectrosc. 2015, 69, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Bank, W.; Lech, G.; Long, H.; Olsen, N.; Chance, B. Correlative magnetic resonance and near-infrared technologies for the evaluation of mitochondrial disease. BioFactors 1998, 7, 253–254. [Google Scholar] [CrossRef] [PubMed]
- van Beekvelt, M.C.; van Engelen, B.G.; Wevers, R.A.; Colier, W.N. Quantitative near-infrared spectroscopy discriminates between mitochondrial myopathies and normal muscle. Ann. Neurol. 1999, 46, 667–670. [Google Scholar] [CrossRef]
- Radda, G.K. Introduction to magnetic resonance spectroscopy (MRS) and positron emission tomography (PET) for the inves-tigation of mitochondrial myopathies. BioFactors 1998, 7, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Kemp, G.J.; Radda, G.K. Quantitative interpretation of bioenergetic data from 31P and 1H magnetic resonance spectroscopic studies of skeletal muscle: An analytical review. Magn. Reson. Q. 1994, 10, 43–63. [Google Scholar] [PubMed]
- Arnold, D.L.; Taylor, D.J.; Radda, G.K. Investigation of human mitochondrial myopathies by phosphorus magnetic resonance spectroscopy. Ann. Neurol. 1985, 18, 189–196. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Quistorff, B.; Wibrand, F.; Vissing, J. 31P-MRS of skeletal muscle is not a sensitive diagnostic test for mitochondrial myopathy. J. Neurol. 2007, 254, 29–37. [Google Scholar] [CrossRef] [PubMed]
- McArdle, B. Myopathy due to a defect in muscle glycogen breakdown. Clin. Sci. 1951, 10, 13–35. [Google Scholar]
- Jeppesen, T.D. Aerobic Exercise Training in Patients With mtDNA-Related Mitochondrial Myopathy. Front. Physiol. 2020, 11, 349. [Google Scholar] [CrossRef]
- Klopstock, T.; Querner, V.; Schmidt, F.; Gekeler, F.; Walter, M.; Hartard, M.; Henning, M.; Gasser, T.; Pongratz, D.; Straube, A.; et al. A placebo-controlled crossover trial of creatine in mitochondrial diseases. Neurology 2000, 55, 1748–1751. [Google Scholar] [CrossRef]
- Kornblum, C.; Schröder, R.; Müller, K.; Vorgerd, M.; Eggers, J.; Bogdanow, M.; Papassotiropoulos, A.; Fabian, K.; Klockgether, T.; Zange, J. Creatine has no beneficial effect on skeletal muscle energy metabolism in patients with single mitochondrial DNA deletions: A placebo-controlled, double-blind 31P-MRS crossover study. Eur. J. Neurol. 2005, 12, 300–309. [Google Scholar] [CrossRef]
- Nabben, M.; Schmitz, J.P.J.; Ciapaite, J.; Le Clercq, C.M.P.; Van Riel, N.A.; Haak, H.R.; Nicolay, K.; De Coo, I.F.M.; Smeets, H.J.M.; Praet, S.F.; et al. Dietary nitrate does not reduce oxygen cost of exercise or improve muscle mitochondrial function in patients with mitochondrial myopathy. Am. J. Physiol. Integr. Comp. Physiol. 2017, 312, R689–R701. [Google Scholar] [CrossRef]
- Scarlato, G.; Bresolin, N.; Moroni, I.; Doriguzzi, C.; Castelli, E.; Comi, G.; Angelini, C.; Carenzi, A. Multicenter trial with ubidecarenone: Treatment of 44 patients with mitochondrial myopathies. Rev. Neurol. 1991, 147, 542–548. [Google Scholar]
- Rodan, L.H.; Wells, G.D.; Banks, L.; Thompson, S.; Schneiderman, J.E.; Tein, I. L-Arginine Affects Aerobic Capacity and Muscle Metabolism in MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-Like Episodes) Syndrome. PLoS ONE 2015, 10, e0127066. [Google Scholar] [CrossRef]
- Madsen, K.L.; Buch, A.E.; Cohen, B.H.; Falk, M.J.; Goldsberry, A.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Muraresku, C.C.; Meyer, C.; et al. Safety and efficacy of omaveloxolone in patients with mitochondrial myopathy: MOTOR trial. Neurology 2020, 94, e687–e698. [Google Scholar] [CrossRef]
- Hunter, S.K. Performance Fatigability: Mechanisms and Task Specificity. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Filler, K.; Lyon, D.; Bennett, J.; McCain, N.; Elswick, R.; Lukkahatai, N.; Saligan, L.N. Association of mitochondrial dysfunction and fatigue: A review of the literature. BBA Clin. 2014, 1, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Lindinger, M.I.; McKelvie, R.S.; Heigenhauser, G.J. K+ and Lac- distribution in humans during and after high-intensity exercise: Role in muscle fatigue attenuation? J. Appl. Physiol. 1995, 78, 765–777. [Google Scholar] [CrossRef]
- Axelson, H.W.; Melberg, A.; Ronquist, G.; Askmark, K. Microdialysis and electromyography of experimental muscle fatigue in healthy volunteers and patients with mitochondrial myopathy. Muscle Nerve 2002, 26, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, E.A.; Johns, T.; Karpati, G. Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy. Hum. Mol. Genet. 1997, 6, 2239–2242. [Google Scholar] [CrossRef] [PubMed]
- Romer, L.M.; Polkey, M.I. Exercise-induced respiratory muscle fatigue: Implications for performance. J. Appl. Physiol. 2008, 104, 879–888. [Google Scholar] [CrossRef]
- Johnson, B.D.; Aaron, E.A.; Babcock, M.A.; Dempsey, J.A. Respiratory muscle fatigue during exercise: Implications for performance. Med. Sci. Sports Exerc. 1996, 28, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Cejudo, P.; Bautista, J.; Montemayor, T.; Villagómez, R.; Jiménez, L.; Ortega, F.; Campos, Y.; Sánchez, H.; Arenas, J. Exercise training in mitochondrial myopathy: A randomized controlled trial. Muscle Nerve 2005, 32, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, T.D.; Schwartz, M.; Olsen, D.B.; Wibrand, F.; Krag, T.; Dunø, M.; Hauerslev, S.; Vissing, J. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 2006, 129 Pt 12, 3402–3412. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Dunø, M.; Schwartz, M.; Krag, T.; Rafiq, J.; Wibrand, F.; Vissing, J. Short- and long-term effects of endurance training in patients with mitochondrial myopathy. Eur. J. Neurol. 2009, 16, 1336–1339. [Google Scholar] [CrossRef] [PubMed]
- Taivassalo, T.; Gardner, J.L.; Taylor, R.W.; Schaefer, A.M.; Newman, J.; Barron, M.J.; Haller, R.G.; Turnbull, D.M. Endurance training and detraining in mi-tochondrial myopathies due to single large-scale mtDNA deletions. Brain J. Neurol. 2006, 129 Pt 12, 3391–3401. [Google Scholar] [CrossRef] [PubMed]
- Taivassalo, T.; De Stefano, N.; Argov, Z.; Matthews, P.M.; Chen, J.; Genge, A.; Karpati, G.; Arnold, D.L. Effects of aerobic training in patients with mitochondrial myopathies. Neurology 1998, 50, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Taivassalo, T.; Shoubridge, E.A.; Chen, J.; Kennaway, N.G.; DiMauro, S.; Arnold, D.L.; Haller, R.G. Aerobic conditioning in patients with mitochondrial myopathies: Physiological, biochemical, and genetic effects. Ann. Neurol. 2001, 50, 133–141. [Google Scholar] [CrossRef]
- Porcelli, S.; Marzorati, M.; Morandi, L.; Grassi, B. Home-based aerobic exercise training improves skeletal muscle oxidative metabolism in patients with metabolic myopathies. J. Appl. Physiol. 2016, 121, 699–708. [Google Scholar] [CrossRef]
- Chelimsky, T.C.; Mcneeley, K.M.; Comfort, B.; Piantadosi, C.A.; LaManna, J.C. Effect of exercise and ischemia on tissue oxi-metry and cytochrome in normal subjects, patients with chronic limb pain, and patients with mitochondrial mitopathies. Adv. Exp. Med. Biol. 1997, 411, 445–451. [Google Scholar]
- Siciliano, G.; Simoncini, C.; Gerfo, A.L.; Orsucci, D.; Ricci, G.; Mancuso, M. Effects of aerobic training on exercise-related oxidative stress in mitochondrial myopathies. Neuromuscul. Disord. 2012, 22, S172–S177. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A. Exercise as a Therapeutic Strategy for Primary Mitochondrial Cytopathies. J. Child Neurol. 2014, 29, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Safdar, A.; Bishop, D.; Tarnopolsky, M.A.; Gibala, M.J. An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1α and activates mitochondrial biogenesis in human skeletal muscle. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R1303–R1310. [Google Scholar] [CrossRef] [PubMed]
- Adhihetty, P.J.; Taivassalo, T.; Haller, R.G.; Walkinshaw, D.R.; Hood, D.A. The effect of training on the expression of mito-chondrial biogenesis- and apoptosis-related proteins in skeletal muscle of patients with mtDNA defects. Am. J. Physiol. Endo-crinol. Metab. 2007, 293, E672–E680. [Google Scholar] [CrossRef] [PubMed]
- Zoladz, J.A.; Grassi, B.; Majerczak, J.; Szkutnik, Z.; Korostyński, M.; Karasiński, J.; Kilarski, W.; Korzeniewski, B. Training induced acceleration of O(2) uptake on-kinetics precedes muscle mitochondrial biogenesis in humans. Exp. Physiol. 2013, 98, 883–898. [Google Scholar] [CrossRef]
- Stefanetti, R.J.; Blain, A.; Jimenez-Moreno, C.; Errington, L.; Ng, Y.S.; McFarland, R.; Turnbull, D.M.; Newman, J.; Gorman, G.S. Measuring the effects of exercise in neuromuscular disorders: A systematic review and meta-analyses. Wellcome Open Res. 2020, 5, 84. [Google Scholar] [CrossRef]
- Murphy, J.L.; Blakely, E.L.; Schaefer, A.M.; He, L.; Wyrick, P.; Haller, R.G.; Taylor, R.W.; Turnbull, U.M.; Taivassalo, T. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 2008, 131, 2832–2840. [Google Scholar] [CrossRef]
- Newman, J.; Galna, B.; Jakovljevic, D.G.; Bates, M.G.; Schaefer, A.M.; McFarland, R.; Turnbull, D.M.; Trenell, M.I.; Taylor, R.W.; Rochester, L.; et al. Preliminary Evaluation of Clinician Rated Outcome Measures in Mitochondrial Disease. J. Neuromuscul. Dis. 2015, 2, 151–155. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | Measure | Method | Characteristics in MM | Diagnostic Potential | Strengths | Weaknesses |
|---|---|---|---|---|---|---|
| Maximal exercise | VO2max | CPET or Douglas bag | ↓ | ÷ ↑ Sensitive ↓ Specific | Equipment available in most hospitals. Directly reflects aerobic energy metabolism. Correlates to mtDNA mutation load. | Requires trained staff Sensitive to day-to-day variations in motivation to reach maximal exercise capacity |
| VE/VO2max | CPET or Douglas bag | ↑ | ÷ ↑ Sensitive ↓ Specific | |||
| CO/VO2max | * Acethylene rebreathing | ↑ | ÷ ↑ Sensitive ↓ Specific | Corrects for circulatory adaptations affecting VO2-measurements during exercise | Requires specialized equipment | |
| Epinephrin/ Workload | Maximal exercise plasma value | ↑ | ÷ ↑ Sensitive ↓ Specific | Standard analysis in most hospitals. | Not specific for MM | |
| Plasma Lactate | Post- exercise sampling | ↑ | ÷ ↑ Sensitive ↓ Specific | Standard analysis | Sensitive to degree of volition to reach maximal effort. Equally specific to resting lactate | |
| Serum GDF-15 | 24h post-exercise sample | ↑ | + ↑ Sensitive ↓ Specific | Correlates with oxidative capacity | Not standard analysis. Further research in the use as outcome measure is required | |
| Submaximal exercise | Plasma Lactate | Sampling during exercise | ↑ | ÷ ↑ Sensitive ↓ Specific | Easily standardized Can reflect changes in oxidative capacity | Requires prior maximal exercise testing. Workload must be selected carefully |
| Heart rate | During exercise | ↓ | ÷ ↑ Sensitive ↓ Specific | |||
| One-extremity exercise | ATP turnover | * 31P-MRS One-legged exercise | ↓ | ÷ ↑ Sensitive ↓ Specific | Indirect real-time measure of oxidative capacity in the tested extremity | Requires specialized equipment, and highly trained staff |
| Oxygen saturation/ content | * Blood gas analyzer/ near infrared spectroscopy | ↓ | + ↑ Sensitive ↑ Specific | Correlates with mtDNA mutation load. | Test arm must be sufficiently warm to ensure venous blood flow | |
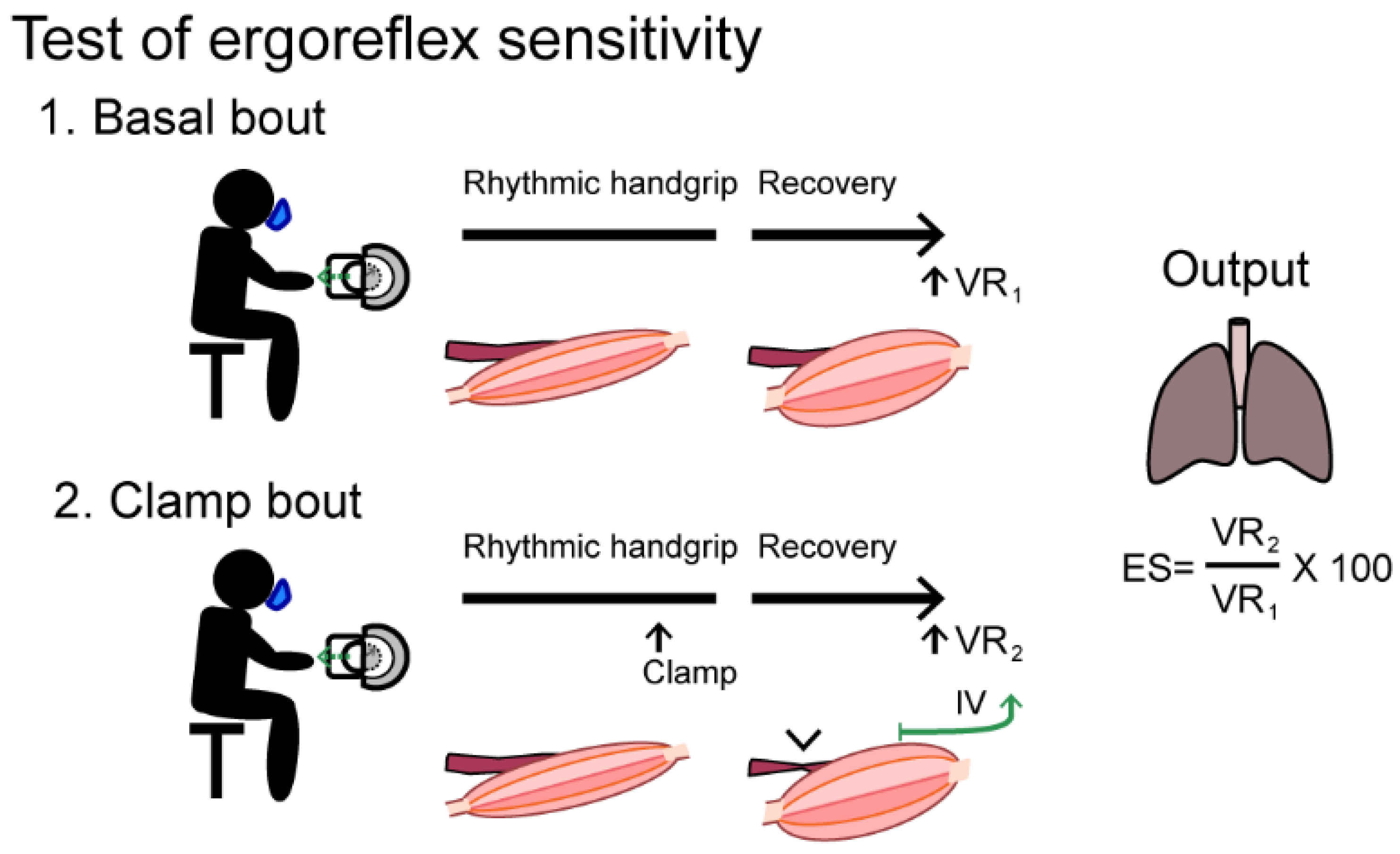
| Ergoreflex sensitivity | Handgrip +/−ischemia | ↑ | ÷ ↑ Sensitive ↓ Specific | Simple test setup. Correlates to degree of cardiac affection | Specificity vs. other myopathies needs to be investigated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeppesen, T.D.; Madsen, K.L.; Poulsen, N.S.; Løkken, N.; Vissing, J. Exercise Testing, Physical Training and Fatigue in Patients with Mitochondrial Myopathy Related to mtDNA Mutations. J. Clin. Med. 2021, 10, 1796. https://doi.org/10.3390/jcm10081796
Jeppesen TD, Madsen KL, Poulsen NS, Løkken N, Vissing J. Exercise Testing, Physical Training and Fatigue in Patients with Mitochondrial Myopathy Related to mtDNA Mutations. Journal of Clinical Medicine. 2021; 10(8):1796. https://doi.org/10.3390/jcm10081796
Chicago/Turabian StyleJeppesen, Tina D., Karen L. Madsen, Nanna S. Poulsen, Nicoline Løkken, and John Vissing. 2021. "Exercise Testing, Physical Training and Fatigue in Patients with Mitochondrial Myopathy Related to mtDNA Mutations" Journal of Clinical Medicine 10, no. 8: 1796. https://doi.org/10.3390/jcm10081796
APA StyleJeppesen, T. D., Madsen, K. L., Poulsen, N. S., Løkken, N., & Vissing, J. (2021). Exercise Testing, Physical Training and Fatigue in Patients with Mitochondrial Myopathy Related to mtDNA Mutations. Journal of Clinical Medicine, 10(8), 1796. https://doi.org/10.3390/jcm10081796