
Movement Disorders in Children with a Mitochondrial Disease: A Cross-Sectional Survey from the Nationwide Italian Collaborative Network of Mitochondrial Diseases

 , ,
, , 
 ,
,  ,
,  , ,
, ,  ,
,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
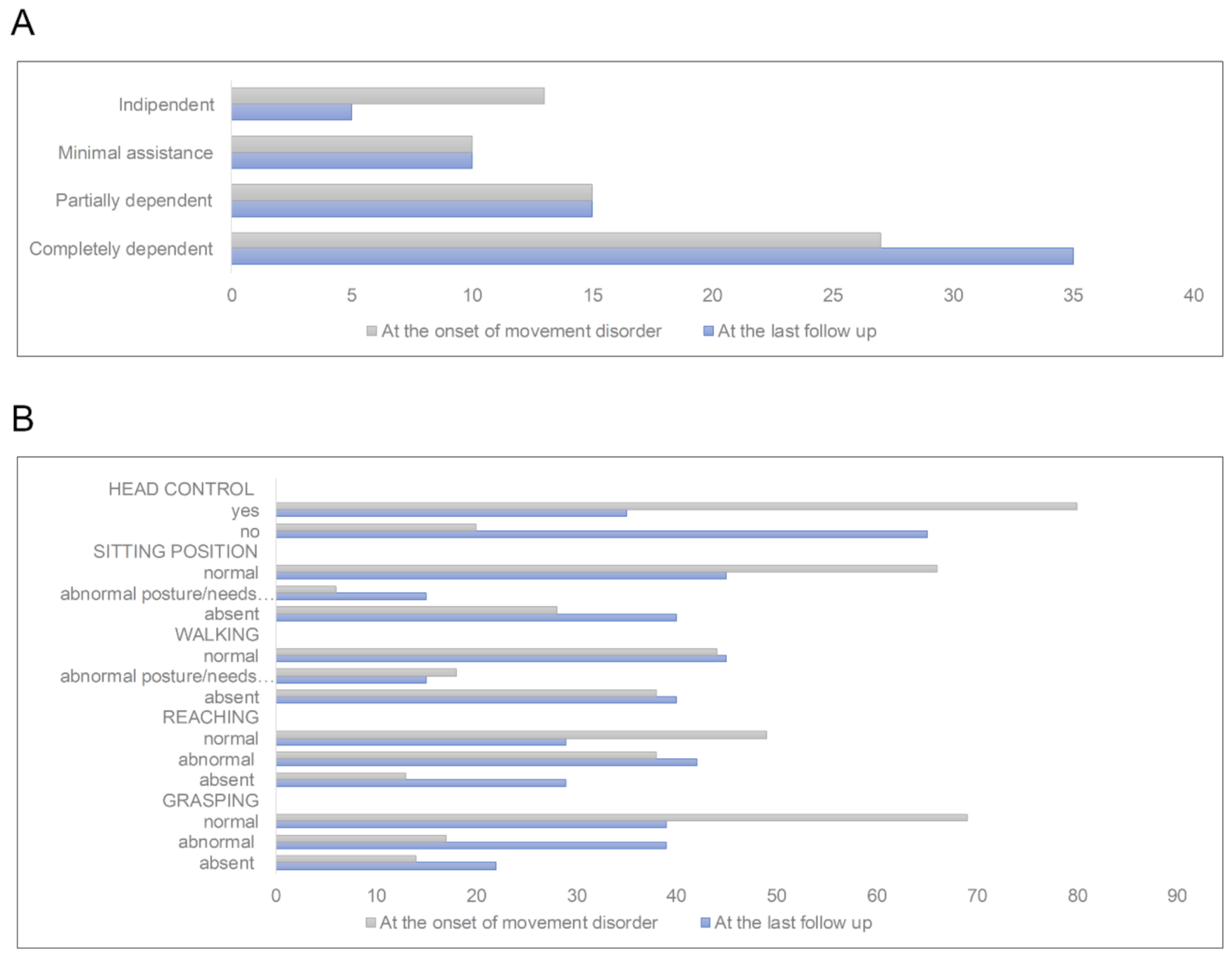
3. Results
3.1. Features of MD Patients with and without Movement Disorders
3.2. Google Survey Results on MD Patients with Movement Disorders
3.2.1. Clinical, Neuroradiological, and Genetic Features
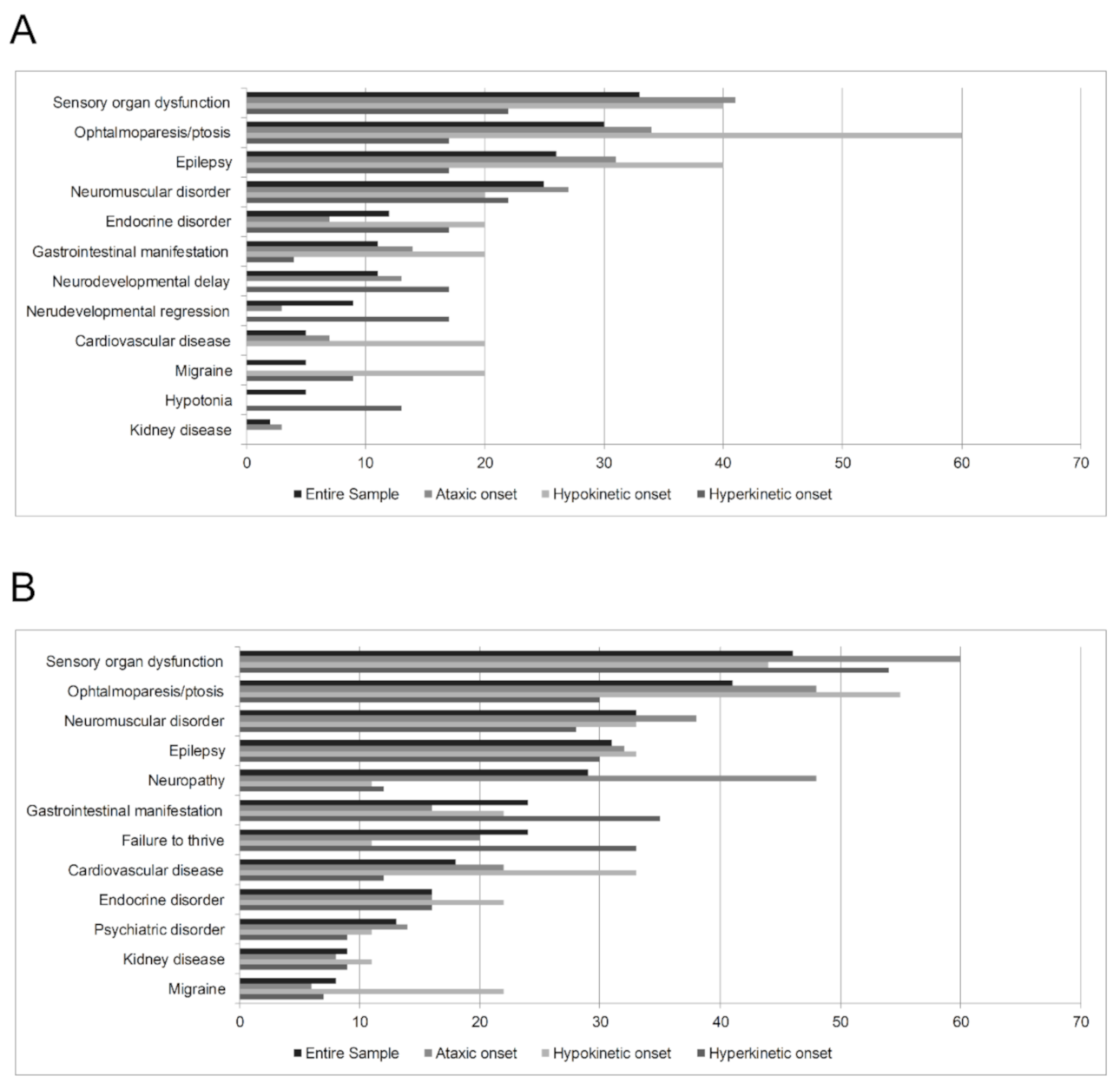
3.2.2. Clinical, Neuroradiological, and Genetic Features According to the Type of Movement Disorder at Onset
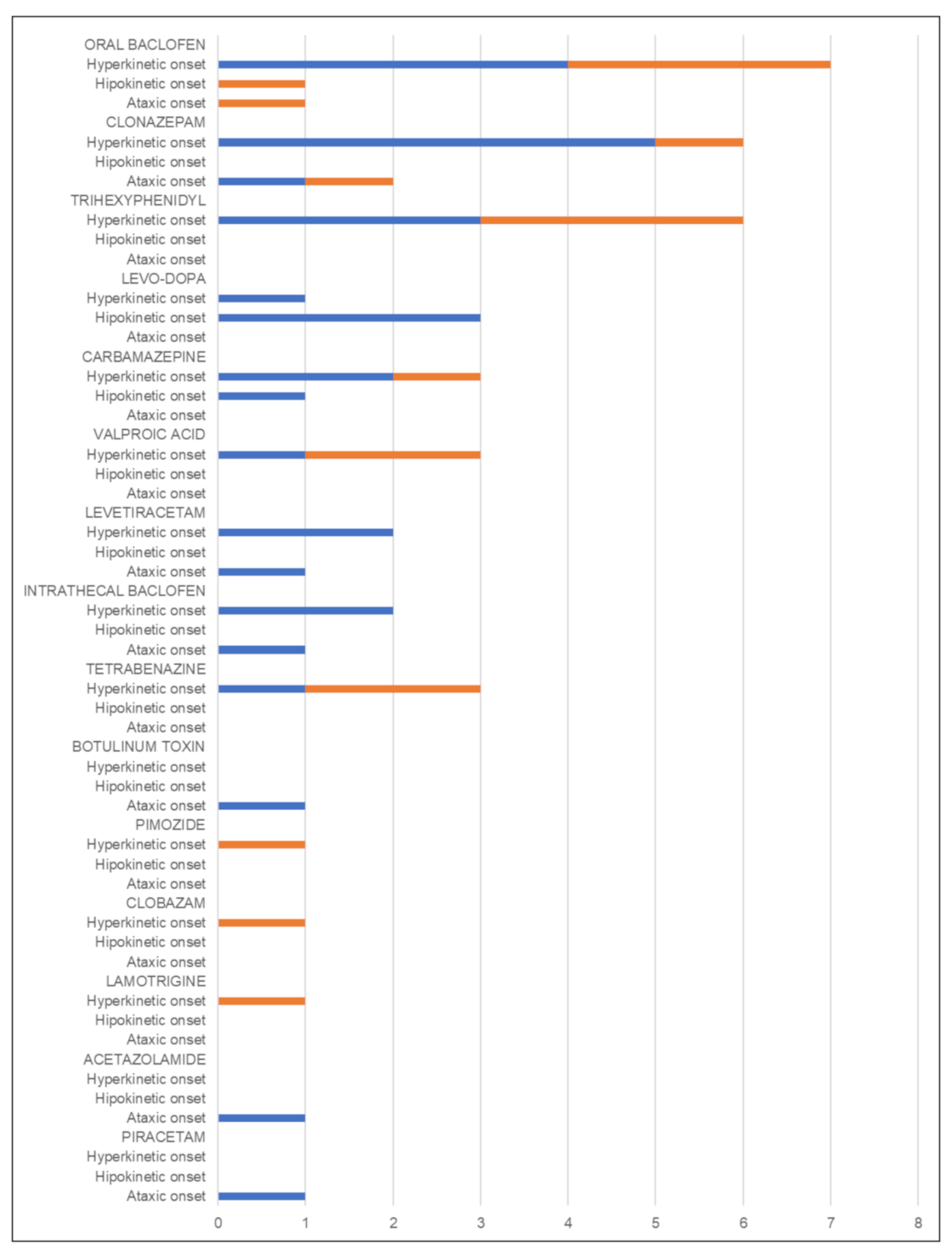
Hyperkinetic Onset Subgroup
Hypokinetic Onset Subgroup
Ataxic Onset Subgroup
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Musumeci, O.; Oteri, R.; Toscano, A. Spectrum of movement disorders in mitochondrial diseases. J. Transl. Genet. Genom. 2020, 4, 221–258. [Google Scholar] [CrossRef]
- Ghaoui, R.; Sue, C.M. Movement disorders in mitochondrial disease. J. Neurol. 2018, 265, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Caer, M.; Viala, K.; Levy, R.; Maisonobe, T.; Chochon, F.; Lombès, A.; Agid, Y. Adult-onset chorea and mitochondrial cytopathy. Mov. Disord. 2004, 20, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.D.; Harding, A.E.; Scaravilli, F.; Smith, S.J.M.; Morgan-Hughes, J.A.; Marsden, C.D. Movement disorders in mitochondrial myopathies. A study of nine cases with two autopsy studies. Mov. Disord. 1990, 5, 109–117. [Google Scholar] [CrossRef]
- Sudarsky, L.; Plotkin, G.M.; Logigian, E.L.; Johns, D.R. Dystonia as a presenting feature of the 3243 mitochondrial DNA mutation. Mov. Disord. 1999, 14, 488–491. [Google Scholar] [CrossRef]
- Baloh, R.H.; Salavaggione, E.; Milbrandt, J.; Pestronk, A. Familial parkinsonism and ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch. Neurol. 2007, 64, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Luoma, P.; Melberg, A.; O Rinne, J.; A Kaukonen, J.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: Clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Martikainen, M.H.; Ng, Y.S.; Gorman, G.S.; Alston, C.L.; Blakely, E.L.; Schaefer, A.M.; Chinnery, P.F.; Burn, D.J.; Taylor, R.W.; McFarland, R.; et al. Clinical, genetic, and radiological features of extrapyramidal movement disorders in mitochondrial disease. JAMA Neurol. 2016, 73, 668–674. [Google Scholar] [CrossRef] [Green Version]
- Schreglmann, S.R.; Riederer, F.; Galovic, M.; Ganos, C.; Kägi, G.; Waldvogel, D.; Jaunmuktane, Z.; Schaller, A.; Hidding, U.; Krasemann, E.; et al. Movement disorders in genetically confirmed mitochondrial disease and the putative role of the cerebellum. Mov. Disord. 2017, 33, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M.; et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar] [CrossRef]
- Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Federico, A.; Minetti, C.; Moggio, M.; Mongini, T.; Santorelli, F.M.; et al. Revisiting mitochondrial ocular myopathies: A study from the Italian Network. J. Neurol. 2017, 264, 1777–1784. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, A.; Minetti, C.; Moggio, M.; Mongini, T.; et al. The m.3243A>G mitochondrial DNA mutation and related phenotypes. A matter of gender? J. Neurol. 2014, 261, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Minetti, C.; Moggio, M.; Mongini, T.; Servidei, S.; et al. Phenotypic heterogeneity of the 8344A>G mtDNA “MERRF” mutation. Neurology 2013, 80, 2049–2054. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Catteruccia, M.; Pegoraro, E.; Carelli, V.; Valentino, M.L.; Comi, G.P.; Minetti, C.; et al. Myoclonus in mitochondrial disorders. Mov. Disord. 2014, 29, 722–728. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Federico, A.; Minetti, C.; Moggio, M.; Mongini, T.; et al. “Mitochondrial neuropathies”: A survey from the large cohort of the Italian Network. Neuromuscul. Disord. 2016, 26, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Ticci, C.; Sicca, F.; Ardissone, A.; Bertini, E.; Carelli, V.; Diodato, D.; Di Vito, L.; Filosto, M.; La Morgia, C.; Lamperti, C.; et al. Mitochondrial epilepsy: A cross-sectional nationwide Italian survey. Neurogenetics 2020, 21, 87–96. [Google Scholar] [CrossRef]
- Filosto, M.; Piccinelli, S.C.; Lamperti, C.; Mongini, T.; Servidei, S.; Musumeci, O.; Tonin, P.; Santorelli, F.M.; Simoncini, C.; Primiano, G.; et al. Muscle pain in mitochondrial diseases: A picture from the Italian network. J. Neurol. 2019, 266, 953–959. [Google Scholar] [CrossRef]
- Musumeci, O.; Barca, E.; Lamperti, C.; Servidei, S.; Comi, G.P.; Moggio, M.; Mongini, T.; Siciliano, G.; Filosto, M.; Pegoraro, E.; et al. Lipomatosis incidence and characteristics in an Italian cohort of mitochondrial patients. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Kurian, M.A.; Dale, R.C. Movement disorders presenting in childhood. Contin. Lifelong Learn. Neurol. 2016, 22, 1159–1185. [Google Scholar] [CrossRef] [Green Version]
- Albanese, A.; Bhatia, K.; Bressman, S.B.; DeLong, M.R.; Fahn, S.; Fung, V.S.; Hallett, M.; Jankovic, J.; Jinnah, H.A.; Klein, C.; et al. Phenomenology and classification of dystonia: A consensus update. Mov. Disord. 2013, 28, 863–873. [Google Scholar] [CrossRef] [Green Version]
- Zutt, R.; Van Egmond, M.E.; Elting, J.W.; Van Laar, P.J.; Brouwer, O.F.; Sival, D.A.; Kremer, H.P.; De Koning, T.J.; Tijssen, M.A. A novel diagnostic approach to patients with myoclonus. Nat. Rev. Neurol. 2015, 11, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.P.; Bain, P.; Bajaj, N.; Elble, R.J.; Hallett, M.; Louis, E.D.; Raethjen, J.; Stamelou, M.; Testa, C.M.; Deuschl, G.; et al. Consensus Statement on the classification of tremors. from the task force on tremor of the International Parkinson and Movement Disorder Society. Mov. Disord. 2018, 33, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Lang, A.; Van De Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the International Parkinson and Movement Disorder Society task force. Mov. Disord. 2017, 32, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Praticò, A.D.; Pavone, V.; Lubrano, R.; Falsaperla, R.; Rizzo, R.; Ruggieri, M. Ataxia in children: Early recognition and clinical evaluation. Ital. J. Pediatr. 2017, 43, 6. [Google Scholar] [CrossRef]
- Koene, S.; Van Bon, L.; Bertini, E.; Jimenez-Moreno, C.; Van Der Giessen, L.; De Groot, I.; McFarland, R.; Parikh, S.; Rahman, S.; Wood, M.; et al. Outcome measures for children with mitochondrial disease: Consensus recommendations for future studies from a Delphi-based international workshop. J. Inherit. Metab. Dis. 2018, 41, 1267–1273. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Hubsch, T.; Du Montcel, S.T.; Baliko, L.; Berciano, J.; Boesch, S.; Depondt, C.; Giunti, P.; Globas, C.; Infante, J.; Kang, J.-S.; et al. Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology 2006, 66, 1717–1720. [Google Scholar] [CrossRef]
- Battini, R.; Sgandurra, G.; Petacchi, E.; Guzzetta, A.; Di Pietro, R.; Giannini, M.T.; Leuzzi, V.; Mercuri, E.; Cioni, G. Movement Disorder-Childhood rating scale: Reliability and validity. Pediatr. Neurol. 2008, 39, 259–265. [Google Scholar] [CrossRef]
- Parikh, S.; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R. A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Scaglia, F. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004, 114, 925–931. [Google Scholar] [CrossRef]
- Filip, P.; Lungu, O.V.; Bareš, M. Dystonia and the cerebellum: A new field of interest in movement disorders? Clin. Neurophysiol. 2013, 124, 1269–1276. [Google Scholar] [CrossRef]
- Rees, E.M.; Farmer, R.; Cole, J.H.; Haider, S.; Durr, A.; Landwehrmeyer, B.; Scahill, R.I.; Tabrizi, S.J.; Hobbs, N.Z. Cerebellar abnormalities in Huntington’s disease: A role in motor and psychiatric impairment? Mov. Disord. 2014, 29, 1648–1654. [Google Scholar] [CrossRef]
- Ganos, C.; Kassavetis, P.; Erro, R.; Edwards, M.J.; Rothwell, J.; Bhatia, K.P. The role of the cerebellum in the pathogenesis of cortical myoclonus. Mov. Disord. 2014, 29, 437–443. [Google Scholar] [CrossRef]
- Shakkottai, V.G.; Batla, A.; Bhatia, K.; Dauer, W.T.; Dresel, C.; Niethammer, M.; Eidelberg, D.; Raike, R.S.; Smith, Y.; Jinnah, H.A.; et al. Current opinions and areas of consensus on the role of the cerebellum in dystonia. Cerebellum 2017, 16, 577–594. [Google Scholar] [CrossRef] [Green Version]
- Koy, A.; Lin, J.-P.; Sanger, T.D.; A Marks, W.; Mink, J.W.; Timmermann, L. Advances in management of movement disorders in children. Lancet Neurol. 2016, 15, 719–735. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S. Mitochondrial extrapyramidal syndromes: Using age and phenomenology to guide genetic testing. JAMA Neurol. 2016, 73, 630–632. [Google Scholar] [CrossRef]
- Tolomeo, D.; Rubegni, A.; Severino, M.; Pochiero, F.; Bruno, C.; Cassandrini, D.; Madeo, A.; Doccini, S.; Pedemonte, M.; Rossi, A.; et al. Clinical and neuroimaging features of the m.10197G>A mtDNA mutation: New case reports and expansion of the phenotype variability. J. Neurol. Sci. 2019, 399, 69–75. [Google Scholar] [CrossRef]
- Ji, K.; Zhao, B.; Lin, Y.; Wang, W.; Liu, F.; Li, W.; Zhao, Y.; Yan, C. “Myo-neuropathy” is commonly associated with mitochondrial tRNALysine mutation. J. Neurol. 2020, 267, 3319–3328. [Google Scholar] [CrossRef]
- Eggink, H.; Kuiper, A.; Peall, K.J.; Contarino, M.F.; Bosch, A.M.; Post, B.; A Sival, D.; Tijssen, M.A.J.; De Koning, T.J. Rare inborn errors of metabolism with movement disorders: A case study to evaluate the impact upon quality of life and adaptive functioning. Orphanet J. Rare Dis. 2014, 9, 177. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.; Galli, R.; Pizzanelli, C.; Filosto, M.; Siciliano, G.; Murri, L. Antimyoclonic effect of levetiracetam in MERRF syndrome. J. Neurol. Sci. 2006, 243, 97–99. [Google Scholar] [CrossRef]
- Tinker, R.J.; Lim, A.Z.; Stefanetti, R.J.; McFarland, R. Current and emerging clinical treatment in mitochondrial disease. Mol. Diagn. Ther. 2021, 25, 181–206. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Phenotype | Movement Disorders: Yes (n = 197) | Movement Disorders: No (n = 383) | Significance Level |
|---|---|---|---|
| Generalized myopathy | 151 (76.6%) | 160 (41.8%) | p< 0.001 |
| Cognitive involvement | 103 (52.3%) | 64 (16.7%) | p< 0.001 |
| Ocular myopathy | 88 (44.7%) | 147 (38.4%) | n.s. |
| Pyramidal involvement | 78 (39.6%) | 39 (10.2%) | p< 0.001 |
| Hearing loss | 73 (37.1%) | 58 (15.1%) | p< 0.001 |
| Failure to thrive and short stature | 70 (35.5%) | 55 (14.4%) | p< 0.001 |
| Epileptic seizures | 58 (29.4%) | 49 (12.8%) | p< 0.001 |
| Peripheral neuropathy | 43 (21.8%) | 19 (5.0%) | p< 0.001 |
| Swallowing impairment | 38 (19.3%) | 31 (8.1%) | p< 0.001 |
| Retinopathy | 34 (17.3%) | 20 (5.2%) | p< 0.001 |
| Migraine | 34 (17.3%) | 31 (8.1%) | p= 0.001 |
| Gastrointestinal dysmotility/vomiting | 31 (15.7%) | 33 (8.6%) | n.s. |
| Respiratory involvement | 29 (14.7%) | 25 (6.5%) | p= 0.002 |
| Cardiac involvement | 27 (13.7%) | 41 (10.7%) | n.s. |
| Optic neuropathy | 26 (13.2%) | 133 (34.7%) | p< 0.001 |
| Entire Sample (n = 98) | Hyperkinetic Onset (n = 41) | Hypokinetic Onset (n = 9) | Ataxic Onset (n = 48) | |
|---|---|---|---|---|
| Basal ganglia abnormalities | 53 (54.1%) | 24 (58.5%) | 6 (66.7%) | 23 (47.9%) |
| Cerebral white matter abnormalities | 47 (48.0%) | 20 (48.8%) | 6 (66.7%) | 21 (43.8%) |
| Cerebellar atrophy | 41 (41.8%) | 10 (24.4%) | 3 (33.3%) | 28 (58.3%) |
| p= 0.002 | ||||
| Cerebral atrophy | 34 (34.7%) | 15 (36.6%) | 3 (33.3%) | 16 (33.3%) |
| Cerebellar white matter abnormalities | 19 (19.4%) | 6 (14.6%) | 1 (11.1%) | 12 (25.0%) |
| Brainstem atrophy | 11 (11.2%) | 5 (12.2%) | 2 (22.2%) | 4 (8.3%) |
| Thalamic/subthalamic involvement | 13 (13.3%) | 5 (12.2%) | 1 (11.1%) | 7 (14.6%) |
| Dentate nucleus alterations | 11 (11.2%) | 5 (12.2%) | 2 (22.2%) | 4 (8.3%) |
| Stroke-like lesions | 5 (5.1%) | 1 (2.4%) | 2 (22.2%) | 2 (4.2%) |
| Cortical laminar necrosis | 4 (4.1%) | 2 (4.9%) | 1 (11.1%) | 1 (2.1%) |
| MUTATION | Entire Sample (n = 92) | Hyperkinetic Onset (n = 39) | Hypokinetic Onset (n = 9) | Ataxic Onset (n = 44) |
|---|---|---|---|---|
| Nuclear DNA mutations | 41 (44.6%) | 17 (43.6%) | 3 (33.3%) | 21 (47.7%) |
| mtDNA rearrangements | 10 (10.9%) | 5 (12.8%) | 2 (22.2%) | 3 (6.8%) |
| m.8993T > G | 8 (8.7%) | 1 (2.6%) | 0 (0.0%) | 7 (15.9%) |
| m.8344A > G | 6 (6.5%) | 4 (10.3%) | 0 (0.0%) | 2 (4.5%) |
| m.3243A > G | 6 (6.5%) | 1 (2.6%) | 1 (11.1%) | 4 (9.1%) |
| MT-ND3 mutations | 6 (6.5%) | 5 (12.8%) | 1 (11.1%) | 0 (0.0%) |
| Additional mtDNA mutations | 15 (16.3%) | 6 (15.4%) | 2 (22.2%) | 7 (15.9%) |
| Ataxia (10 years) | Subtypes cerebellar (32/50), spino-cerebellar (16/50), sensory (7/50) |
| Body sites / functions involved trunk (18/50), limbs (33/50), walking (39/50) | |
| Features associated dysmetria 35/50, dysarthria (26/50), adiadokinesia (16/50), nystagmus (9/50), hyporeflexia/areflexia (20/50), hyperreflexia (2/50), hypotonia (15/50), hypertonia (3/50) | |
| Dystonia (1 year) | Body distribution focal (2/18), multifocal (4/18), generalized (10/18), hemidystonia (2/18) |
| Temporal pattern persistent (16/18), action specific (1/18), paroxysmal (1/18) | |
| Tremor (8 years) | Body distribution focal (5/14), segmental (7/14), generalized (2/14) |
| Activation conditions rest-tremor (3/14), postural tremor (4/14), simple kinetic tremor (2/14), intention tremor (3/14), task specific tremor (2/14) | |
| Hypokinetic disorder (10 years) | Features associated bradykinesia (9/9), stiffness (6/9), hypomimia (3/9), postural instability (2/9) |
| Chorea (1 year) | Body distribution limbs (5/5), trunk (1/5), face (1/5 |
| Myoclonus (11 years) | Body distribution multifocal (1/4), generalized (3/4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ticci, C.; Orsucci, D.; Ardissone, A.; Bello, L.; Bertini, E.; Bonato, I.; Bruno, C.; Carelli, V.; Diodato, D.; Doccini, S.; et al. Movement Disorders in Children with a Mitochondrial Disease: A Cross-Sectional Survey from the Nationwide Italian Collaborative Network of Mitochondrial Diseases. J. Clin. Med. 2021, 10, 2063. https://doi.org/10.3390/jcm10102063
Ticci C, Orsucci D, Ardissone A, Bello L, Bertini E, Bonato I, Bruno C, Carelli V, Diodato D, Doccini S, et al. Movement Disorders in Children with a Mitochondrial Disease: A Cross-Sectional Survey from the Nationwide Italian Collaborative Network of Mitochondrial Diseases. Journal of Clinical Medicine. 2021; 10(10):2063. https://doi.org/10.3390/jcm10102063
Chicago/Turabian StyleTicci, Chiara, Daniele Orsucci, Anna Ardissone, Luca Bello, Enrico Bertini, Irene Bonato, Claudio Bruno, Valerio Carelli, Daria Diodato, Stefano Doccini, and et al. 2021. "Movement Disorders in Children with a Mitochondrial Disease: A Cross-Sectional Survey from the Nationwide Italian Collaborative Network of Mitochondrial Diseases" Journal of Clinical Medicine 10, no. 10: 2063. https://doi.org/10.3390/jcm10102063
APA StyleTicci, C., Orsucci, D., Ardissone, A., Bello, L., Bertini, E., Bonato, I., Bruno, C., Carelli, V., Diodato, D., Doccini, S., Donati, M. A., Dosi, C., Filosto, M., Fiorillo, C., La Morgia, C., Lamperti, C., Marchet, S., Martinelli, D., Minetti, C., ... Santorelli, F. M. (2021). Movement Disorders in Children with a Mitochondrial Disease: A Cross-Sectional Survey from the Nationwide Italian Collaborative Network of Mitochondrial Diseases. Journal of Clinical Medicine, 10(10), 2063. https://doi.org/10.3390/jcm10102063