
Neuropathology of the Brainstem to Mechanistically Understand and to Treat Alzheimer’s Disease

{kind=link}
Abstract
1. Introduction
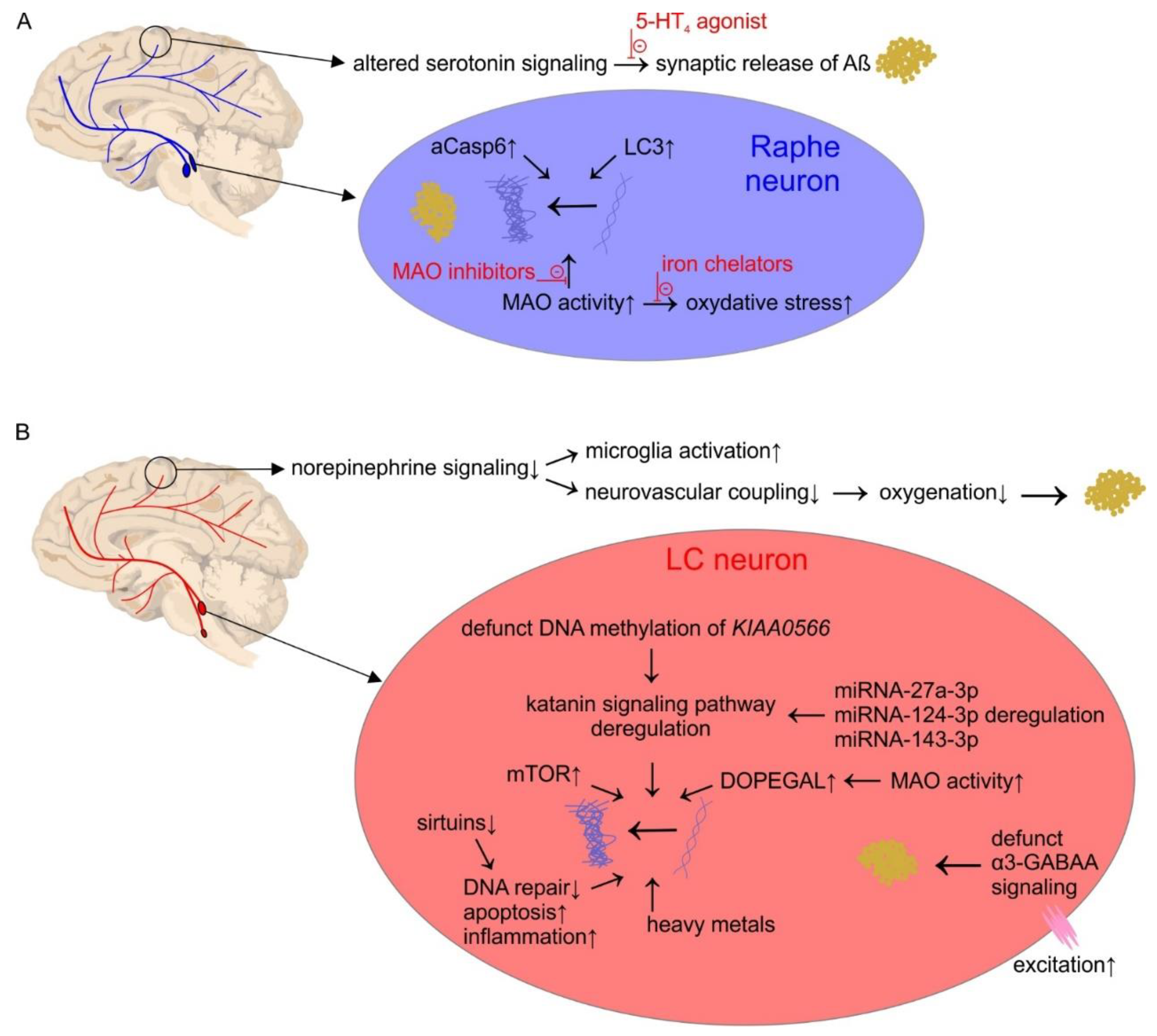
2. The Serotonin System
Pathophysiology
3. The Norepinephrine System
Pathophysiology
4. The Dopamine System
5. Co-Release of Biogenic Amines
6. Brainstem Monoaminergic Nuclei in AD: Interconnections and Afferent Loss
7. Other Brainstem Pathologies in AD
8. Diagnostic Possibilities of Brainstem Pathology in AD
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.C.; Gong, C.X.; Khatoon, S.; Pei, J.J.; Wang, J.Z.; Grundke-Iqbal, I. Mechanisms of neurofibrillary degeneration and the formation of neurofibrillary tangles. J. Neural Transm. Suppl. 1998, 53, 169–180. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E.; Bohl, J. Staging of Alzheimer-related cortical destruction. Eur. Neurol. 1993, 33, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Gold, G.; von Gunten, A.; Hof, P.R.; Bouras, C. Pathological substrates of cognitive decline in Alzheimer’s disease. Front. Neurol. Neurosci. 2009, 24, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.; Strijkstra, A.M.; Hut, R.A.; Rudiger, J.; Van der Zee, E.A.; Harkány, T.; Holzer, M.; Härtig, W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci. 2003, 23, 6972–6981. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012, 123, 157–171. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Kharitonova, E.K.; Bacskai, B.J. Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease. Cells 2020, 9, 2513. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Simic, G.; Babic Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buee, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef] [PubMed]
- Green, J.P. Histamin and serotonin. In Basic Neurochemistry, 4th ed.; Siegel, G.J., Agranoff, B., Albers, R.W., Molinoff, P., Eds.; Raven Press: New York, NY, USA, 1987; pp. 253–269. [Google Scholar]
- Walther, D.J.; Peter, J.U.; Bashammakh, S.; Hortnagl, H.; Voits, M.; Fink, H.; Bader, M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 2003, 299, 76. [Google Scholar] [CrossRef]
- Levitt, P.; Pintar, J.E.; Breakefield, X.O. Immunocytochemical demonstration of monoamine oxidase B in brain astrocytes and serotonergic neurons. Proc. Natl. Acad. Sci. USA 1982, 79, 6385–6389. [Google Scholar] [CrossRef]
- Edmondson, D.E. Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: Biological implications. Curr. Pharm. Des. 2014, 20, 155–160. [Google Scholar] [CrossRef]
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Blakely, R.D.; Berson, H.E.; Fremeau, R.T., Jr.; Caron, M.G.; Peek, M.M.; Prince, H.K.; Bradley, C.C. Cloning and expression of a functional serotonin transporter from rat brain. Nature 1991, 354, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.J.; Mezey, E.; Brownstein, M.J. Cloning of a serotonin transporter affected by antidepressants. Science 1991, 254, 579–580. [Google Scholar] [CrossRef]
- Kupfer, D.J.; Frank, E.; Phillips, M.L. Major Depressive Disorder: New Clinical, Neurobiological, and Treatment Perspectives. Focus (Am. Psychiatr. Publ.) 2016, 14, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Spies, M.; Knudsen, G.M.; Lanzenberger, R.; Kasper, S. The serotonin transporter in psychiatric disorders: Insights from PET imaging. Lancet Psychiatry 2015, 2, 743–755. [Google Scholar] [CrossRef]
- Fuxe, K.; Dahlstrom, A.B.; Jonsson, G.; Marcellino, D.; Guescini, M.; Dam, M.; Manger, P.; Agnati, L. The discovery of central monoamine neurons gave volume transmission to the wired brain. Prog. Neurobiol. 2010, 90, 82–100. [Google Scholar] [CrossRef] [PubMed]
- De-Miguel, F.F.; Trueta, C. Synaptic and extrasynaptic secretion of serotonin. Cell. Mol. Neurobiol. 2005, 25, 297–312. [Google Scholar] [CrossRef]
- Darmon, M.; Al Awabdh, S.; Emerit, M.B.; Masson, J. Insights into Serotonin Receptor Trafficking: Cell Membrane Targeting and Internalization. Prog. Mol. Biol. Transl. Sci. 2015, 132, 97–126. [Google Scholar] [CrossRef]
- Dahlstroem, A.; Fuxe, K. Evidence for the Existence of Monoamine-Containing Neurons in the Central Nervous System. I. Demonstration of Monoamines in the Cell Bodies of Brain Stem Neurons. Acta Physiol. Scand. Suppl. 1964, 232, 231–255. [Google Scholar]
- Steinbusch, H.W. Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience 1981, 6, 557–618. [Google Scholar] [CrossRef]
- Baker, K.G.; Halliday, G.M.; Hornung, J.P.; Geffen, L.B.; Cotton, R.G.; Tork, I. Distribution, morphology and number of monoamine-synthesizing and substance P-containing neurons in the human dorsal raphe nucleus. Neuroscience 1991, 42, 757–775. [Google Scholar] [CrossRef]
- Hornung, J.P. The human raphe nuclei and the serotonergic system. J. Chem. Neuroanat. 2003, 26, 331–343. [Google Scholar] [CrossRef]
- Aitken, A.R.; Tork, I. Early development of serotonin-containing neurons and pathways as seen in wholemount preparations of the fetal rat brain. J. Comp. Neurol. 1988, 274, 32–47. [Google Scholar] [CrossRef]
- Kosofsky, B.E.; Molliver, M.E. The serotoninergic innervation of cerebral cortex: Different classes of axon terminals arise from dorsal and median raphe nuclei. Synapse 1987, 1, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Raghanti, M.A.; Stimpson, C.D.; Marcinkiewicz, J.L.; Erwin, J.M.; Hof, P.R.; Sherwood, C.C. Differences in cortical serotonergic innervation among humans, chimpanzees, and macaque monkeys: A comparative study. Cereb. Cortex 2008, 18, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Marklund, P.; Fransson, P.; Cabeza, R.; Petersson, K.M.; Ingvar, M.; Nyberg, L. Sustained and transient neural modulations in prefrontal cortex related to declarative long-term memory, working memory, and attention. Cortex 2007, 43, 22–37. [Google Scholar] [CrossRef]
- Le Verche, V.; Kaindl, A.M.; Verney, C.; Csaba, Z.; Peineau, S.; Olivier, P.; Adle-Biassette, H.; Leterrier, C.; Vitalis, T.; Renaud, J.; et al. The somatostatin 2A receptor is enriched in migrating neurons during rat and human brain development and stimulates migration and axonal outgrowth. PLoS ONE 2009, 4, e5509. [Google Scholar] [CrossRef] [PubMed]
- Cases, O.; Lebrand, C.; Giros, B.; Vitalis, T.; De Maeyer, E.; Caron, M.G.; Price, D.J.; Gaspar, P.; Seif, I. Plasma membrane transporters of serotonin, dopamine, and norepinephrine mediate serotonin accumulation in atypical locations in the developing brain of monoamine oxidase A knock-outs. J. Neurosci. 1998, 18, 6914–6927. [Google Scholar] [CrossRef]
- Dayer, A. Serotonin-related pathways and developmental plasticity: Relevance for psychiatric disorders. Dialogues Clin. Neurosci. 2014, 16, 29–41. [Google Scholar]
- Kraus, C.; Castren, E.; Kasper, S.; Lanzenberger, R. Serotonin and neuroplasticity—Links between molecular, functional and structural pathophysiology in depression. Neurosci. Biobehav. Rev. 2017, 77, 317–326. [Google Scholar] [CrossRef]
- Whitaker-Azmitia, P.M. The discovery of serotonin and its role in neuroscience. Neuropsychopharmacology 1999, 21, 2S–8S. [Google Scholar] [CrossRef]
- Pytliak, M.; Vargova, V.; Mechirova, V.; Felsoci, M. Serotonin receptors—From molecular biology to clinical applications. Physiol. Res. 2011, 60, 15–25. [Google Scholar] [CrossRef]
- Barrett, F.S.; Workman, C.I.; Sair, H.I.; Savonenko, A.V.; Kraut, M.A.; Sodums, D.J.; Joo, J.J.; Nassery, N.; Marano, C.M.; Munro, C.A.; et al. Association between serotonin denervation and resting-state functional connectivity in mild cognitive impairment. Hum. Brain Mapp. 2017, 38, 3391–3401. [Google Scholar] [CrossRef]
- Smith, G.S.; Barrett, F.S.; Joo, J.H.; Nassery, N.; Savonenko, A.; Sodums, D.J.; Marano, C.M.; Munro, C.A.; Brandt, J.; Kraut, M.A.; et al. Molecular imaging of serotonin degeneration in mild cognitive impairment. Neurobiol. Dis. 2017, 105, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Simic, G.; Stanic, G.; Mladinov, M.; Jovanov-Milosevic, N.; Kostovic, I.; Hof, P.R. Does Alzheimer’s disease begin in the brainstem? Neuropathol. Appl. Neurobiol. 2009, 35, 532–554. [Google Scholar] [CrossRef]
- Deakin, J.F. Depression and antisocial personality disorder: Two contrasting disorders of 5HT function. J. Neural Transm. Suppl. 2003, 79–93. [Google Scholar] [CrossRef]
- Lei, S. Serotonergic modulation of Neural activities in the entorhinal cortex. Int. J. Physiol. Pathophysiol. Pharmacol. 2012, 4, 201–210. [Google Scholar]
- Gompf, H.S.; Anaclet, C. The neuroanatomy and neurochemistry of sleep-wake control. Curr. Opin. Physiol. 2020, 15, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Saper, C.B. The central circadian timing system. Curr. Opin. Neurobiol. 2013, 23, 747–751. [Google Scholar] [CrossRef]
- Todd, W.D.; Fenselau, H.; Wang, J.L.; Zhang, R.; Machado, N.L.; Venner, A.; Broadhurst, R.Y.; Kaur, S.; Lynagh, T.; Olson, D.P.; et al. A hypothalamic circuit for the circadian control of aggression. Nat. Neurosci. 2018, 21, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Scarmeas, N.; Brandt, J.; Blacker, D.; Albert, M.; Hadjigeorgiou, G.; Dubois, B.; Devanand, D.; Honig, L.; Stern, Y. Disruptive behavior as a predictor in Alzheimer disease. Arch. Neurol. 2007, 64, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.M.; Gerstner, J.R.; Holtzman, D.M. The sleep-wake cycle and Alzheimer’s disease: What do we know? Neurodegener. Dis. Manag. 2014, 4, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Schneider, L. Treatment Options for Agitation in Dementia. Curr. Treat. Options Neurol. 2019, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Amyloid-beta may be released from non-junctional varicosities of axons generated from abnormal tau-containing brainstem nuclei in sporadic Alzheimer’s disease: A hypothesis. Acta Neuropathol. 2013, 126, 303–306. [Google Scholar] [CrossRef]
- Rub, U.; Del Tredici, K.; Schultz, C.; Thal, D.R.; Braak, E.; Braak, H. The evolution of Alzheimer’s disease-related cytoskeletal pathology in the human raphe nuclei. Neuropathol. Appl. Neurobiol. 2000, 26, 553–567. [Google Scholar] [CrossRef]
- Ehrenberg, A.J.; Nguy, A.K.; Theofilas, P.; Dunlop, S.; Suemoto, C.K.; Di Lorenzo Alho, A.T.; Leite, R.P.; Diehl Rodriguez, R.; Mejia, M.B.; Rub, U.; et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: The pathological building blocks of early Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Kemper, T. Nucleus raphe dorsalis in dementia of the Alzheimer type: Neurofibrillary changes and neuronal packing density. J. Neuropathol. Exp. Neurol. 1984, 43, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Webster, M.T.; Chessell, I.P.; Holmes, C.; Stratmann, G.C.; Procter, A.W.; Cross, A.J.; Green, A.R.; Bowen, D.M. Neurotransmitters and second messengers in aging and Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1993, 695, 19–26. [Google Scholar] [CrossRef]
- Chan-Palay, V.; Asan, E. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson’s disease with and without dementia and depression. J. Comp. Neurol. 1989, 287, 373–392. [Google Scholar] [CrossRef]
- Chen, C.P.; Eastwood, S.L.; Hope, T.; McDonald, B.; Francis, P.T.; Esiri, M.M. Immunocytochemical study of the dorsal and median raphe nuclei in patients with Alzheimer’s disease prospectively assessed for behavioural changes. Neuropathol. Appl. Neurobiol. 2000, 26, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Wilcock, G.K.; Esiri, M.M.; Francis, P.T.; Bowen, D.M. Monoaminergic innervation of the frontal and temporal lobes in Alzheimer’s disease. Brain Res. 1987, 401, 231–238. [Google Scholar] [CrossRef]
- Trillo, L.; Das, D.; Hsieh, W.; Medina, B.; Moghadam, S.; Lin, B.; Dang, V.; Sanchez, M.M.; De Miguel, Z.; Ashford, J.W.; et al. Ascending monoaminergic systems alterations in Alzheimer’s disease. Translating basic science into clinical care. Neurosci. Biobehav. Rev. 2013, 37, 1363–1379. [Google Scholar] [CrossRef]
- Gottfries, C.G.; Bartfai, T.; Carlsson, A.; Eckernas, S.; Svennerholm, L. Multiple biochemical deficits in both gray and white matter of Alzheimer brains. Prog. Neuropsychopharmacol. Biol. Psychiatry 1986, 10, 405–413. [Google Scholar] [CrossRef]
- Ramon y Cajal, S. Histologie du Systeme Nerveux de l’Homme et des Vertébrés; Maloine: Paris, France, 1955; Volume II. [Google Scholar]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Kepe, V.; Barrio, J.R.; Huang, S.C.; Ercoli, L.; Siddarth, P.; Shoghi-Jadid, K.; Cole, G.M.; Satyamurthy, N.; Cummings, J.L.; Small, G.W.; et al. Serotonin 1A receptors in the living brain of Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2006, 103, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.K.; Tsang, S.W.; Francis, P.T.; Esiri, M.M.; Keene, J.; Hope, T.; Chen, C.P. Reduced serotonin 5-HT1A receptor binding in the temporal cortex correlates with aggressive behavior in Alzheimer disease. Brain Res. 2003, 974, 82–87. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Hirst, W.D.; Chen, C.P.; Lasheras, B.; Francis, P.T.; Ramirez, M.J. Differential involvement of 5-HT(1B/1D) and 5-HT6 receptors in cognitive and non-cognitive symptoms in Alzheimer’s disease. Neuropsychopharmacology 2004, 29, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Marner, L.; Frokjaer, V.G.; Kalbitzer, J.; Lehel, S.; Madsen, K.; Baare, W.F.; Knudsen, G.M.; Hasselbalch, S.G. Loss of serotonin 2A receptors exceeds loss of serotonergic projections in early Alzheimer’s disease: A combined [11C]DASB and [18F]altanserin-PET study. Neurobiol. Aging 2012, 33, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Van der Schyf, C.J. Role of serotonin in Alzheimer’s disease: A new therapeutic target? CNS Drugs 2011, 25, 765–781. [Google Scholar] [CrossRef]
- Schechter, L.E.; Smith, D.L.; Rosenzweig-Lipson, S.; Sukoff, S.J.; Dawson, L.A.; Marquis, K.; Jones, D.; Piesla, M.; Andree, T.; Nawoschik, S.; et al. Lecozotan (SRA-333): A selective serotonin 1A receptor antagonist that enhances the stimulated release of glutamate and acetylcholine in the hippocampus and possesses cognitive-enhancing properties. J. Pharmacol. Exp. Ther. 2005, 314, 1274–1289. [Google Scholar] [CrossRef]
- Shen, F.; Smith, J.A.; Chang, R.; Bourdet, D.L.; Tsuruda, P.R.; Obedencio, G.P.; Beattie, D.T. 5-HT(4) receptor agonist mediated enhancement of cognitive function in vivo and amyloid precursor protein processing in vitro: A pharmacodynamic and pharmacokinetic assessment. Neuropharmacology 2011, 61, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Marcos, B.; Gil-Bea, F.J.; Hirst, W.D.; Garcia-Alloza, M.; Ramirez, M.J. Lack of localization of 5-HT6 receptors on cholinergic neurons: Implication of multiple neurotransmitter systems in 5-HT6 receptor-mediated acetylcholine release. Eur. J. Neurosci. 2006, 24, 1299–1306. [Google Scholar] [CrossRef]
- Ramirez, B.G.; Blazquez, C.; Gomez del Pulgar, T.; Guzman, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef]
- Berthoux, C.; Hamieh, A.M.; Rogliardo, A.; Doucet, E.L.; Coudert, C.; Ango, F.; Grychowska, K.; Chaumont-Dubel, S.; Zajdel, P.; Maldonado, R.; et al. Early 5-HT6 receptor blockade prevents symptom onset in a model of adolescent cannabis abuse. EMBO Mol. Med. 2020, 12, e10605. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Where, when, and in what form does sporadic Alzheimer’s disease begin? Curr. Opin. Neurol. 2012, 25, 708–714. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Tipton, K.F. Enzymology of monoamine oxidase. Cell Biochem. Funct. 1986, 4, 79–87. [Google Scholar] [CrossRef]
- Manzoor, S.; Hoda, N. A comprehensive review of monoamine oxidase inhibitors as Anti-Alzheimer’s disease agents: A review. Eur. J. Med. Chem. 2020, 206, 112787. [Google Scholar] [CrossRef]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Weiner, L.M.; Bar-Am, O.; Epsztejn, S.; Cabantchik, Z.I.; Warshawsky, A.; Youdim, M.B.; Fridkin, M. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg. Med. Chem. 2005, 13, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Ruffolo, R.R., Jr. alpha-Adrenoceptors. Monogr. Neural Sci. 1984, 10, 224–252. [Google Scholar] [PubMed]
- Pacholczyk, T.; Blakely, R.D.; Amara, S.G. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature 1991, 350, 350–354. [Google Scholar] [CrossRef]
- Descarries, L.; Mechawar, N. Ultrastructural evidence for diffuse transmission by monoamine and acetylcholine neurons of the central nervous system. Prog. Brain Res. 2000, 125, 27–47. [Google Scholar] [CrossRef]
- Foote, S.L.; Bloom, F.E.; Aston-Jones, G. Nucleus locus ceruleus: New evidence of anatomical and physiological specificity. Physiol. Rev. 1983, 63, 844–914. [Google Scholar] [CrossRef] [PubMed]
- Poe, G.R.; Foote, S.; Eschenko, O.; Johansen, J.P.; Bouret, S.; Aston-Jones, G.; Harley, C.W.; Manahan-Vaughan, D.; Weinshenker, D.; Valentino, R.; et al. Locus coeruleus: A new look at the blue spot. Nat. Rev. Neurosci. 2020, 21, 644–659. [Google Scholar] [CrossRef]
- Paspalas, C.D.; Papadopoulos, G.C. Ultrastructural relationships between noradrenergic nerve fibers and non-neuronal elements in the rat cerebral cortex. Glia 1996, 17, 133–146. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Negoro, R.; Kadoi, H.; Motegi, T.; Shibagaki, F.; Yamamuro, A.; Ishimaru, Y.; Maeda, S. Noradrenaline protects neurons against H2 O2 -induced death by increasing the supply of glutathione from astrocytes via beta3-adrenoceptor stimulation. J. Neurosci. Res. 2020. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Biagioni, F.; Galgani, A.; Pavese, N.; Lazzeri, G.; Fornai, F. Locus Coeruleus Modulates Neuroinflammation in Parkinsonism and Dementia. Int. J. Mol. Sci. 2020, 21, 8630. [Google Scholar] [CrossRef]
- Hartman, B.K.; Zide, D.; Udenfriend, S. The use of dopamine -hydroxylase as a marker for the central noradrenergic nervous system in rat brain. Proc. Natl. Acad. Sci. USA 1972, 69, 2722–2726. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Galgani, A.; Puglisi-Allegra, S.; Limanaqi, F.; Busceti, C.L.; Fornai, F. Locus Coeruleus and neurovascular unit: From its role in physiology to its potential role in Alzheimer’s disease pathogenesis. J. Neurosci. Res. 2020, 98, 2406–2434. [Google Scholar] [CrossRef] [PubMed]
- Swanson, L.W.; Hartman, B.K. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J. Comp. Neurol. 1975, 163, 467–505. [Google Scholar] [CrossRef]
- Huang, H.P.; Zhu, F.P.; Chen, X.W.; Xu, Z.Q.; Zhang, C.X.; Zhou, Z. Physiology of quantal norepinephrine release from somatodendritic sites of neurons in locus coeruleus. Front. Mol. Neurosci. 2012, 5, 29. [Google Scholar] [CrossRef]
- Starke, K. Presynaptic autoreceptors in the third decade: Focus on alpha2-adrenoceptors. J. Neurochem. 2001, 78, 685–693. [Google Scholar] [CrossRef]
- Szabadi, E. Functional neuroanatomy of the central noradrenergic system. J. Psychopharmacol. 2013, 27, 659–693. [Google Scholar] [CrossRef]
- Moore, R.Y.; Bloom, F.E. Central catecholamine neuron systems: Anatomy and physiology of the norepinephrine and epinephrine systems. Annu. Rev. Neurosci. 1979, 2, 113–168. [Google Scholar] [CrossRef]
- Domyancic, A.V.; Morilak, D.A. Distribution of alpha1A adrenergic receptor mRNA in the rat brain visualized by in situ hybridization. J. Comp. Neurol. 1997, 386, 358–378. [Google Scholar] [CrossRef]
- Pieribone, V.A.; Nicholas, A.P.; Dagerlind, A.; Hokfelt, T. Distribution of alpha 1 adrenoceptors in rat brain revealed by in situ hybridization experiments utilizing subtype-specific probes. J. Neurosci. 1994, 14, 4252–4268. [Google Scholar] [CrossRef] [PubMed]
- Gritti, I.; Mainville, L.; Mancia, M.; Jones, B.E. GABAergic and other noncholinergic basal forebrain neurons, together with cholinergic neurons, project to the mesocortex and isocortex in the rat. J. Comp. Neurol. 1997, 383, 163–177. [Google Scholar] [CrossRef]
- Hermann, D.M.; Luppi, P.H.; Peyron, C.; Hinckel, P.; Jouvet, M. Afferent projections to the rat nuclei raphe magnus, raphe pallidus and reticularis gigantocellularis pars alpha demonstrated by iontophoretic application of choleratoxin (subunit b). J. Chem. Neuroanat. 1997, 13, 1–21. [Google Scholar] [CrossRef]
- Samuels, E.R.; Szabadi, E. Functional neuroanatomy of the noradrenergic locus coeruleus: Its roles in the regulation of arousal and autonomic function part II: Physiological and pharmacological manipulations and pathological alterations of locus coeruleus activity in humans. Curr. Neuropharmacol. 2008, 6, 254–285. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.B.; Ray, B.; Lahiri, D.K.; Tilmont, E.M.; Tinkler, G.P.; Herbert, R.L.; Greig, N.H.; Ingram, D.K.; Ottinger, M.A.; Mattison, J.A. Effects of Reducing Norepinephrine Levels via DSP4 Treatment on Amyloid-beta Pathology in Female Rhesus Macaques (Macaca Mulatta). J. Alzheimers Dis. 2019, 68, 115–126. [Google Scholar] [CrossRef]
- Grudzien, A.; Shaw, P.; Weintraub, S.; Bigio, E.; Mash, D.C.; Mesulam, M.M. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging 2007, 28, 327–335. [Google Scholar] [CrossRef]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M.; Rossor, M.N.; Iversen, L.L.; Reynolds, G.P.; Hauser, D.L. Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1987, 1, 256–262. [Google Scholar] [CrossRef]
- Heneka, M.T.; Nadrigny, F.; Regen, T.; Martinez-Hernandez, A.; Dumitrescu-Ozimek, L.; Terwel, D.; Jardanhazi-Kurutz, D.; Walter, J.; Kirchhoff, F.; Hanisch, U.K.; et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. USA 2010, 107, 6058–6063. [Google Scholar] [CrossRef]
- Andres-Benito, P.; Fernandez-Duenas, V.; Carmona, M.; Escobar, L.A.; Torrejon-Escribano, B.; Aso, E.; Ciruela, F.; Ferrer, I. Locus coeruleus at asymptomatic early and middle Braak stages of neurofibrillary tangle pathology. Neuropathol. Appl. Neurobiol. 2017, 43, 373–392. [Google Scholar] [CrossRef]
- Bekar, L.K.; Wei, H.S.; Nedergaard, M. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J. Cereb. Blood Flow Metab. 2012, 32, 2135–2145. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Heneka, M.T.; Gavrilyuk, V.; Dello Russo, C.; Weinberg, G.; Galea, E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem. Int. 2002, 41, 357–365. [Google Scholar] [CrossRef]
- Pamphlett, R.; Kum Jew, S. Different Populations of Human Locus Ceruleus Neurons Contain Heavy Metals or Hyperphosphorylated Tau: Implications for Amyloid-beta and Tau Pathology in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 45, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Mak, R.; Lee, J.; Buckland, M.E.; Harding, A.J.; Kum Jew, S.; Paterson, D.J.; Jones, M.W.M.; Lay, P.A. Concentrations of toxic metals and essential trace elements vary among individual neurons in the human locus ceruleus. PLoS ONE 2020, 15, e0233300. [Google Scholar] [CrossRef] [PubMed]
- Mravec, B.; Lejavova, K.; Cubinkova, V. Locus (coeruleus) minoris resistentiae in pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 992–1001. [Google Scholar] [CrossRef]
- Andres-Benito, P.; Delgado-Morales, R.; Ferrer, I. Altered Regulation of KIAA0566, and Katanin Signaling Expression in the Locus Coeruleus With Neurofibrillary Tangle Pathology. Front. Cell. Neurosci. 2018, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Thune, K.; Andres-Benito, P.; Tahir, W.; Ansoleaga, B.; Hernandez-Ortega, K.; Marti, E.; Zerr, I.; Ferrer, I. MicroRNA Expression in the Locus Coeruleus, Entorhinal Cortex, and Hippocampus at Early and Middle Stages of Braak Neurofibrillary Tangle Pathology. J. Mol. Neurosci. 2017, 63, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Liu, X.; Ahn, E.H.; Xiang, J.; Manfredsson, F.P.; Yang, X.; Luo, H.R.; Liles, L.C.; Weinshenker, D.; Ye, K. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J. Clin. Investig. 2020, 130, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Magri, A.; Medina, D.X.; Wisely, E.V.; Lopez-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Spiers, J.G.; Chen, H.C.; Bourgognon, J.M.; Steinert, J.R. Dysregulation of stress systems and nitric oxide signaling underlies neuronal dysfunction in Alzheimer’s disease. Free Radic Biol. Med. 2019, 134, 468–483. [Google Scholar] [CrossRef]
- Kelly, L.; Seifi, M.; Ma, R.; Mitchell, S.J.; Rudolph, U.; Viola, K.L.; Klein, W.L.; Lambert, J.J.; Swinny, J.D. Identification of intraneuronal amyloid beta oligomers in locus coeruleus neurons of Alzheimer’s patients and their potential impact on inhibitory neurotransmitter receptors and neuronal excitability. Neuropathol. Appl. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Monsma, F.J., Jr.; Mahan, L.C.; McVittie, L.D.; Gerfen, C.R.; Sibley, D.R. Molecular cloning and expression of a D1 dopamine receptor linked to adenylyl cyclase activation. Proc. Natl. Acad. Sci. USA 1990, 87, 6723–6727. [Google Scholar] [CrossRef]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef]
- Bonoldi, I.; Howes, O.D. Presynaptic dopaminergic function: Implications for understanding treatment response in psychosis. CNS Drugs 2014, 28, 649–663. [Google Scholar] [CrossRef]
- Wei, W.; Ding, S.; Zhou, F.M. Dopaminergic treatment weakens medium spiny neuron collateral inhibition in the parkinsonian striatum. J. Neurophysiol. 2017, 117, 987–999. [Google Scholar] [CrossRef]
- Kilty, J.E.; Lorang, D.; Amara, S.G. Cloning and expression of a cocaine-sensitive rat dopamine transporter. Science 1991, 254, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Kitayama, S.; Lin, C.L.; Patel, A.; Nanthakumar, E.; Gregor, P.; Kuhar, M.; Uhl, G. Cloning and expression of a cocaine-sensitive dopamine transporter complementary DNA. Science 1991, 254, 576–578. [Google Scholar] [CrossRef]
- Isaacson, J.R.; Brillman, S.; Chhabria, N.; Isaacson, S.H. Impact of DaTscan Imaging on Clinical Decision Making in Clinically Uncertain Parkinson’s Disease. J. Parkinsons Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kalaba, P.; Ilic, M.; Aher, N.Y.; Dragacevic, V.; Wieder, M.; Zehl, M.; Wackerlig, J.; Beyl, S.; Sartori, S.B.; Ebner, K.; et al. Structure-Activity Relationships of Novel Thiazole-Based Modafinil Analogues Acting at Monoamine Transporters. J. Med. Chem. 2020, 63, 391–417. [Google Scholar] [CrossRef]
- Lindvall, O.; Bjorklund, A. Anatomy of the dopaminergic neuron systems in the rat brain. Adv. Biochem. Psychopharmacol. 1978, 19, 1–23. [Google Scholar] [PubMed]
- Stern, G. The effects of lesions in the substantia nigra. Brain 1966, 89, 449–478. [Google Scholar] [CrossRef]
- Bereczki, D. The description of all four cardinal signs of Parkinson’s disease in a Hungarian medical text published in 1690. Parkinsonism Relat. Disord. 2010, 16, 290–293. [Google Scholar] [CrossRef]
- Fields, H.L.; Hjelmstad, G.O.; Margolis, E.B.; Nicola, S.M. Ventral tegmental area neurons in learned appetitive behavior and positive reinforcement. Annu. Rev. Neurosci. 2007, 30, 289–316. [Google Scholar] [CrossRef]
- Murray, A.M.; Weihmueller, F.B.; Marshall, J.F.; Hurtig, H.I.; Gottleib, G.L.; Joyce, J.N. Damage to dopamine systems differs between Parkinson’s disease and Alzheimer’s disease with parkinsonism. Ann. Neurol. 1995, 37, 300–312. [Google Scholar] [CrossRef]
- Joyce, J.N.; Smutzer, G.; Whitty, C.J.; Myers, A.; Bannon, M.J. Differential modification of dopamine transporter and tyrosine hydroxylase mRNAs in midbrain of subjects with Parkinson’s, Alzheimer’s with parkinsonism, and Alzheimer’s disease. Mov. Disord. 1997, 12, 885–897. [Google Scholar] [CrossRef]
- Ceravolo, R.; Volterrani, D.; Gambaccini, G.; Bernardini, S.; Rossi, C.; Logi, C.; Tognoni, G.; Manca, G.; Mariani, G.; Bonuccelli, U.; et al. Presynaptic nigro-striatal function in a group of Alzheimer’s disease patients with parkinsonism: Evidence from a dopamine transporter imaging study. J. Neural Transm. (Vienna) 2004, 111, 1065–1073. [Google Scholar] [CrossRef]
- Perez, S.E.; Lazarov, O.; Koprich, J.B.; Chen, E.Y.; Rodriguez-Menendez, V.; Lipton, J.W.; Sisodia, S.S.; Mufson, E.J. Nigrostriatal dysfunction in familial Alzheimer’s disease-linked APPswe/PS1DeltaE9 transgenic mice. J. Neurosci. 2005, 25, 10220–10229. [Google Scholar] [CrossRef]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J.; Logan, J.; Schlyer, D.; MacGregor, R.; Hitzemann, R.; Wolf, A.P. Decreased dopamine transporters with age in health human subjects. Ann. Neurol. 1994, 36, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Gibb, W.R.; Mountjoy, C.Q.; Mann, D.M.; Lees, A.J. The substantia nigra and ventral tegmental area in Alzheimer’s disease and Down’s syndrome. J. Neurol. Neurosurg. Psychiatry 1989, 52, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Storga, D.; Vrecko, K.; Birkmayer, J.G.; Reibnegger, G. Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci. Lett. 1996, 203, 29–32. [Google Scholar] [CrossRef]
- Nobili, A.; Latagliata, E.C.; Viscomi, M.T.; Cavallucci, V.; Cutuli, D.; Giacovazzo, G.; Krashia, P.; Rizzo, F.R.; Marino, R.; Federici, M.; et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat. Commun. 2017, 8, 14727. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, Y.; Li, M.; Pan, D.; Li, Y.; Wang, Y.; Wang, L.; Wu, Q.; Yang, M. Multi-target PET evaluation in APP/PS1/tau mouse model of Alzheimer’s disease. Neurosci. Lett. 2020, 728, 134938. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Smith, H.; Ganderton, R.; Arranz, M.; Collier, D.; Powell, J.; Lovestone, S. Psychosis and aggression in Alzheimer’s disease: The effect of dopamine receptor gene variation. J. Neurol. Neurosurg. Psychiatry 2001, 71, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Jost, B.C.; Grossberg, G.T. The evolution of psychiatric symptoms in Alzheimer’s disease: A natural history study. J. Am. Geriatr. Soc. 1996, 44, 1078–1081. [Google Scholar] [CrossRef]
- Bozzali, M.; Serra, L.; Cercignani, M. Quantitative MRI to understand Alzheimer’s disease pathophysiology. Curr. Opin. Neurol. 2016, 29, 437–444. [Google Scholar] [CrossRef]
- Bozzali, M.; D’Amelio, M.; Serra, L. Ventral tegmental area disruption in Alzheimer’s disease. Aging (Albany N. Y.) 2019, 11, 1325–1326. [Google Scholar] [CrossRef]
- D’Amelio, M.; Serra, L.; Bozzali, M. Ventral Tegmental Area in Prodromal Alzheimer’s Disease: Bridging the Gap between Mice and Humans. J. Alzheimers Dis. 2018, 63, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, L.; Sala, A.; Caminiti, S.P.; Presotto, L.; Perani, D.; Alzheimer’s Disease Neuroimaging Initiative. In vivo MRI Structural and PET Metabolic Connectivity Study of Dopamine Pathways in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 75, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Udo, N.; Hashimoto, N.; Toyonaga, T.; Isoyama, T.; Oyanagi, Y.; Narita, H.; Shiga, T.; Nakagawa, S.; Kusumi, I. Apathy in Alzheimer’s Disease Correlates with the Dopamine Transporter Level in the Caudate Nuclei. Dement. Geriatr. Cogn. Dis. Extra 2020, 10, 86–93. [Google Scholar] [CrossRef]
- Vorobyov, V.; Bakharev, B.; Medvinskaya, N.; Nesterova, I.; Samokhin, A.; Deev, A.; Tatarnikova, O.; Ustyugov, A.A.; Sengpiel, F.; Bobkova, N. Loss of Midbrain Dopamine Neurons and Altered Apomorphine EEG Effects in the 5xFAD Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 70, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Cordella, A.; Krashia, P.; Nobili, A.; Pignataro, A.; La Barbera, L.; Viscomi, M.T.; Valzania, A.; Keller, F.; Ammassari-Teule, M.; Mercuri, N.B.; et al. Dopamine loss alters the hippocampus-nucleus accumbens synaptic transmission in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2018, 116, 142–154. [Google Scholar] [CrossRef]
- Paul, A.; Viswanathan, G.K.; Huber, A.; Arad, E.; Engel, H.; Jelinek, R.; Gazit, E.; Segal, D. Inhibition of tau amyloid formation and disruption of its preformed fibrils by Naphthoquinone-Dopamine hybrid. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, R.; Chia, S.; Pisani, K.; Ruggeri, F.S.; Xu, C.K.; Sneideris, T.; Perni, M.; Sarwat, S.; Joshi, P.; Kumita, J.R.; et al. A dopamine metabolite stabilizes neurotoxic amyloid-beta oligomers. Commun. Biol. 2021, 4, 19. [Google Scholar] [CrossRef]
- Ziu, I.; Rettig, I.; Luo, D.; Dutta, A.; McCormick, T.M.; Wu, C.; Martic, S. The multifunctional dopamine D2/D3 receptor agonists also possess inhibitory activity against the full-length tau441 protein aggregation. Bioorg. Med. Chem. 2020, 28, 115667. [Google Scholar] [CrossRef]
- Ramesh, M.; Makam, P.; Voshavar, C.; Khare, H.; Rajasekhar, K.; Ramakumar, S.; Govindaraju, T. l-Dopa and dopamine conjugated naphthalenediimides modulate amyloid beta toxicity. Org. Biomol. Chem. 2018, 16, 7682–7692. [Google Scholar] [CrossRef]
- Nam, E.; Derrick, J.S.; Lee, S.; Kang, J.; Han, J.; Lee, S.J.C.; Chung, S.W.; Lim, M.H. Regulatory Activities of Dopamine and Its Derivatives toward Metal-Free and Metal-Induced Amyloid-beta Aggregation, Oxidative Stress, and Inflammation in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 2655–2666. [Google Scholar] [CrossRef]
- Montoya, A.; Elgueta, D.; Campos, J.; Chovar, O.; Falcon, P.; Matus, S.; Alfaro, I.; Bono, M.R.; Pacheco, R. Dopamine receptor D3 signalling in astrocytes promotes neuroinflammation. J. Neuroinflamm. 2019, 16, 258. [Google Scholar] [CrossRef]
- Cheng, Z.Y.; Xia, Q.P.; Hu, Y.H.; Wang, C.; He, L. Dopamine D1 receptor agonist A-68930 ameliorates Abeta1-42-induced cognitive impairment and neuroinflammation in mice. Int. Immunopharmacol. 2020, 88, 106963. [Google Scholar] [CrossRef] [PubMed]
- Jonas, P.; Bischofberger, J.; Sandkuhler, J. Corelease of two fast neurotransmitters at a central synapse. Science 1998, 281, 419–424. [Google Scholar] [CrossRef]
- Boulland, J.L.; Qureshi, T.; Seal, R.P.; Rafiki, A.; Gundersen, V.; Bergersen, L.H.; Fremeau, R.T., Jr.; Edwards, R.H.; Storm-Mathisen, J.; Chaudhry, F.A. Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J. Comp. Neurol. 2004, 480, 264–280. [Google Scholar] [CrossRef]
- Trudeau, L.E. Glutamate co-transmission as an emerging concept in monoamine neuron function. J. Psychiatry Neurosci. 2004, 29, 296–310. [Google Scholar] [PubMed]
- Ranjbar-Slamloo, Y.; Fazlali, Z. Dopamine and Noradrenaline in the Brain; Overlapping or Dissociate Functions? Front. Mol. Neurosci. 2019, 12, 334. [Google Scholar] [CrossRef]
- Devoto, P.; Sagheddu, C.; Santoni, M.; Flore, G.; Saba, P.; Pistis, M.; Gessa, G.L. Noradrenergic Source of Dopamine Assessed by Microdialysis in the Medial Prefrontal Cortex. Front. Pharmacol. 2020, 11, 588160. [Google Scholar] [CrossRef] [PubMed]
- Kempadoo, K.A.; Mosharov, E.V.; Choi, S.J.; Sulzer, D.; Kandel, E.R. Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Proc. Natl. Acad. Sci. USA 2016, 113, 14835–14840. [Google Scholar] [CrossRef]
- Takeuchi, T.; Duszkiewicz, A.J.; Sonneborn, A.; Spooner, P.A.; Yamasaki, M.; Watanabe, M.; Smith, C.C.; Fernandez, G.; Deisseroth, K.; Greene, R.W.; et al. Locus coeruleus and dopaminergic consolidation of everyday memory. Nature 2016, 537, 357–362. [Google Scholar] [CrossRef]
- Burnstock, G. Cotransmission. Curr. Opin. Pharmacol. 2004, 4, 47–52. [Google Scholar] [CrossRef]
- Campbell, G. Cotransmission. Annu. Rev. Pharmacol. Toxicol. 1987, 27, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Kupfermann, I. Functional studies of cotransmission. Physiol. Rev. 1991, 71, 683–732. [Google Scholar] [CrossRef] [PubMed]
- Hokfelt, T. Neuropeptides in perspective: The last ten years. Neuron 1991, 7, 867–879. [Google Scholar] [CrossRef]
- Burnstock, G. Autonomic neurotransmission: 60 years since sir Henry Dale. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.M.; Terenius, L.; Hokfelt, T.; Martling, C.R.; Tatemoto, K.; Mutt, V.; Polak, J.; Bloom, S.; Goldstein, M. Neuropeptide Y (NPY)-like immunoreactivity in peripheral noradrenergic neurons and effects of NPY on sympathetic function. Acta Physiol. Scand. 1982, 116, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Holets, V.R.; Hokfelt, T.; Rokaeus, A.; Terenius, L.; Goldstein, M. Locus coeruleus neurons in the rat containing neuropeptide Y, tyrosine hydroxylase or galanin and their efferent projections to the spinal cord, cerebral cortex and hypothalamus. Neuroscience 1988, 24, 893–906. [Google Scholar] [CrossRef]
- Fung, S.J.; Reddy, V.K.; Zhuo, H.; Liu, R.H.; Wang, Z.; Barnes, C.D. Anatomical evidence for the presence of glutamate or enkephalin in noradrenergic projection neurons of the locus coeruleus. Microsc. Res. Tech. 1994, 29, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Le Maitre, E.; Barde, S.S.; Palkovits, M.; Diaz-Heijtz, R.; Hokfelt, T.G. Distinct features of neurotransmitter systems in the human brain with focus on the galanin system in locus coeruleus and dorsal raphe. Proc. Natl. Acad. Sci. USA 2013, 110, E536–E545. [Google Scholar] [CrossRef]
- Weinshenker, D.; Holmes, P.V. Regulation of neurological and neuropsychiatric phenotypes by locus coeruleus-derived galanin. Brain Res. 2016, 1641, 320–337. [Google Scholar] [CrossRef]
- Fung, S.J.; Reddy, V.K.; Liu, R.H.; Wang, Z.; Barnes, C.D. Existence of glutamate in noradrenergic locus coeruleus neurons of rodents. Brain Res. Bull. 1994, 35, 505–512. [Google Scholar] [CrossRef]
- Stornetta, R.L.; Sevigny, C.P.; Guyenet, P.G. Vesicular glutamate transporter DNPI/VGLUT2 mRNA is present in C1 and several other groups of brainstem catecholaminergic neurons. J. Comp. Neurol. 2002, 444, 191–206. [Google Scholar] [CrossRef]
- Herring, N.; Paterson, D.J. Neuromodulators of peripheral cardiac sympatho-vagal balance. Exp. Physiol. 2009, 94, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Gupta, G.; Shrivastava, B.; Dahiya, R.; Tiwari, J.; Ashwathanarayana, M.; Sharma, R.K.; Agrawal, M.; Mishra, A.; Dua, K. Calcitonin gene-related peptide (CGRP): A novel target for Alzheimer’s disease. CNS Neurosci. Ther. 2017, 23, 457–461. [Google Scholar] [CrossRef]
- Robertson, I.H. A noradrenergic theory of cognitive reserve: Implications for Alzheimer’s disease. Neurobiol. Aging 2013, 34, 298–308. [Google Scholar] [CrossRef]
- Hokfelt, T.; Xu, Z.Q.; Shi, T.J.; Holmberg, K.; Zhang, X. Galanin in ascending systems. Focus on coexistence with 5-hydroxytryptamine and noradrenaline. Ann. N. Y. Acad. Sci. 1998, 863, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Q.; Shi, T.J.; Hokfelt, T. Galanin/GMAP- and NPY-like immunoreactivities in locus coeruleus and noradrenergic nerve terminals in the hippocampal formation and cortex with notes on the galanin-R1 and -R2 receptors. J. Comp. Neurol. 1998, 392, 227–251. [Google Scholar] [CrossRef]
- Counts, S.E.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J. Neuroprotective role for galanin in Alzheimer’s disease. Exp. Suppl. 2010, 102, 143–162. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, D.; Ahmad, S.; Wahlestedt, C.; Walker, P. Expression of the novel galanin receptor subtype GALR2 in the adult rat CNS: Distinct distribution from GALR1. J. Comp. Neurol. 1999, 409, 469–481. [Google Scholar] [CrossRef]
- Lang, R.; Gundlach, A.L.; Holmes, F.E.; Hobson, S.A.; Wynick, D.; Hokfelt, T.; Kofler, B. Physiology, signaling, and pharmacology of galanin peptides and receptors: Three decades of emerging diversity. Pharmacol. Rev. 2015, 67, 118–175. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, Y.; Wu, L.; Zhang, S. Evolution of galanin receptor genes: Insights from the deuterostome genomes. J. Biomol. Struct. Dyn. 2010, 28, 97–106. [Google Scholar] [CrossRef]
- Mennicken, F.; Hoffert, C.; Pelletier, M.; Ahmad, S.; O’Donnell, D. Restricted distribution of galanin receptor 3 (GalR3) mRNA in the adult rat central nervous system. J. Chem. Neuroanat. 2002, 24, 257–268. [Google Scholar] [CrossRef]
- Belfer, I.; Hipp, H.; Bollettino, A.; McKnight, C.; Evans, C.; Virkkunen, M.; Albaugh, B.; Max, M.B.; Goldman, D.; Enoch, M.A. Alcoholism is associated with GALR3 but not two other galanin receptor genes. Genes Brain Behav. 2007, 6, 473–481. [Google Scholar] [CrossRef]
- Weiss, J.M.; Boss-Williams, K.A.; Moore, J.P.; Demetrikopoulos, M.K.; Ritchie, J.C.; West, C.H.K. Testing the hypothesis that locus coeruleus hyperactivity produces depression-related changes via galanin. Neuropeptides 2005, 39, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Vila-Porcile, E.; Xu, Z.Q.; Mailly, P.; Nagy, F.; Calas, A.; Hokfelt, T.; Landry, M. Dendritic synthesis and release of the neuropeptide galanin: Morphological evidence from studies on rat locus coeruleus neurons. J. Comp. Neurol. 2009, 516, 199–212. [Google Scholar] [CrossRef]
- Foster, S.L.; Galaj, E.; Karne, S.L.; Ferré, S.; Weinshenker, D. Cell-type specific expression and behavioral impact of galanin and GalR1 in the locus coeruleus during opioid withdrawal. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kuteeva, E.; Hokfelt, T.; Ogren, S.O. Behavioural characterisation of transgenic mice overexpressing galanin under the PDGF-B promoter. Neuropeptides 2005, 39, 299–304. [Google Scholar] [CrossRef]
- Hokfelt, T.; Barde, S.; Xu, Z.D.; Kuteeva, E.; Ruegg, J.; Le Maitre, E.; Risling, M.; Kehr, J.; Ihnatko, R.; Theodorsson, E.; et al. Neuropeptide and Small Transmitter Coexistence: Fundamental Studies and Relevance to Mental Illness. Front. Neural Circuits 2018, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Kolb, P.E.; Leverenz, J.B.; Peskind, E.R.; Raskind, M.A. Preservation of noradrenergic neurons in the locus ceruleus that coexpress galanin mRNA in Alzheimer’s disease. J. Neurochem. 1999, 73, 2028–2036. [Google Scholar] [PubMed]
- Cheng, Y.; Yu, L.C. Galanin protects amyloid-beta-induced neurotoxicity on primary cultured hippocampal neurons of rats. J. Alzheimers Dis. 2010, 20, 1143–1157. [Google Scholar] [CrossRef]
- Pirondi, S.; Fernandez, M.; Schmidt, R.; Hokfelt, T.; Giardino, L.; Calza, L. The galanin-R2 agonist AR-M1896 reduces glutamate toxicity in primary neural hippocampal cells. J. Neurochem. 2005, 95, 821–833. [Google Scholar] [CrossRef]
- Ding, X.; MacTavish, D.; Kar, S.; Jhamandas, J.H. Galanin attenuates beta-amyloid (Abeta) toxicity in rat cholinergic basal forebrain neurons. Neurobiol. Dis. 2006, 21, 413–420. [Google Scholar] [CrossRef]
- Elliott-Hunt, C.R.; Holmes, F.E.; Hartley, D.M.; Perez, S.; Mufson, E.J.; Wynick, D. Endogenous galanin protects mouse hippocampal neurons against amyloid toxicity in vitro via activation of galanin receptor-2. J. Alzheimers Dis. 2011, 25, 455–462. [Google Scholar] [CrossRef]
- Alexandris, A.S.; Walker, L.; Liu, A.K.L.; McAleese, K.E.; Johnson, M.; Pearce, R.K.B.; Gentleman, S.M.; Attems, J. Cholinergic deficits and galaninergic hyperinnervation of the nucleus basalis of Meynert in Alzheimer’s disease and Lewy body disorders. Neuropathol. Appl. Neurobiol. 2020, 46, 264–278. [Google Scholar] [CrossRef]
- Freimann, K.; Kurrikoff, K.; Langel, U. Galanin receptors as a potential target for neurological disease. Expert Opin. Ther. Targets 2015, 19, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.H.; Jones, E.G.; DeFelipe, J.; Schmechel, D.; Brandon, C.; Emson, P.C. Neuropeptide-containing neurons of the cerebral cortex are also GABAergic. Proc. Natl. Acad. Sci. USA 1984, 81, 6526–6530. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Shiosaka, S.; Emson, P.C.; Tohyama, M. Distribution of neuropeptide Y in the forebrain and diencephalon: An immunohistochemical analysis. Brain Res. 1985, 361, 52–60. [Google Scholar] [CrossRef]
- Larhammar, D. Structural diversity of receptors for neuropeptide Y, peptide YY and pancreatic polypeptide. Regul. Pept. 1996, 65, 165–174. [Google Scholar] [CrossRef]
- Dumont, Y.; Chabot, J.G.; Quirion, R. Receptor autoradiography as mean to explore the possible functional relevance of neuropeptides: Focus on new agonists and antagonists to study natriuretic peptides, neuropeptide Y and calcitonin gene-related peptides. Peptides 2004, 25, 365–391. [Google Scholar] [CrossRef]
- Clark, J.T.; Kalra, P.S.; Crowley, W.R.; Kalra, S.P. Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 1984, 115, 427–429. [Google Scholar] [CrossRef]
- Dyzma, M.; Boudjeltia, K.Z.; Faraut, B.; Kerkhofs, M. Neuropeptide Y and sleep. Sleep Med. Rev. 2010, 14, 161–165. [Google Scholar] [CrossRef]
- Everitt, B.J.; Hokfelt, T.; Terenius, L.; Tatemoto, K.; Mutt, V.; Goldstein, M. Differential co-existence of neuropeptide Y (NPY)-like immunoreactivity with catecholamines in the central nervous system of the rat. Neuroscience 1984, 11, 443–462. [Google Scholar] [CrossRef]
- Sabban, E.L.; Serova, L.I.; Newman, E.; Aisenberg, N.; Akirav, I. Changes in Gene Expression in the Locus Coeruleus-Amygdala Circuitry in Inhibitory Avoidance PTSD Model. Cell. Mol. Neurobiol. 2018, 38, 273–280. [Google Scholar] [CrossRef]
- Kohler, C.; Smialowska, M.; Eriksson, L.G.; Chanpalay, V.; Davies, S. Origin of the neuropeptide Y innervation of the rat retrohippocampal region. Neurosci. Lett. 1986, 65, 287–292. [Google Scholar] [CrossRef]
- Wilcox, B.J.; Unnerstall, J.R. Identification of a subpopulation of neuropeptide Y-containing locus coeruleus neurons that project to the entorhinal cortex. Synapse 1990, 6, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Mazurek, M.F.; Chattha, G.K.; Svendsen, C.N.; Bird, E.D.; Martin, J.B. Neuropeptide Y immunoreactivity is reduced in cerebral cortex in Alzheimer’s disease. Ann. Neurol. 1986, 20, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Neves, J.; Pereira de Almeida, L.; Cavadas, C. Neuropeptide Y (NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol. Dis. 2016, 95, 210–224. [Google Scholar] [CrossRef]
- Wee, J.J.; Kumar, S. Prediction of hub genes of Alzheimer’s disease using a protein interaction network and functional enrichment analysis. Genom. Inform. 2020, 18, e39. [Google Scholar] [CrossRef]
- Li, C.; Wu, X.; Liu, S.; Zhao, Y.; Zhu, J.; Liu, K. Roles of Neuropeptide Y in Neurodegenerative and Neuroimmune Diseases. Front. Neurosci. 2019, 13, 869. [Google Scholar] [CrossRef] [PubMed]
- Croce, N.; Ciotti, M.T.; Gelfo, F.; Cortelli, S.; Federici, G.; Caltagirone, C.; Bernardini, S.; Angelucci, F. Neuropeptide Y protects rat cortical neurons against beta-amyloid toxicity and re-establishes synthesis and release of nerve growth factor. ACS Chem. Neurosci. 2012, 3, 312–318. [Google Scholar] [CrossRef]
- Rose, J.B.; Crews, L.; Rockenstein, E.; Adame, A.; Mante, M.; Hersh, L.B.; Gage, F.H.; Spencer, B.; Potkar, R.; Marr, R.A.; et al. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer’s disease. J. Neurosci. 2009, 29, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Croce, N.; Dinallo, V.; Ricci, V.; Federici, G.; Caltagirone, C.; Bernardini, S.; Angelucci, F. Neuroprotective effect of neuropeptide Y against beta-amyloid 25-35 toxicity in SH-SY5Y neuroblastoma cells is associated with increased neurotrophin production. Neurodegener. Dis. 2011, 8, 300–309. [Google Scholar] [CrossRef]
- Croce, N.; Gelfo, F.; Ciotti, M.T.; Federici, G.; Caltagirone, C.; Bernardini, S.; Angelucci, F. NPY modulates miR-30a-5p and BDNF in opposite direction in an in vitro model of Alzheimer disease: A possible role in neuroprotection? Mol. Cell. Biochem. 2013, 376, 189–195. [Google Scholar] [CrossRef]
- Qian, J.; Colmers, W.F.; Saggau, P. Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: Modulation of presynaptic Ca2+ entry. J. Neurosci. 1997, 17, 8169–8177. [Google Scholar] [CrossRef]
- Santos-Carvalho, A.; Elvas, F.; Alvaro, A.R.; Ambrosio, A.F.; Cavadas, C. Neuropeptide Y receptors activation protects rat retinal neural cells against necrotic and apoptotic cell death induced by glutamate. Cell Death Dis. 2013, 4, e636. [Google Scholar] [CrossRef]
- Guiard, B.P.; El Mansari, M.; Merali, Z.; Blier, P. Functional interactions between dopamine, serotonin and norepinephrine neurons: An in-vivo electrophysiological study in rats with monoaminergic lesions. Int. J. Neuropsychopharmacol. 2008, 11, 625–639. [Google Scholar] [CrossRef]
- Brown, R.E.; Sergeeva, O.A.; Eriksson, K.S.; Haas, H.L. Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline). J. Neurosci. 2002, 22, 8850–8859. [Google Scholar] [CrossRef]
- Morilak, D.A.; Barrera, G.; Echevarria, D.J.; Garcia, A.S.; Hernandez, A.; Ma, S.; Petre, C.O. Role of brain norepinephrine in the behavioral response to stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Mejias-Aponte, C.A.; Drouin, C.; Aston-Jones, G. Adrenergic and noradrenergic innervation of the midbrain ventral tegmental area and retrorubral field: Prominent inputs from medullary homeostatic centers. J. Neurosci. 2009, 29, 3613–3626. [Google Scholar] [CrossRef] [PubMed]
- Serra, L.; D’Amelio, M.; Di Domenico, C.; Dipasquale, O.; Marra, C.; Mercuri, N.B.; Caltagirone, C.; Cercignani, M.; Bozzali, M. In vivo mapping of brainstem nuclei functional connectivity disruption in Alzheimer’s disease. Neurobiol. Aging 2018, 72, 72–82. [Google Scholar] [CrossRef]
- Strong, R.; Huang, J.S.; Huang, S.S.; Chung, H.D.; Hale, C.; Burke, W.J. Degeneration of the cholinergic innervation of the locus ceruleus in Alzheimer’s disease. Brain Res. 1991, 542, 23–28. [Google Scholar] [CrossRef]
- Yeung, L.Y.; Kung, H.F.; Yew, D.T. Localization of 5-HT1A and 5-HT2A positive cells in the brainstems of control age-matched and Alzheimer individuals. Age (Dordr.) 2010, 32, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.R.; Szabadi, E. Functional neuroanatomy of the noradrenergic locus coeruleus: Its roles in the regulation of arousal and autonomic function part I: Principles of functional organisation. Curr. Neuropharmacol. 2008, 6, 235–253. [Google Scholar] [CrossRef]
- Uys, M.M.; Shahid, M.; Harvey, B.H. Therapeutic Potential of Selectively Targeting the alpha2C-Adrenoceptor in Cognition, Depression, and Schizophrenia-New Developments and Future Perspective. Front. Psychiatry 2017, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Youssofzadeh, V.; Vemana, V.; McGinnity, T.M.; Prasad, G.; Wong-Lin, K. An integrated modelling framework for neural circuits with multiple neuromodulators. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef]
- Jodo, E.; Aston-Jones, G. Activation of locus coeruleus by prefrontal cortex is mediated by excitatory amino acid inputs. Brain Res. 1997, 768, 327–332. [Google Scholar] [CrossRef]
- Jodo, E.; Chiang, C.; Aston-Jones, G. Potent excitatory influence of prefrontal cortex activity on noradrenergic locus coeruleus neurons. Neuroscience 1998, 83, 63–79. [Google Scholar] [CrossRef]
- Carpentier, V.; Vaudry, H.; Laquerriere, A.; Tayot, J.; Leroux, P. Distribution of somatostatin receptors in the adult human brainstem. Brain Res. 1996, 734, 135–148. [Google Scholar] [CrossRef]
- Burgos-Ramos, E.; Hervas-Aguilar, A.; Aguado-Llera, D.; Puebla-Jimenez, L.; Hernandez-Pinto, A.M.; Barrios, V.; Arilla-Ferreiro, E. Somatostatin and Alzheimer’s disease. Mol. Cell. Endocrinol. 2008, 286, 104–111. [Google Scholar] [CrossRef]
- Kowall, N.W.; Beal, M.F. Cortical somatostatin, neuropeptide Y, and NADPH diaphorase neurons: Normal anatomy and alterations in Alzheimer’s disease. Ann. Neurol. 1988, 23, 105–114. [Google Scholar] [CrossRef]
- Adori, C.; Gluck, L.; Barde, S.; Yoshitake, T.; Kovacs, G.G.; Mulder, J.; Magloczky, Z.; Havas, L.; Bolcskei, K.; Mitsios, N.; et al. Critical role of somatostatin receptor 2 in the vulnerability of the central noradrenergic system: New aspects on Alzheimer’s disease. Acta Neuropathol. 2015, 129, 541–563. [Google Scholar] [CrossRef]
- Bostanciklioglu, M. Neuromodulation of Memory Formation and Extinction. Curr. Neurovasc. Res. 2020, 17, 319–326. [Google Scholar] [CrossRef]
- Rub, U.; Schultz, C.; Del Tredici, K.; Braak, H. Early involvement of the tegmentopontine reticular nucleus during the evolution of Alzheimer’s disease-related cytoskeletal pathology. Brain Res. 2001, 908, 107–112. [Google Scholar] [CrossRef]
- Rub, U.; Del Tredici, K.; Schultz, C.; Buttner-Ennever, J.A.; Braak, H. The premotor region essential for rapid vertical eye movements shows early involvement in Alzheimer’s disease-related cytoskeletal pathology. Vis. Res. 2001, 41, 2149–2156. [Google Scholar] [CrossRef]
- Mavroudis, I.A.; Manani, M.G.; Petrides, F.; Petsoglou, C.; Njau, S.N.; Costa, V.G.; Baloyannis, S.J. Dendritic and spinal alterations of neurons from Edinger-Westphal nucleus in Alzheimer’s disease. Folia Neuropathol. 2014, 52, 197–204. [Google Scholar] [CrossRef]
- Rub, U.; Del Tredici, K.; Schultz, C.; Thal, D.R.; Braak, E.; Braak, H. The autonomic higher order processing nuclei of the lower brain stem are among the early targets of the Alzheimer’s disease-related cytoskeletal pathology. Acta Neuropathol. 2001, 101, 555–564. [Google Scholar] [CrossRef]
- Cole, L.J.; Gavrilescu, M.; Johnston, L.A.; Gibson, S.J.; Farrell, M.J.; Egan, G.F. The impact of Alzheimer’s disease on the functional connectivity between brain regions underlying pain perception. Eur. J. Pain 2011, 15, 568.e1–568.e11. [Google Scholar] [CrossRef]
- Priovoulos, N.; Poser, B.A.; Ivanov, D.; Verhey, F.R.J.; Jacobs, H.I.L. In vivo imaging of the nucleus of the solitary tract with Magnetization Transfer at 7 Tesla. Neuroimage 2019, 201, 116071. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Dysfunctional nucleus tractus solitarius: Its crucial role in promoting neuropathogenetic cascade of Alzheimer’s dementia--a novel hypothesis. Neurochem. Res. 2012, 37, 846–868. [Google Scholar] [CrossRef]
- Rub, U.; Stratmann, K.; Heinsen, H.; Turco, D.D.; Seidel, K.; Dunnen, W.; Korf, H.W. The Brainstem Tau Cytoskeletal Pathology of Alzheimer’s Disease: A Brief Historical Overview and Description of its Anatomical Distribution Pattern, Evolutional Features, Pathogenetic and Clinical Relevance. Curr. Alzheimer Res. 2016, 13, 1178–1197. [Google Scholar] [CrossRef] [PubMed]
- Sumners, C.; Alleyne, A.; Rodriguez, V.; Pioquinto, D.J.; Ludin, J.A.; Kar, S.; Winder, Z.; Ortiz, Y.; Liu, M.; Krause, E.G.; et al. Brain angiotensin type-1 and type-2 receptors: Cellular locations under normal and hypertensive conditions. Hypertens. Res. 2020, 43, 281–295. [Google Scholar] [CrossRef]
- Phillips, M.I.; de Oliveira, E.M. Brain renin angiotensin in disease. J. Mol. Med. 2008, 86, 715–722. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. Neuroinflammation and sympathetic overactivity: Mechanisms and implications in hypertension. Auton. Neurosci. 2018, 210, 10–17. [Google Scholar] [CrossRef]
- Fazal, K.; Perera, G.; Khondoker, M.; Howard, R.; Stewart, R. Associations of centrally acting ACE inhibitors with cognitive decline and survival in Alzheimer’s disease. BJPsych Open 2017, 3, 158–164. [Google Scholar] [CrossRef]
- Skoog, I.; Lernfelt, B.; Landahl, S.; Palmertz, B.; Andreasson, L.A.; Nilsson, L.; Persson, G.; Oden, A.; Svanborg, A. 15-year longitudinal study of blood pressure and dementia. Lancet 1996, 347, 1141–1145. [Google Scholar] [CrossRef]
- Nelson, L.; Gard, P.; Tabet, N. Hypertension and inflammation in Alzheimer’s disease: Close partners in disease development and progression! J. Alzheimers Dis. 2014, 41, 331–343. [Google Scholar] [CrossRef]
- Grinberg, L.T.; Rub, U.; Ferretti, R.E.; Nitrini, R.; Farfel, J.M.; Polichiso, L.; Gierga, K.; Jacob-Filho, W.; Heinsen, H.; Brazilian Brain Bank Study Group. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol. Appl. Neurobiol. 2009, 35, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Theofilas, P.; Ehrenberg, A.J.; Dunlop, S.; Di Lorenzo Alho, A.T.; Nguy, A.; Leite, R.E.P.; Rodriguez, R.D.; Mejia, M.B.; Suemoto, C.K.; Ferretti-Rebustini, R.E.L.; et al. Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dement. 2017, 13, 236–246. [Google Scholar] [CrossRef]
- Kremen, W.S.; Panizzon, M.S.; Elman, J.A.; Granholm, E.L.; Andreassen, O.A.; Dale, A.M.; Gillespie, N.A.; Gustavson, D.E.; Logue, M.W.; Lyons, M.J.; et al. Pupillary dilation responses as a midlife indicator of risk for Alzheimer’s disease: Association with Alzheimer’s disease polygenic risk. Neurobiol. Aging 2019, 83, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018, 41, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Raskind, M.A.; Peskind, E.R.; Halter, J.B.; Jimerson, D.C. Norepinephrine and MHPG levels in CSF and plasma in Alzheimer’s disease. Arch. Gen. Psychiatry 1984, 41, 343–346. [Google Scholar] [CrossRef]
- Pillet, L.E.; Taccola, C.; Cotoni, J.; Thiriez, H.; Andre, K.; Verpillot, R. Correlation between cognition and plasma noradrenaline level in Alzheimer’s disease: A potential new blood marker of disease evolution. Transl. Psychiatry 2020, 10, 213. [Google Scholar] [CrossRef]
- Galgani, A.; Lombardo, F.; Della Latta, D.; Martini, N.; Bonuccelli, U.; Fornai, F.; Giorgi, F.S. Locus Coeruleus Magnetic Resonance Imaging in Neurological Diseases. Curr. Neurol. Neurosci. Rep. 2020, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Liebe, T.; Kaufmann, J.; Li, M.; Skalej, M.; Wagner, G.; Walter, M. In vivo anatomical mapping of human locus coeruleus functional connectivity at 3 T MRI. Hum. Brain Mapp. 2020, 41, 2136–2151. [Google Scholar] [CrossRef]
- Olivieri, P.; Lagarde, J.; Lehericy, S.; Valabregue, R.; Michel, A.; Mace, P.; Caille, F.; Gervais, P.; Bottlaender, M.; Sarazin, M. Early alteration of the locus coeruleus in phenotypic variants of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Kirilina, E.; Otaduy, M.C.G.; Ivanov, D.; Acosta-Cabronero, J.; Callaghan, M.F.; Lambert, C.; Cardenas-Blanco, A.; Pine, K.; Passamonti, L.; et al. Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain 2019, 142, 2558–2571. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, P.; Petersen, K.J.; Cronin, M.J.; Lin, Y.C.; Kang, H.; Donahue, M.J.; Smith, S.A.; Claassen, D.O. Quantitative magnetization transfer imaging of the human locus coeruleus. Neuroimage 2019, 200, 191–198. [Google Scholar] [CrossRef]
- Sun, W.; Tang, Y.; Qiao, Y.; Ge, X.; Mather, M.; Ringman, J.M.; Shi, Y.; for Alzheimer’s Disease Neuroimaging Initiative. A probabilistic atlas of locus coeruleus pathways to transentorhinal cortex for connectome imaging in Alzheimer’s disease. Neuroimage 2020, 223, 117301. [Google Scholar] [CrossRef]
- Del Cerro, I.; Villarreal, M.F.; Abulafia, C.; Duarte-Abritta, B.; Sanchez, S.M.; Castro, M.N.; Bocaccio, H.; Ferrer, I.; Menchon, J.M.; Sevlever, G.; et al. Disrupted functional connectivity of the locus coeruleus in healthy adults with parental history of Alzheimer’s disease. J. Psychiatr. Res. 2020, 123, 81–88. [Google Scholar] [CrossRef]
- Jacobs, H.I.; Priovoulos, N.; Poser, B.A.; Pagen, L.H.; Ivanov, D.; Verhey, F.R.; Uludag, K. Dynamic behavior of the locus coeruleus during arousal-related memory processing in a multi-modal 7T fMRI paradigm. eLife 2020, 9. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Tabuchi, K.; Ojika, M.; Zucca, F.A.; Zecca, L.; Ito, S. Norepinephrine and its metabolites are involved in the synthesis of neuromelanin derived from the locus coeruleus. J. Neurochem. 2015, 135, 768–776. [Google Scholar] [CrossRef]
- Betts, M.J.; Cardenas-Blanco, A.; Kanowski, M.; Jessen, F.; Duzel, E. In vivo MRI assessment of the human locus coeruleus along its rostrocaudal extent in young and older adults. Neuroimage 2017, 163, 150–159. [Google Scholar] [CrossRef]
- Clewett, D.V.; Lee, T.H.; Greening, S.; Ponzio, A.; Margalit, E.; Mather, M. Neuromelanin marks the spot: Identifying a locus coeruleus biomarker of cognitive reserve in healthy aging. Neurobiol. Aging 2016, 37, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Dordevic, M.; Muller-Fotti, A.; Muller, P.; Schmicker, M.; Kaufmann, J.; Muller, N.G. Optimal Cut-Off Value for Locus Coeruleus-to-Pons Intensity Ratio as Clinical Biomarker for Alzheimer’s Disease: A Pilot Study. J. Alzheimers Dis. Rep. 2017, 1, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ryan, J.; Andreescu, C.; Aizenstein, H.; Lim, H.K. Brainstem morphological changes in Alzheimer’s disease. Neuroreport 2015, 26, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Zhu, M.; He, Y.; Zhang, H.; Chen, X.; Gao, W.; Fu, Y.; Alzheimer’s Disease Neuroimaging Initiative. Brainstem atrophy in the early stage of Alzheimer’s disease: A voxel-based morphometry study. Brain Imaging Behav. 2021, 15, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Li, Y.; Mather, M.; Nation, D.A. Alzheimer’s Disease Neuroimaging, I. Brainstem Volumetric Integrity in Preclinical and Prodromal Alzheimer’s Disease. J. Alzheimers Dis. 2020, 77, 1579–1594. [Google Scholar] [CrossRef]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Cedazo-Minguez, A.; Kenigsberg, P.A.; Page, G.; Duarte, A.I.; Giusti, P.; Zusso, M.; Robert, P.; Frisoni, G.B.; Cattaneo, A.; et al. Current and emerging avenues for Alzheimer’s disease drug targets. J. Intern. Med. 2019, 286, 398–437. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patthy, Á.; Murai, J.; Hanics, J.; Pintér, A.; Zahola, P.; Hökfelt, T.G.M.; Harkany, T.; Alpár, A. Neuropathology of the Brainstem to Mechanistically Understand and to Treat Alzheimer’s Disease. J. Clin. Med. 2021, 10, 1555. https://doi.org/10.3390/jcm10081555
Patthy Á, Murai J, Hanics J, Pintér A, Zahola P, Hökfelt TGM, Harkany T, Alpár A. Neuropathology of the Brainstem to Mechanistically Understand and to Treat Alzheimer’s Disease. Journal of Clinical Medicine. 2021; 10(8):1555. https://doi.org/10.3390/jcm10081555
Chicago/Turabian StylePatthy, Ágoston, János Murai, János Hanics, Anna Pintér, Péter Zahola, Tomas G. M. Hökfelt, Tibor Harkany, and Alán Alpár. 2021. "Neuropathology of the Brainstem to Mechanistically Understand and to Treat Alzheimer’s Disease" Journal of Clinical Medicine 10, no. 8: 1555. https://doi.org/10.3390/jcm10081555
APA StylePatthy, Á., Murai, J., Hanics, J., Pintér, A., Zahola, P., Hökfelt, T. G. M., Harkany, T., & Alpár, A. (2021). Neuropathology of the Brainstem to Mechanistically Understand and to Treat Alzheimer’s Disease. Journal of Clinical Medicine, 10(8), 1555. https://doi.org/10.3390/jcm10081555