
Alpha-1 Antitrypsin Screening in a Selected Cohort of Patients Affected by Chronic Pulmonary Diseases in Naples, Italy

 ,
, 
 and
and
Abstract
1. Introduction
2. Materials and Methods
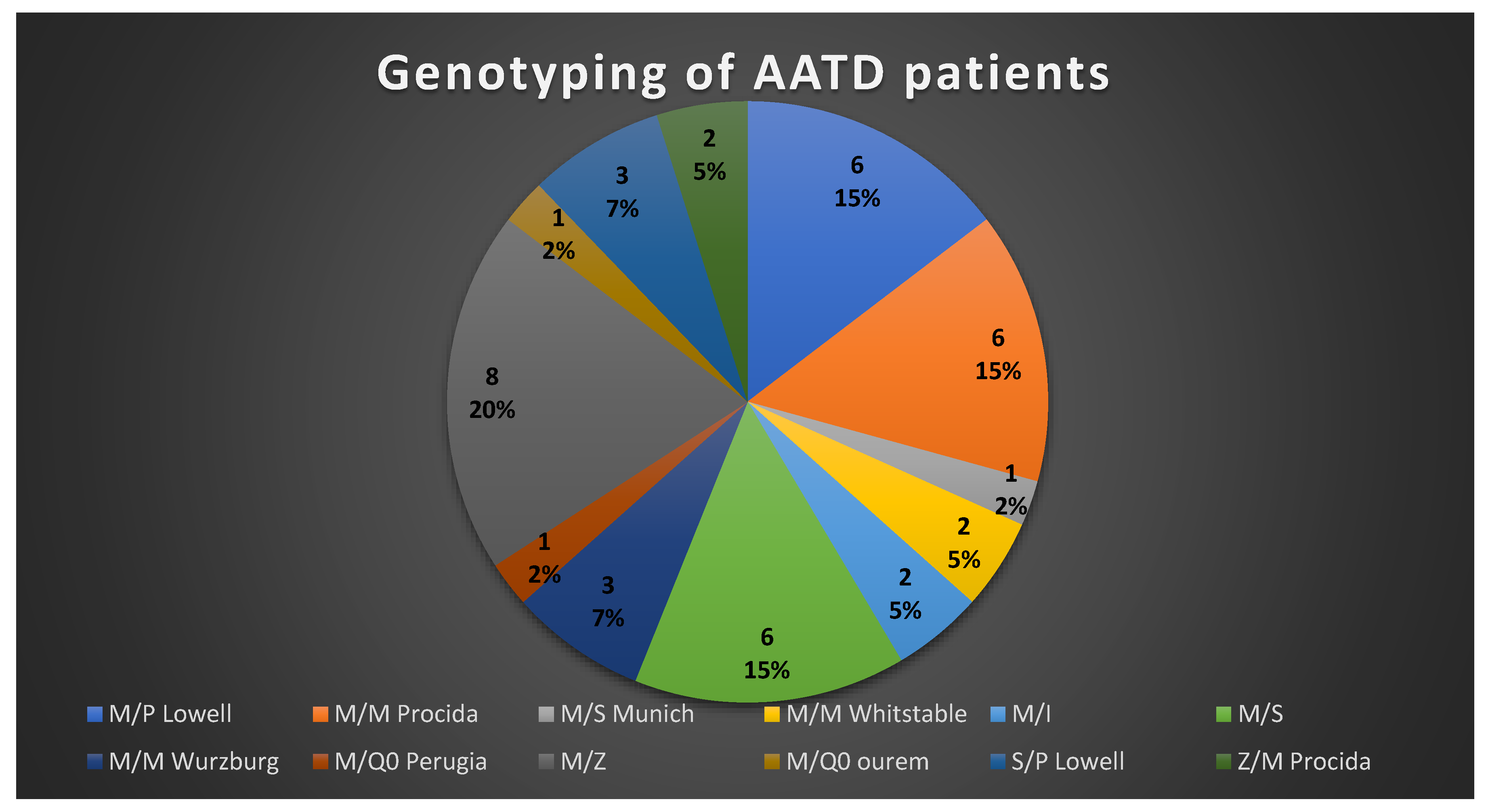
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohlsson, K. Neutral Leucocyte Proteases and Elastase Inhibited by Plasma Alpha 1 -Antitrypsin. Scand. J. Clin. Lab. Investig. 1971, 28, 251–253. [Google Scholar] [CrossRef]
- Beatty, K.; Bieth, J.; Travis, J. Kinetics of Association of Serine Proteinases with Native and Oxidized Alpha-1-Proteinase Inhibitor and Alpha-1-Antichymotrypsin. J. Biol. Chem. 1980, 255, 3931–3934. [Google Scholar] [CrossRef]
- Rao, N.V.; Wehner, N.G.; Marshall, B.C.; Gray, W.R.; Gray, B.H.; Hoidal, J.R. Characterization of Proteinase-3 (PR-3), a Neutrophil Serine Proteinase. Structural and Functional Properties. J. Biol. Chem. 1991, 266, 9540–9548. [Google Scholar] [CrossRef]
- Laurell, C.-B.; Eriksson, S. The Electrophoretic Α1-Globulin Pattern of Serum in Α1-Antitrypsin Deficiency. 1963. COPD 2013, 10 (Suppl. 1), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bornhorst, J.A.; Calderon, F.R.O.; Procter, M.; Tang, W.; Ashwood, E.R.; Mao, R. Genotypes and Serum Concentrations of Human Alpha-1-Antitrypsin “P” Protein Variants in a Clinical Population. J. Clin. Pathol. 2007, 60, 1124–1128. [Google Scholar] [CrossRef]
- Abboud, R.T.; Nelson, T.N.; Jung, B.; Mattman, A. Alpha1-Antitrypsin Deficiency: A Clinical-Genetic Overview. Appl. Clin. Genet. 2011, 4, 55–65. [Google Scholar] [CrossRef]
- de Serres, F.J.; Blanco, I. Prevalence of Α1-Antitrypsin Deficiency Alleles PI*S and PI*Z Worldwide and Effective Screening for Each of the Five Phenotypic Classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: A Comprehensive Review. Ther. Adv. Respir. Dis. 2012, 6, 277–295. [Google Scholar] [CrossRef]
- Ottaviani, S.; Barzon, V.; Buxens, A.; Gorrini, M.; Larruskain, A.; El Hamss, R.; Balderacchi, A.M.; Corsico, A.G.; Ferrarotti, I. Molecular Diagnosis of Alpha1-Antitrypsin Deficiency: A New Method Based on Luminex Technology. J. Clin. Lab. Anal. 2020, 34, e23279. [Google Scholar] [CrossRef]
- Giacopuzzi, E.; Laffranchi, M.; Berardelli, R.; Ravasio, V.; Ferrarotti, I.; Gooptu, B.; Borsani, G.; Fra, A. Real-World Clinical Applicability of Pathogenicity Predictors Assessed on SERPINA1 Mutations in Alpha-1-Antitrypsin Deficiency. Hum. Mutat. 2018, 39, 1203–1213. [Google Scholar] [CrossRef]
- American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society Statement: Standards for the Diagnosis and Management of Individuals with Alpha-1 Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2003, 168, 818–900. [Google Scholar] [CrossRef]
- Stoller, J.K.; Aboussouan, L.S. A Review of Α1-Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259. [Google Scholar] [CrossRef]
- de Serres, F.; Blanco, I. Role of Alpha-1 Antitrypsin in Human Health and Disease. J. Intern. Med. 2014, 276, 311–335. [Google Scholar] [CrossRef]
- Brantly, M.L.; Wittes, J.T.; Vogelmeier, C.F.; Hubbard, R.C.; Fells, G.A.; Crystal, R.G. Use of a Highly Purified Alpha 1-Antitrypsin Standard to Establish Ranges for the Common Normal and Deficient Alpha 1-Antitrypsin Phenotypes. Chest 1991, 100, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotti, I.; Ottaviani, S. Laboratory diagnosis. In α1-Antitrypsin Deficiency (ERS Monograph); Strnad, P., Brantly, M.L., Bals, R., Eds.; European Respiratory Society: Sheffield, UK, 2019; pp. 39–51. [Google Scholar] [CrossRef]
- Faber, J.P.; Weidinger, S.; Olek, K. Sequence Data of the Rare Deficient Alpha 1-Antitrypsin Variant PI Zaugsburg. Am. J. Hum. Genet. 1990, 46, 1158–1162. [Google Scholar]
- Takahashi, H.; Nukiwa, T.; Satoh, K.; Ogushi, F.; Brantly, M.; Fells, G.; Stier, L.; Courtney, M.; Crystal, R.G. Characterization of the Gene and Protein of the Alpha 1-Antitrypsin “Deficiency” Allele Mprocida. J. Biol. Chem. 1988, 263, 15528–15534. [Google Scholar] [CrossRef]
- Faber, J.P.; Poller, W.; Weidinger, S.; Kirchgesser, M.; Schwaab, R.; Bidlingmaier, F.; Olek, K. Identification and DNA Sequence Analysis of 15 New Alpha 1-Antitrypsin Variants, Including Two PI*Q0 Alleles and One Deficient PI*M Allele. Am. J. Hum. Genet. 1994, 55, 1113–1121. [Google Scholar]
- Poller, W.; Merklein, F.; Schneider-Rasp, S.; Haack, A.; Fechner, H.; Wang, H.; Anagnostopoulos, I.; Weidinger, S. Molecular Characterisation of the Defective Alpha 1-Antitrypsin Alleles PI Mwurzburg (Pro369Ser), Mheerlen (Pro369Leu), and Q0lisbon (Thr68Ile). Eur. J. Hum. Genet. EJHG 1999, 7, 321–331. [Google Scholar] [CrossRef]
- Ambrose, H.J.; Chambers, S.M.; Mieli-Vergani, G.; Ferrie, R.; Newton, C.R.; Robertson, N.H. Molecular Characterization of a New Alpha-1-Antitrypsin M Variant Allele, Mwhitstable: Implications for DNA-Based Diagnosis. Diagn. Mol. Pathol. Am. J. Surg. Pathol. Part B 1999, 8, 205–210. [Google Scholar] [CrossRef]
- Graham, A.; Kalsheker, N.A.; Newton, C.R.; Bamforth, F.J.; Powell, S.J.; Markham, A.F. Molecular Characterisation of Three Alpha-1-Antitrypsin Deficiency Variants: Proteinase Inhibitor (Pi) Nullcardiff (Asp256----Val); PiMmalton (Phe51----Deletion) and PiI (Arg39-Cys). Hum. Genet. 1989, 84, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Vaz Rodrigues, L.; Costa, F.; Marques, P.; Mendonça, C.; Rocha, J.; Seixas, S. Severe α-1 Antitrypsin Deficiency Caused by Q0(Ourém) Allele: Clinical Features, Haplotype Characterization and History. Clin. Genet. 2012, 81, 462–469. [Google Scholar] [CrossRef]
- Ferrarotti, I.; Carroll, T.P.; Ottaviani, S.; Fra, A.M.; O’Brien, G.; Molloy, K.; Corda, L.; Medicina, D.; Curran, D.R.; McElvaney, N.G.; et al. Identification and Characterisation of Eight Novel SERPINA1 Null Mutations. Orphanet J. Rare Dis. 2014, 9, 172. [Google Scholar] [CrossRef]
- Tsechkovski, M.; Boulyjenkov, V.; Heuck, C. Alpha 1-Antitrypsin Deficiency: Memorandum from a WHO Meeting. Bull. World Health Organ. 1997, 75, 397–415. [Google Scholar]
- Greulich, T.; Weist, B.J.D.; Koczulla, A.R.; Janciauskiene, S.; Klemmer, A.; Lux, W.; Alter, P.; Vogelmeier, C.F. Prevalence of Comorbidities in COPD Patients by Disease Severity in a German Population. Respir. Med. 2017, 132, 132–138. [Google Scholar] [CrossRef]
- Miravitlles, M.; Dirksen, A.; Ferrarotti, I.; Koblizek, V.; Lange, P.; Mahadeva, R.; McElvaney, N.G.; Parr, D.; Piitulainen, E.; Roche, N.; et al. European Respiratory Society Statement: Diagnosis and Treatment of Pulmonary Disease in Α1-Antitrypsin Deficiency. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Balderacchi, A.M.; Barzon, V.; Ottaviani, S.; Corino, A.; Zorzetto, M.; Wencker, M.; Corsico, A.G.; Ferrarotti, I. Comparison of Different Algorithms in Laboratory Diagnosis of Alpha1-Antitrypsin Deficiency. Clin. Chem. Lab. Med. 2021. [Google Scholar] [CrossRef]
- Prinsen, J.H.; Schweisfurth, H.; Rasche, B.; Breuer, J. Comparison of Three Methods for the Determination of Serum Alpha-1-Antitrypsin in Patients with Pulmonary Diseases. Clin. Physiol. Biochem. 1989, 7, 198–202. [Google Scholar]
- Lopes, A.P.; Mineiro, M.A.; Costa, F.; Gomes, J.; Santos, C.; Antunes, C.; Maia, D.; Melo, R.; Canotilho, M.; Magalhães, E.; et al. Portuguese Consensus Document for the Management of Alpha-1-Antitrypsin Deficiency. Pulmonology 2018, 24 (Suppl. 1), 1–21. [Google Scholar] [CrossRef]
- da Costa, C.H.; Noronha Filho, A.J.; Marques, E.; Silva, R.M.F.; da Cruz, T.F.; de Oliveira Monteiro, V.; Pio, M.; Rufino, R.L. Alpha 1-Antitrypsin Deficiency in Patients with Chronic Obstructive Pulmonary Disease Patients: Is Systematic Screening Necessary? BMC Res. Notes 2019, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Menga, G.; Fernandez Acquier, M.; Echazarreta, A.L.; Sorroche, P.B.; Lorenzon, M.V.; Fernández, M.E.; Saez, M.S.; grupo de estudio DAAT.AR. Prevalence of Alpha-1 Antitrypsin Deficiency in COPD Patients in Argentina. The DAAT.AR Study. Arch. Bronconeumol. 2020, 56, 571–577. [Google Scholar] [CrossRef]
- Carroll, T.P.; O’Connor, C.A.; Floyd, O.; McPartlin, J.; Kelleher, D.P.; O’Brien, G.; Dimitrov, B.D.; Morris, V.B.; Taggart, C.C.; McElvaney, N.G. The Prevalence of Alpha-1 Antitrypsin Deficiency in Ireland. Respir. Res. 2011, 12, 91. [Google Scholar] [CrossRef]
- Veith, M.; Tüffers, J.; Peychev, E.; Klemmer, A.; Kotke, V.; Janciauskiene, S.; Wilhelm, S.; Bals, R.; Koczulla, A.R.; Vogelmeier, C.F.; et al. The Distribution of Alpha-1 Antitrypsin Genotypes Between Patients with COPD/Emphysema, Asthma and Bronchiectasis. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 2827–2836. [Google Scholar] [CrossRef]
- de Serres, F.J.; Blanco, I.; Fernández-Bustillo, E. Genetic Epidemiology of Alpha-1 Antitrypsin Deficiency in Southern Europe: France, Italy, Portugal and Spain. Clin. Genet. 2003, 63, 490–509. [Google Scholar] [CrossRef]
- Ferrarotti, I.; Baccheschi, J.; Zorzetto, M.; Tinelli, C.; Corda, L.; Balbi, B.; Campo, I.; Pozzi, E.; Faa, G.; Coni, P.; et al. Prevalence and Phenotype of Subjects Carrying Rare Variants in the Italian Registry for Alpha1-Antitrypsin Deficiency. J. Med. Genet. 2005, 42, 282–287. [Google Scholar] [CrossRef]
- Corda, L.; Medicina, D.; La Piana, G.E.; Bertella, E.; Moretti, G.; Bianchi, L.; Pinelli, V.; Savoldi, G.; Baiardi, P.; Facchetti, F.; et al. Population Genetic Screening for Alpha1-Antitrypsin Deficiency in a High-Prevalence Area. Respir. Int. Rev. Thorac. Dis. 2011, 82, 418–425. [Google Scholar] [CrossRef]
- Mosella, M.; Accardo, M.; Molino, A.; Maniscalco, M.; Zamparelli, A.S. Description of a New Rare Alpha-1 Antitrypsin Mutation in Naples (Italy): PI*M S-Napoli. Ann. Thorac. Med. 2018, 13, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, A.; Ferrarotti, I.; Lanza, M.; Cauteruccio, R.; Spirito, V.D.; Fiorentino, G. Alpha 1 antitrypsin deficiency and intermediate risk: The case of a heterozygote for the MWurzburg allele. Ital. Rev. Respir. Dis. 2020, 35, 115–117. [Google Scholar] [CrossRef]
- Aiello, M.; Fantin, A.; Longo, C.; Ferrarotti, I.; Bertorelli, G.; Chetta, A. Clinical Manifestations in Patients with PI*MMMalton Genotypes. A Matter Still Unsolved in Alpha-1 Antitrypsin Deficiency. Respirol. Case Rep. 2020, 8, e00528. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Female/Male, n (%) | 15/26 (36.5%/63.5%) |
|---|---|
| Age (years, mean ± SD) | 56.6 ± 15.7 |
| Smoking History | |
| Active Smokers, n (%) | 1 (2.4%) |
| Former Smokers, n (%) | 19 (46.3%) |
| Never Smokers, n (%) | 21 (51.0%) |
| Clinical pattern | |
| Dyspnea, n (%) | 4 (9.7%) |
| COPD, n (%) | 16 (39.0%) |
| Bronchiectasis, n (%) | 1 (2.4%) |
| Asthma, n (%) | 3 (7.0%) |
| Emphysema, n (%) | 18 (44%) |
| CT Features | |
| No findings, n (%) | 12 (29.0%) |
| Panlobular Emphysema, n (%) | 19 (46.0%) |
| Centrilobular Emphysema, n (%) | 7 (17.0%) |
| Bronchiectasis, n (%) | 6 (14.6%) |
| Fibrosis, n (%) | 2 (2.8%) |
| Variant Name | Mutation | Consequences | Clinical Manifestation |
|---|---|---|---|
| Z | Glu 342 > Lys c. 1096G > A | Polymerization, decreased inhibitory activity; protein deficiency | Emphysema, COPD |
| S | Glu 264 > Val c. 863A > T | increased turnover; decreased inhibitory activity; mild protein deficiency | Emphysema, Asthma |
| PLowell | Asp 256 > Val c. 839A > T | Polymerization, degraded in liver cells; protein deficiency | Emphysema, COPD |
| MProcida | Leu 41 > Pro c.194T > C | Intracellular proteolysis, decreased inhibitory activity; protein deficiency | COPD, Asthma |
| SMunich | Ser330Phe c. 1061C > T | Mild protein deficiency | COPD |
| MWhitstable | Intron mutation, 26 bp detection and 2 bp insertion in intron IV | Truncated protein, protein deficiency | Emphysema and fibrosis |
| I | Arg 39 > Cys c. 187C > T | Polymerization, slightly decreased inhibitory activity; mild protein deficiency | Emphysema, bronchiectasis |
| MWurzburg | Pro369Ser c. 1177C > T | Degradation, protein deficiency | Emphysema |
| Q0Perugia | Val239→ DelG→STOP CODON241 V239GTG−delG > T ter241TGA | No detectable protein | Emphysema |
| Q0ourem | IVS1C + 1G→A exon5L352TTA, insT > Ter376TGA | No detectable protein | COPD, Lung cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Annunziata, A.; Ferrarotti, I.; Coppola, A.; Lanza, M.; Imitazione, P.; Spinelli, S.; Micco, P.D.; Fiorentino, G. Alpha-1 Antitrypsin Screening in a Selected Cohort of Patients Affected by Chronic Pulmonary Diseases in Naples, Italy. J. Clin. Med. 2021, 10, 1546. https://doi.org/10.3390/jcm10081546
Annunziata A, Ferrarotti I, Coppola A, Lanza M, Imitazione P, Spinelli S, Micco PD, Fiorentino G. Alpha-1 Antitrypsin Screening in a Selected Cohort of Patients Affected by Chronic Pulmonary Diseases in Naples, Italy. Journal of Clinical Medicine. 2021; 10(8):1546. https://doi.org/10.3390/jcm10081546
Chicago/Turabian StyleAnnunziata, Anna, Ilaria Ferrarotti, Antonietta Coppola, Maurizia Lanza, Pasquale Imitazione, Sara Spinelli, Pierpaolo Di Micco, and Giuseppe Fiorentino. 2021. "Alpha-1 Antitrypsin Screening in a Selected Cohort of Patients Affected by Chronic Pulmonary Diseases in Naples, Italy" Journal of Clinical Medicine 10, no. 8: 1546. https://doi.org/10.3390/jcm10081546
APA StyleAnnunziata, A., Ferrarotti, I., Coppola, A., Lanza, M., Imitazione, P., Spinelli, S., Micco, P. D., & Fiorentino, G. (2021). Alpha-1 Antitrypsin Screening in a Selected Cohort of Patients Affected by Chronic Pulmonary Diseases in Naples, Italy. Journal of Clinical Medicine, 10(8), 1546. https://doi.org/10.3390/jcm10081546