Abstract
Myofibrillar myopathies (MFM) are heterogeneous hereditary muscle diseases with characteristic myopathological features of Z-disk dissolution and aggregates of its degradation products. The onset and progression of the disease are variable, with an elusive genetic background, and around half of the cases lacking molecular diagnosis. Here, we attempted to establish possible genetic foundations of MFM by performing whole exome sequencing (WES) in eleven unrelated families of 13 patients clinically diagnosed as MFM spectrum. A filtering strategy aimed at identification of variants related to the disease was used and included integrative analysis of WES data and human phenotype ontology (HPO) terms, analysis of muscle-expressed genes, and analysis of the disease-associated interactome. Genetic diagnosis was possible in eight out of eleven cases. Putative causative mutations were found in the DES (two cases), CRYAB, TPM3, and SELENON (four cases) genes, the latter typically presenting with a rigid spine syndrome. Moreover, a variety of additional, possibly phenotype-affecting variants were found. These findings indicate a markedly heterogeneous genetic background of MFM and show the usefulness of next generation sequencing in the identification of disease-associated mutations. Finally, we discuss the emerging concept of variant load as the basis of phenotypic heterogeneity.
1. Introduction
Myofibrillar myopathy (MFM) is a heterogeneous group of hereditary diseases of the skeletal muscles characterized by pathological myofibrillar disintegration and abnormal protein deposits. The myofibrils dissolution is centered around the Z-disc and is described as Z-disc streaming. This leads to ectopic aggregation of degraded granulofilamentous material in diverse patterns in the myofibril-free regions in proximity of the nuclei and sarcolemma. The accumulated proteins include but are not limited to those encoded by genes most commonly associated with the disease. Autophagic vacuoles containing degraded fragments of membranous organelles are also observed [].
MFM should be diagnosed based on clinical presentation, electromyography, and especially on muscle biopsy findings [], however, genetic results are a coveted confirmation of the diagnosis, and identification of the MFM type requires uncovering of the causative mutation.
The genetic basis of MFM is only partially understood. The majority of genetically diagnosed patients harbor pathogenic mutations in one of six genes: DES, CRYAB, MYOT, LDB3, FLNC, or BAG3. In addition, mutations in genes typically associated with other muscular diseases can result in MFM-like phenotypes. The list of such genes includes FHL, TTN, DNAJB6, PLEC, ACTA1, HSPB8, LMNA, KY, PYROXD1, SQSTM1, and TIA1 and is still expanding []. Mutations in the SELENON gene are found among other disease entities in desmin-related myopathy with Mallory body-like inclusions (small clusters of accumulated proteins in the muscle fibers). SELENON-related muscle disorders are characterized by axial muscle weakness, spine stiffness, scoliosis, and serious breathing problems (https://ghr.nlm.nih.gov/gene/SELENON#conditions (accessed on 18 October 2020)).
Since many clinically MFM-like cases are left without genetic diagnosis, further efforts to uncover a genetic basis of the disease are justified. Next generation sequencing (NGS) followed by bioinformatic analyses of the obtained lists of genomic variants provide a wide, potentially non-biased approach towards discovering variants involved in pathogenesis []. This approach can identify novel mutations in known causative genes or even in genes not previously known to be related to a specific phenotype. Mutations in genes previously linked with other disorders may explain the observed clinical overlap between diverse muscle diseases. In addition to the causative genotype, some of the detected variants could also be phenotype-affecting, and some of them might even be deemed as co-causative.
Here, we report a whole exome sequencing study on a group of Polish patients with clinically diagnosed MFM. We identified pathogenic mutations in 73% of cases. These were found in genes already associated with myopathies, but not always typically with MFM. In some cases, we suggest additional, possible phenotype-affecting variants.
2. Materials and Methods
2.1. Patient Recruitment
The study involved thirteen patients from eleven unrelated families with clinically diagnosed myofibrillar myopathy from a single neuromuscular diagnosis and treatment medical center (see Table 1). The mean age of the patients at onset was 14 years (range 0–46), with nine patients exhibiting their first symptoms in childhood (0–9 years), two patients with the disease onset in early adulthood (21 and 26 years), and two patients with the disease onset in the fourth to fifth decade of life (39 and 46 years).

Table 1.
Red flags characteristic for myofibrillar myopathies (MFM) and their frequency in patients. CK: creatine kinase.
2.2. Ethics Approval and Consent to Participate
The study was approved by the Ethics Committee of the Warsaw Medical University and MSW Hospital (Warsaw, Poland) in compliance with national legislation and the Code of Ethical Principles for Medical Research Involving Human Subjects of the World Medical Association. Written consent was obtained from all study subjects according to the Declaration of Helsinki (BMJ 1991; 302:1194). The authors are very grateful to all for their participation in this study.
2.3. Clinical Assessment
The patients were initially diagnosed, studied, and documented in the years 2003–2019 at the Department of Neurology, Medical University of Warsaw. The following were carried out for each patient: pedigree analysis; neurological exam including muscle strength assessment according to the Medical Research Council’s (MRC) grading scale; muscle biopsy; serum creatine kinase (CK) level determination; electrophysiological studies including nerve conduction tests and concentric needle EMG and, when indicated, cardiological examination with electrocardiography (EKG) and echocardiography; and respiratory function tests. In selected cases, an MRI scan of affected muscles was performed.
Genetic and clinical findings without reference to whole exome data have been published for patients 1a, 1b, 2, and 3 [,].
2.4. Genetic Analyses
DNA was extracted from venous blood using standard methods []. Genomic DNA was captured using a SureSelect Human All Exon v5+UTR or SureSelect Human All Exon 6 (pts 7 and 8a) enrichment kit (Agilent Technologies, Santa Clara, CA, USA) and paired-end 100-nt sequencing was performed on an Illumina HiSeq2000 or HiSeq1500 platform (Illumina, San Diego, CA, USA). Fast read files were generated from the sequencing platform via the Illumina pipeline. Adapter sequences in the raw data were removed and low quality reads with low base quality were discarded. Paired-end reads from FASTQ files were aligned to the human reference genome hs38DH using the Burrows-Wheeler Alignment (BWA) package MEM algorithm. Resulting Binary Alignment Map (BAM) files were post-processed with Sentieon DNAseq software (Sentieon, Inc., San Jose, CA, USA), implementing the GATK 3.5 Best Practices protocol, and variant call format (VCF) files were generated. The obtained 15 Gb of aligned sequence data resulted in approximately 75× the median coverage of the target capture regions with 97.5% of target bases covered at least 10×. Capture performance statistics were calculated using CollectHsMetrics in Picard 2.17.10. The alignments were viewed with the Integrative Genomics Viewer []. SNVs (single-nucleotide variants) and indels (small insertion/deletion) variants were called using the GATK Unified Genotyper. Annovar was used for initial variant annotation [].
2.5. Bioinformatic Analyses
Exome sequencing identified on average 156,000 SNVs and 29,000 indels per exome, of which 89,000 and 17,000, respectively, were intergenic or intronic (but not affecting splice sites) and removed from further consideration. Only variants with an impact on protein secondary structure (missense, nonsense, and frameshift) and on splicing were retained. Further filtering was based on Phred quality scores, minor allele frequency in the ExAC (Exome Aggregation Consortium) and gnomAD (The Genome Aggregation Database) databases (below 1% were retained), and association with human phenotype ontology (HPO) terms []. The HPO term used was HP:0011805 Abnormal muscle morphology. Prioritization was based on the predicted effect on the protein, predicted pathogenicity (assessed with Mutation Taster, Mutation Assessor, PolyPhen2, Provean, CADD, and SIFT), and known association with myopathic phenotypes. The variants on the final list were individually assessed together by both geneticists and clinicians according to the guidelines of the American College of Medical Genetics and Genomics [], with emphasis on the possible match with the actual phenotype of each patient.
Selected variants (including all putative causative mutations and discussed phenotype variants) were confirmed with direct fluorescence-based sequencing (ABI 3130 Genetic Analyzer, Applied Biosystems, Waltham, MA, USA).
3. Results
3.1. Whole Exome Sequencing
Using exome sequencing, we identified putative pathogenic mutations in known myopathy-related genes in eight of the eleven MFM cases (73%). These were pathogenic or likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) criteria and matched the respective patient’s phenotype (see Table 2).
Table 2.
Putative causative mutations and genes with potentially phenotype-influencing variants in MFM patients.
Dominant mutations were identified in DES (two cases), CRYAB (one case), and TPM3 (one case) and found to segregate with the phenotype in a family or to occur de novo (patient 4). Five further patients (5-8b) had mutations in both copies of the SELENON gene, and family analysis confirmed that they were inherited from both parents (biparental inheritance).
Filtering of the variants annotated based on the HPO terms associated with genes returned between 32 and 64 variants per genome (47.3 on average) that could be related to muscle abnormality. These variants were found in various myopathy-associated genes, in genes associated with myofibrillar myopathy, limb-girdle muscular dystrophy, rigid spine congenital muscular dystrophy, nemaline myopathy, multiminicore myopathy, collagen myopathy, tubular myopathy, or Ehlers-Danlos syndrome. Full results including details of all the variants identified after filtering for each patient and results of the pathway mapping analysis are provided in a supplementary table (see Supplementary Table S1).
3.2. Clinical Data
The clinical data of the patients are shown in Table 3.
Table 3.
Clinical features of the patients.
Histological analysis documented myopathic features in all cases (Selected specimens—Figure 1).
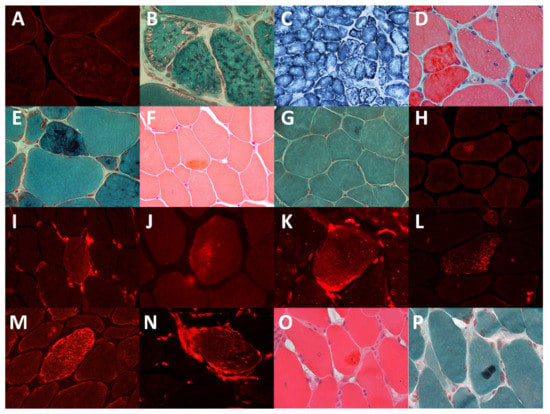
Figure 1.
Immunohistochemistry and light microscope images of muscles of the patients. (A). Patient 2: desmin aggregates (B). Patient 2: small reddish or dark inclusions in the Gomori trichrome staining. (C). Patient 2: lobulated fibers visible in the succinate dehydrogenase staining. (D). Patient 3: dark red material in the hematoxylin and eosin staining. (E). Patient 3: dark blue material in the trichrome staining. (F). Patient 5: acidophilic inclusions in the hematoxylin and eosin staining, (G) Patient 5: acidophilic inclusions in the trichrome staining. (H). Patient 5: desmin aggregates. (I). Patient 5: vimentin aggregates. (J). Patient 6: CRYAB aggregates. (K). Patient 6: vimentin aggregates. (L). Patient 11: CRYAB aggregates. (M). Patient 11: desmin aggregates. (N). Patient 11: vimentin aggregates. (O). Patient 11: large acidophilic inclusions in the hematoxylin and eosin staining, (P). Patient 11: acidophilic inclusions in the trichrome staining.
In addition, MRI was performed for two patients (7, 8a) with SELENON mutations (Figure 2). It showed an absence of the semimembranosus muscle, characteristic of SELENON-related cases, moderate-to-severe changes in the sartorius muscle, and sparing of the gracilis muscle.
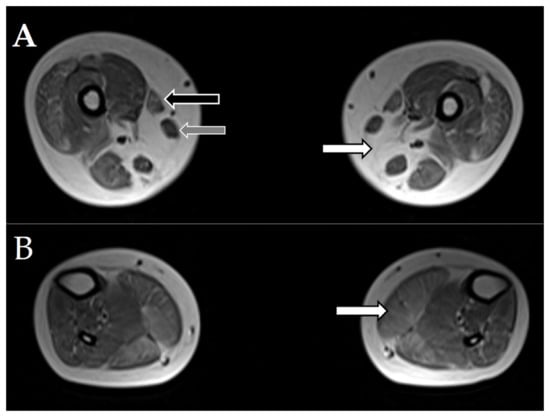
Figure 2.
Magnetic resonance imaging (MRI) of muscles of patients 7 and 8a with SELENON mutations. Selected axial T1W images of the muscles. (A). White arrow points to the absence of the semimembranosus muscle. Black arrow points to the moderately affected sartorius muscle. Grey arrow points to the intact gracilis muscle. (B). White arrow points to moderate, diffuse changes in the gastrocnemius muscle.
The MRI of the patient 1a with desminopathy (previously reported by Fichna et al., 2014 []) showed characteristic severe changes in the semitendinosus muscle and moderate-to-severe changes in the adductor magnus, vastus intermedius, and gracilis muscles with relative sparing of the rectus femoris, vastus lateralis, adductor longus, and the long head of biceps femoris (Figure 3).
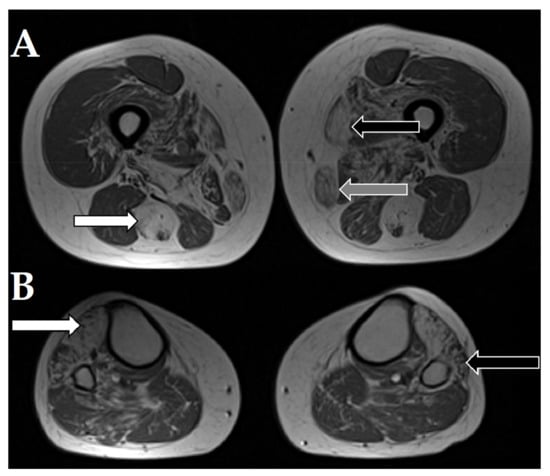
Figure 3.
Magnetic resonance imaging of selected axial T1W images of muscles of the patient 1a with desminopathy. (A). White arrow points to severe fatty atrophy in the semitendinosus muscle. Black arrow points to the sartorius muscle. Grey arrow points to the gracilis muscle. (B). White arrow points to severe changes in the tibialis anterior muscle. Black arrow points to the affected peroneus muscles.
4. Discussion
We completed and reported on the first exome-wide analysis in a group of MFM patients and their families from the Polish population. As a result of bioinformatic filtering we found, on average, 47.4 rare variants per patient in myopathy-related genes. These genes were already associated with diverse diseases of the skeletal muscle, but not necessarily with MFM.
Among the identified variants, causative variants were identified in eight out of eleven cases (73%). They were pathogenic according to the ACMG criteria and are consistent with the clinical characteristics of the patients.
In cases with typical dominant inheritance, the identified causative mutations were mostly in the known MFM-related genes (DES and CRYAB), but in one dominant case, a known causative mutation in TPM3, not commonly associated with MFM, was found. Variants in the TPM3 gene encoding γ-tropomyosin expressed in type 1 slow muscle fibers have been mostly reported with three other types of muscle disease: cap myopathy, congenital myopathy with fiber type disproportion, and nemaline myopathy.
Interestingly, the apparently sporadic cases appeared to be recessive rather than de novo dominant forms of MFM. In three such cases, causative mutations were found in the SELENON gene commonly associated with rigid spine syndrome but also with congenital myopathy with fiber type disproportion. However, some SELENON mutations have also been reported in MFM-like cases [] and were considered in similar phenotypes []. All three variants identified in the present study have already been reported, with p.Arg466Gln being one of the first SELENON mutations associated with a muscle disease []. The c.713dupA variant most common in our cohort has been reported at least five times, most recently in three cases in the Czech population []. The same study also reported three cases of the c.997_1000delGTGC mutation found by us in a single patient. The distribution of MFM subtypes suggests SELENON cases to be fairly common in the Polish and Czech populations. The gene should be of special interest in cases with a childhood onset, rigid spine phenotype, and/or floppy baby syndrome. In addition, an MRI scan indicating an absence of the semimembranosus muscle and sparing of the gracilis muscle is strongly indicative of SELENON-related cases (Figure 1). Magnetic resonance imaging is increasingly being used to characterize the severity and pattern of individual muscle involvement. It could provide a very useful noninvasive diagnostic tool for the detection and quantification of myopathic changes during the clinical workup.
In the three unsolved cases of our study, the pathogenic mutations could likely be located in noncoding (regulatory or deep intronic) regions. Copy number variation would be missed by exome sequencing, but could influence the phenotype. Moreover, in sporadic or first cases in a family, a post-zygotic mutation affecting muscle but not the blood cells could cause the disease.
Additional variants that at the present state of knowledge could not be identified as causative still could explain both the phenotypic overlap, and the inter- and intrafamilial differences.
In a few cases, the patient’s clinical phenotype could be plausibly explained by mutations in more than one gene. Patient no. 2 had a much earlier onset and a more severe disease course than his also-affected mother. They both have a p.Gln348Pro causal missense mutation in the DES gene encoding desmin. Mutations in DES are common cause of MFM, especially with cardiac involvement [], however, the son additionally carries a private DNAJB6 c.962C>T (p.Ser321Leu) mutation, which could explain his more severe course of the disease. The ACMG criteria classify the variant as likely benign, mainly due to its common frequency in the general population (gnomAd MAF = 0.0004437). Verdicts of the prediction programs are inconclusive (SIFT—deleterious, Polyphen, MutationAssessor and MutationTaster—benign). In addition, most of the known pathogenic mutations in DNAJB6 were reported in exon 5, not exon 10, and affect the G/F domain characteristic for the DNAJA and DNAJB proteins. On the other hand, missense mutations in DNAJB6 are rarely benign but are a common cause of myofibrillar disintegration. The DNAJB6 protein is localized primarily at the Z-disk where it is involved in the maintenance of keratin filament organization []. Keratins 18 and 19 seem to play a role complementary to desmin in assembling intermediate filament networks and transducing the contractile force []. Even if the identified DNAJB6 variant is not pathogenic in itself, it could be responsible for the earlier and more severe progression of the desmin-related disease.
In family 1, with a clearly causative DES mutation, a slightly more severe disease course of patient 1a compared to her cousin, patient 1b, could be an effect of phenotype-affecting variants. Mutations in the RYR1 gene have been reported in mild clinical forms of rigid spine syndrome [] and in cases similar to myofibrillar myopathy []. Possibly the novel variant NM_000540.3:c.12809C>A in this gene identified in patient 1a could be responsible for the difference in the clinical manifestation between patients from the same family.
The TPM3 p.Arg168Cys variant identified in patient no. 4 was reported several times in patients presenting a full spectrum of diseases associated with TPM3 mutations [,,]. In addition, other mutations in the same codon, p.Arg168Gly and p.Arg168Lys, have been reported. Although some TPM3 variants are associated with a specific form of myopathy [], the fiber-type distribution pattern and the pattern of protein inclusions often vary widely, even among patients with the same TPM3 mutation. Therefore, it is likely that the phenotype is modified by other genomic variants. In the case of patient no. 4, the LDB3(NM_007078.3):c.1300G>T (p.Ala434Ser) variant seems particularly interesting as it affects a gene already associated with MFM. The ACMG criteria suggest the variant is likely benign as out of 38 missense LDB3 variants of unknown significance, 35 appeared to be benign, and most of the pathogenic mutations reported were in exon 6, not in exon 10. However, the prediction programs do not agree on the variant pathogenicity (SIFT, Polyphen—benign, MutationAssessor and MutationTaster—damaging). In addition, the variant is not reported in the gnomAd database. Nevertheless, even if not pathogenic by itself, this variant could affect the phenotype of a patient carrying a causative mutation in another gene.
In other cases, the copious variants found in many muscle-related genes seem likely to modify or exacerbate phenotypic manifestation of the major pathogenic mutations, however, selecting co-causative variants from among those classified as potentially modifying was not possible. Discrimination between genuine phenotype-affecting variants and the thousands of insignificant variants harbored by each individual is one of the most challenging tasks posed by the introduction of genomic-scale analyses. Indeed, in all the cases studied here we encountered novel rare variants that could be related to myopathies. Some of these genes could be grouped together with known MFM-associated genes according to the structural or functional association of their products. However, their relevance could not be established based on the inheritance mode, patient’s phenotype, and known effects of other mutations in these genes.
Numerous muscular disorders overlap phenotypically with MFM. NGS-based genetic analyses can resolve clinical dilemmas and facilitate exact diagnosis [,]. Additionally, with the reporting of new cases, the reported clinical manifestations of mutations in a given gene are likely to expand. Recent large sequencing studies show that the genetic background of muscle diseases is more complex than previously appreciated [,,]. In addition, our data suggest that mutations in more than one gene in a patient can result in a more severe phenotype. The observed phenotypic variability within a given MFM subtype, particularly between patients with the same causative mutation, indicate a strong influence of disease-modifying genes. Indeed, a common polymorphism in the TIA1 gene has been shown to be a necessary factor in SQSTM-related MFM []. Digenic inheritance has been proven in other muscular disorders, such as a subtype of facioscapulohumeral muscular dystrophy [,] and congenital myasthenic syndrome []. Unequivocal identification of risk, phenotype-modifying or co-causative factors, is a major challenge that requires comprehensive bioinformatic analysis of combined genomic and clinical data followed by functional in vitro studies [].
Myofibril proteins form a complex machinery whose functional or structural impairment can lead to progressive dysfunction of the muscle. The number of rare variants identified in genes already associated with muscle diseases was higher than in patients with non-muscular diseases (i.e., neurodevelopmental) studied by our group. While it suggests their aggregate influence on the phenotype, this conclusion must be verified on a larger cohort of both patients and controls. The variant burden in many genes associated with muscular diseases must not be overlooked, as accumulation of minor changes, even those without a sufficient influence when present in isolation, could result in dramatic phenotype changes. Such oligogenic or even polygenic etiology seems most likely in unsolved cases, where the main causative mutation cannot be identified. On the other hand, some of the mutations deemed disease-causing prior to the advent of NGS might turn out to have a modifying effect only or incomplete penetrance and require additional variants in other genes to result in pathology.
5. Conclusions
The widespread application of next generation sequencing in studies on the genetic basis of various diseases, including muscle disorders, has brought ample data challenging the classical “one gene—one phenotype” paradigm [] and underscores the heterogeneity and complexity of the genome [,].
Our results emphasize the continuum of genetic causality as they show that mutations in many genes can result in MFM-like diseases. The causative mutations found in TPM3 and SELENON—genes associated not only with MFM—highlight the phenotypic heterogeneity and clinical overlap between muscular disorders. Mutations in genes originally identified in patients with other muscle diseases, such as cap myopathy or minicore syndromes, produce a wide spectrum of phenotypes.
Although it is frequently not possible to evaluate the weight of additional putative phenotype-affecting mutations, comprehensive analysis of variants suggests that cumulative “variant load” may likely contribute to the observed phenotypic variability. This could explain the difference in clinical manifestation among cases with the same causative mutations, sometimes even between related patients. Even though the majority of the MFM cases presented here could be explained by mutations in a single gene, the identification of numerous variants in genes associated with various myopathies, some of which could even be related to specific symptoms of the patients, is intriguing. The oligogenic background of MFM is visible also in some of the patients harboring a clearly pathogenic mutation.
Supplementary Materials
The following is available online at https://www.mdpi.com/2077-0383/10/5/914/s1, Table S1: Rare variants in genes associated with myopathies in MFM patients.
Author Contributions
Conceptualization, A.P.-C. and J.P.F.; data curation J.P.F.; formal analysis, M.G.; funding acquisition, A.M. and M.J.; investigation, M.J., E.R., B.K., and J.P.F.; methodology, A.P.-C. and E.R.; project administration, J.P.F.; resources, A.P.-C. and M.J.; software M.G. and J.P.F.; supervision, A.O., C.Z., and J.P.F.; validation, C.Z.; visualization, A.P.-C. and E.R., writing—original draft preparation, A.P.-C. and J.P.F.; writing—review and editing, A.P.-C., M.J., M.G., E.R., B.K., A.M., A.O., C.Z., and J.P.F. All authors have read and agreed to the published version of the manuscript.
Funding
The study on myofibrillar myopathy was funded by the National Science Centre grant NCN 2012/05/D/NZ4/02978 to J.P.F. and A.M. Whole exome sequencing of two cases with SELENON mutations was supported by the National Science Centre grant NCN 2015/17/B/NZ5/01368 to M.J. and M.G. The funding sources had no involvement in the experiment design, analysis, data interpretation, or manuscript preparation.
Institutional Review Board Statement
The study was approved by the Bioethics Committee of the Central Clinical Hospital of the Ministry of the Interior and Administration (no 108/2017, 04.10.2017, Warsaw, Poland) in compliance with national legislation and the Code of Ethical Principles for Medical Research Involving Human Subjects of the World Medical Association.
Informed Consent Statement
Written informed consent was obtained from all study subjects according to the Declaration of Helsinki (BMJ 1991; 302:1194). The authors are very grateful to all for their participation in this study.
Data Availability Statement
All data generated during this study are included in the supplementary file. The raw data analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The Department of Neurology is a member of the European Reference Network of Rare Diseases: EURO-NMD. The authors are very grateful to all the patients and their family members for participation in the study.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Selcen, D. Myofibrillar Myopathies. Neuromuscul. Disord. 2011, 21, 161–171. [Google Scholar] [CrossRef]
- Selcen, D.; Engel, A.G. Myofibrillar Myopathy. In GeneReviews(®); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Fichna, J.P.; Maruszak, A.; Żekanowski, C. Myofibrillar Myopathy in the Genomic Context. J. Appl. Genet. 2018, 59, 431–439. [Google Scholar] [CrossRef]
- Lek, M.; MacArthur, D. The Challenge of Next Generation Sequencing in the Context of Neuromuscular Diseases. J. Neuromuscul. Dis. 2014, 1, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Fichna, J.P.; Karolczak, J.; Potulska-Chromik, A.; Miszta, P.; Berdynski, M.; Sikorska, A.; Filipek, S.; Redowicz, M.J.; Kaminska, A.; Zekanowski, C. Two Desmin Gene Mutations Associated with Myofibrillar Myopathies in Polish Families. PLoS ONE 2014, 9, e115470. [Google Scholar] [CrossRef]
- Fichna, J.P.; Potulska-Chromik, A.; Miszta, P.; Redowicz, M.J.; Kaminska, A.M.; Zekanowski, C.; Filipek, S. A Novel Dominant D109A CRYAB Mutation in a Family with Myofibrillar Myopathy Affects ΑB-Crystallin Structure. BBA Clin. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A Simple Salting out Procedure for Extracting DNA from Human Nucleated Cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Masino, A.J.; Dechene, E.T.; Dulik, M.C.; Wilkens, A.; Spinner, N.B.; Krantz, I.D.; Pennington, J.W.; Robinson, P.N.; White, P.S. Clinical Phenotype-Based Gene Prioritization: An Initial Study Using Semantic Similarity and the Human Phenotype Ontology. BMC Bioinform. 2014, 15, 248. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, A.; Ceuterick-de Groote, C.; Marks, J.J.; Goemans, N.; Schreiber, G.; Hanefeld, F.; Fardeau, M.; Martin, J.-J.; Goebel, H.H.; Richard, P.; et al. Desmin-Related Myopathy with Mallory Body-like Inclusions Is Caused by Mutations of the Selenoprotein N Gene. Ann. Neurol. 2004, 55, 676–686. [Google Scholar] [CrossRef]
- Kostera-Pruszczyk, A.; Goudeau, B.; Ferreiro, A.; Richard, P.; Simon, S.; Vicart, P.; Fidzianska, A. Myofibrillar Myopathy with Congenital Cataract and Skeletal Anomalies without Mutations in the Desmin, AlphaB-Crystallin, Myotilin, LMNA or SEPN1 Genes. Neuromuscul. Disord. 2006, 16, 759–762. [Google Scholar] [CrossRef]
- Moghadaszadeh, B.; Petit, N.; Jaillard, C.; Brockington, M.; Quijano Roy, S.; Merlini, L.; Romero, N.; Estournet, B.; Desguerre, I.; Chaigne, D.; et al. Mutations in SEPN1 Cause Congenital Muscular Dystrophy with Spinal Rigidity and Restrictive Respiratory Syndrome. Nat. Genet. 2001, 29, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Stehlíková, K.; Skálová, D.; Zídková, J.; Haberlová, J.; Voháňka, S.; Mazanec, R.; Mrázová, L.; Vondráček, P.; Ošlejšková, H.; Zámečník, J.; et al. Muscular Dystrophies and Myopathies: The Spectrum of Mutated Genes in the Czech Republic. Clin. Genet. 2017, 91, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kubánek, M.; Schimerová, T.; Piherová, L.; Brodehl, A.; Krebsová, A.; Ratnavadivel, S.; Stanasiuk, C.; Hansíková, H.; Zeman, J.; Paleček, T.; et al. Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction. J. Clin. Med. 2020, 9, 937. [Google Scholar] [CrossRef] [PubMed]
- Izawa, I.; Nishizawa, M.; Ohtakara, K.; Ohtsuka, K.; Inada, H.; Inagaki, M. Identification of Mrj, a DnaJ/Hsp40 Family Protein, as a Keratin 8/18 Filament Regulatory Protein. J. Biol. Chem. 2000, 275, 34521–34527. [Google Scholar] [CrossRef] [PubMed]
- Muriel, J.M.; O’Neill, A.; Kerr, J.P.; Kleinhans-Welte, E.; Lovering, R.M.; Bloch, R.J. Keratin 18 Is an Integral Part of the Intermediate Filament Network in Murine Skeletal Muscle. Am. J. Physiol. Cell Physiol. 2020, 318, C215–C224. [Google Scholar] [CrossRef]
- Jungbluth, H.; Sewry, C.A.; Muntoni, F. Core Myopathies. Semin. Pediatr. Neurol. 2011, 18, 239–249. [Google Scholar] [CrossRef]
- Bevilacqua, J.A.; Monnier, N.; Bitoun, M.; Eymard, B.; Ferreiro, A.; Monges, S.; Lubieniecki, F.; Taratuto, A.L.; Laquerrière, A.; Claeys, K.G.; et al. Recessive RYR1 Mutations Cause Unusual Congenital Myopathy with Prominent Nuclear Internalization and Large Areas of Myofibrillar Disorganization. Neuropathol. Appl. Neurobiol. 2011, 37, 271–284. [Google Scholar] [CrossRef]
- Clarke, N.F. Skeletal Muscle Disease Due to Mutations in Tropomyosin, Troponin and Cofilin. Adv. Exp. Med. Biol. 2008, 642, 40–54. [Google Scholar] [CrossRef]
- Ohlsson, M.; Fidzianska, A.; Tajsharghi, H.; Oldfors, A. TPM3 Mutation in One of the Original Cases of Cap Disease. Neurology 2009, 72, 1961–1963. [Google Scholar] [CrossRef]
- Waddell, L.B.; Kreissl, M.; Kornberg, A.; Kennedy, P.; McLean, C.; Labarre-Vila, A.; Monnier, N.; North, K.N.; Clarke, N.F. Evidence for a Dominant Negative Disease Mechanism in Cap Myopathy Due to TPM3. Neuromuscul. Disord. 2010, 20, 464–466. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Karpicheva, O.E.; Simonyan, A.O.; Avrova, S.V.; Rogozovets, E.A.; Sirenko, V.V.; Redwood, C.S. The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. Int. J. Mol. Sci. 2018, 19, 3975. [Google Scholar] [CrossRef]
- Gómez-López, G.; Dopazo, J.; Cigudosa, J.C.; Valencia, A.; Al-Shahrour, F. Precision Medicine Needs Pioneering Clinical Bioinformaticians. Brief. Bioinform. 2019, 20, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.; Riazul Islam, S.M.; Hussain, M.; Lee, S. Precision Medicine Informatics: Principles, Prospects, and Challenges. IEEE Access 2020, 8, 13593–13612. [Google Scholar] [CrossRef]
- Fichna, J.P.; Macias, A.; Piechota, M.; Korostyński, M.; Potulska-Chromik, A.; Redowicz, M.J.; Zekanowski, C. Whole-Exome Sequencing Identifies Novel Pathogenic Mutations and Putative Phenotype-Influencing Variants in Polish Limb-Girdle Muscular Dystrophy Patients. Hum. Genom. 2018, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Zaganas, I.; Mastorodemos, V.; Spilioti, M.; Mathioudakis, L.; Latsoudis, H.; Michaelidou, K.; Kotzamani, D.; Notas, K.; Dimitrakopoulos, K.; Skoula, I.; et al. Genetic Cause of Heterogeneous Inherited Myopathies in a Cohort of Greek Patients. Mol. Genet. Metab. Rep. 2020, 25. [Google Scholar] [CrossRef]
- Gonzalez-Quereda, L.; Rodriguez, M.J.; Diaz-Manera, J.; Alonso-Perez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Natera-de Benito, D.; Olive, M.; Gonzalez-Mera, L.; et al. Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes 2020, 11, 539. [Google Scholar] [CrossRef]
- Niu, Z.; Pontifex, C.S.; Berini, S.; Hamilton, L.E.; Naddaf, E.; Wieben, E.; Aleff, R.A.; Martens, K.; Gruber, A.; Engel, A.G.; et al. Myopathy with SQSTM1 and TIA1 Variants: Clinical and Pathological Features. Front. Neurol. 2018, 9, 147. [Google Scholar] [CrossRef]
- Lemmers, R.J.L.F.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.E.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic Inheritance of an SMCHD1 Mutation and an FSHD-Permissive D4Z4 Allele Causes Facioscapulohumeral Muscular Dystrophy Type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 729. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.-W.; Wong, K.-S.; Leung, H.-W.; Law, C.-Y. Limb Girdle Myasthenia with Digenic RAPSN and a Novel Disease Gene AK9 Mutations. Eur. J. Hum. Genet. 2017, 25, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Capriotti, E.; Carter, H. VarI-COSI 2018: A Forum for Research Advances in Variant Interpretation and Diagnostics. BMC Genom. 2019, 20, 550. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N. The Continuum of Causality in Human Genetic Disorders. Genome Biol. 2016, 17, 233. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yang, S.; Nykamp, K.; Garcia, J.; Lincoln, S.E.; Topper, S.E. Pathogenic Variant Burden in the ExAC Database: An Empirical Approach to Evaluating Population Data for Clinical Variant Interpretation. Genome Med. 2017, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Spataro, N.; Rodríguez, J.A.; Navarro, A.; Bosch, E. Properties of Human Disease Genes and the Role of Genes Linked to Mendelian Disorders in Complex Disease Aetiology. Hum. Mol. Genet. 2017, 26, 489–500. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).