
Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications




Abstract
1. Introduction
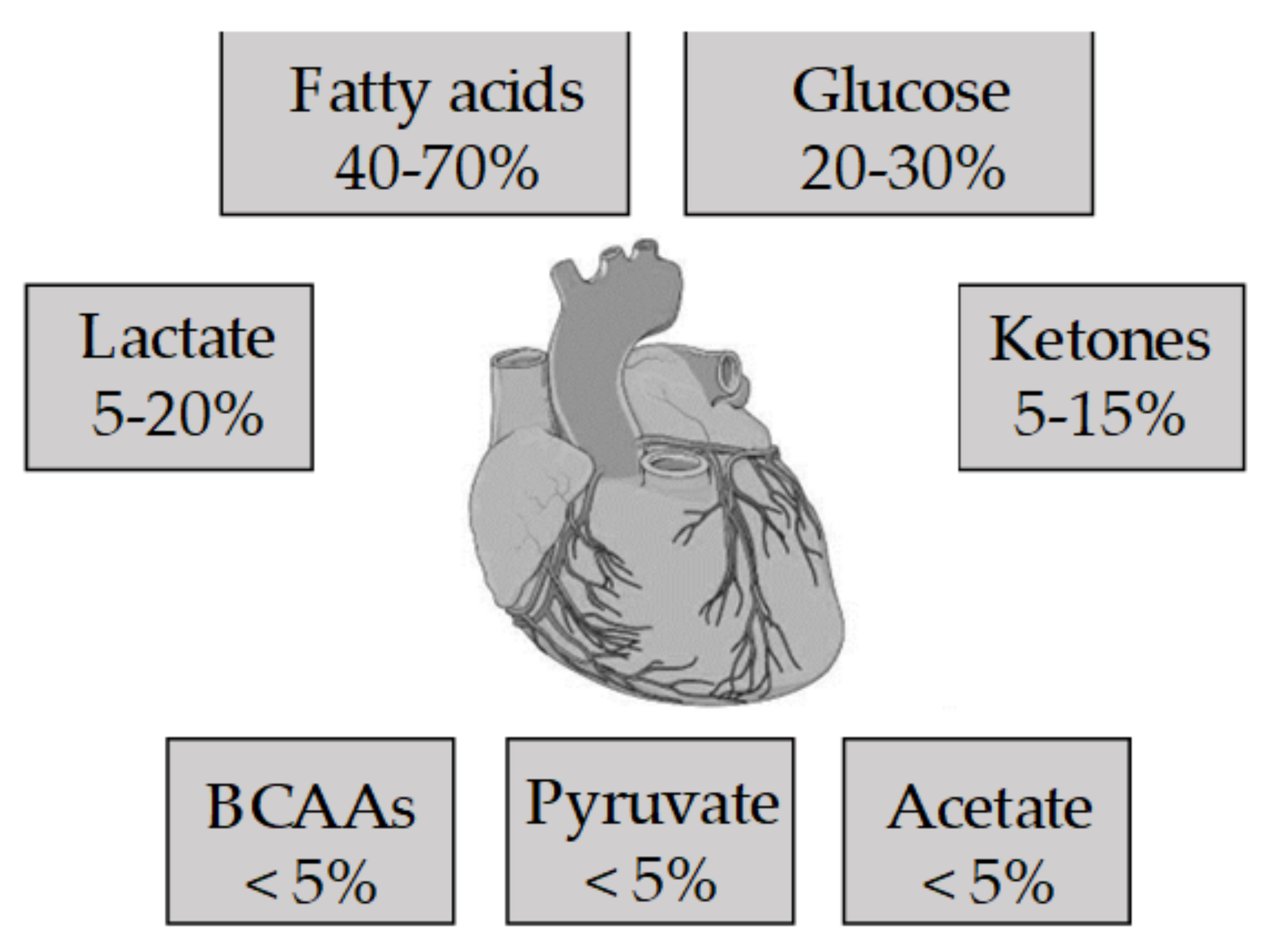
2. The Metabolism of the Healthy Heart
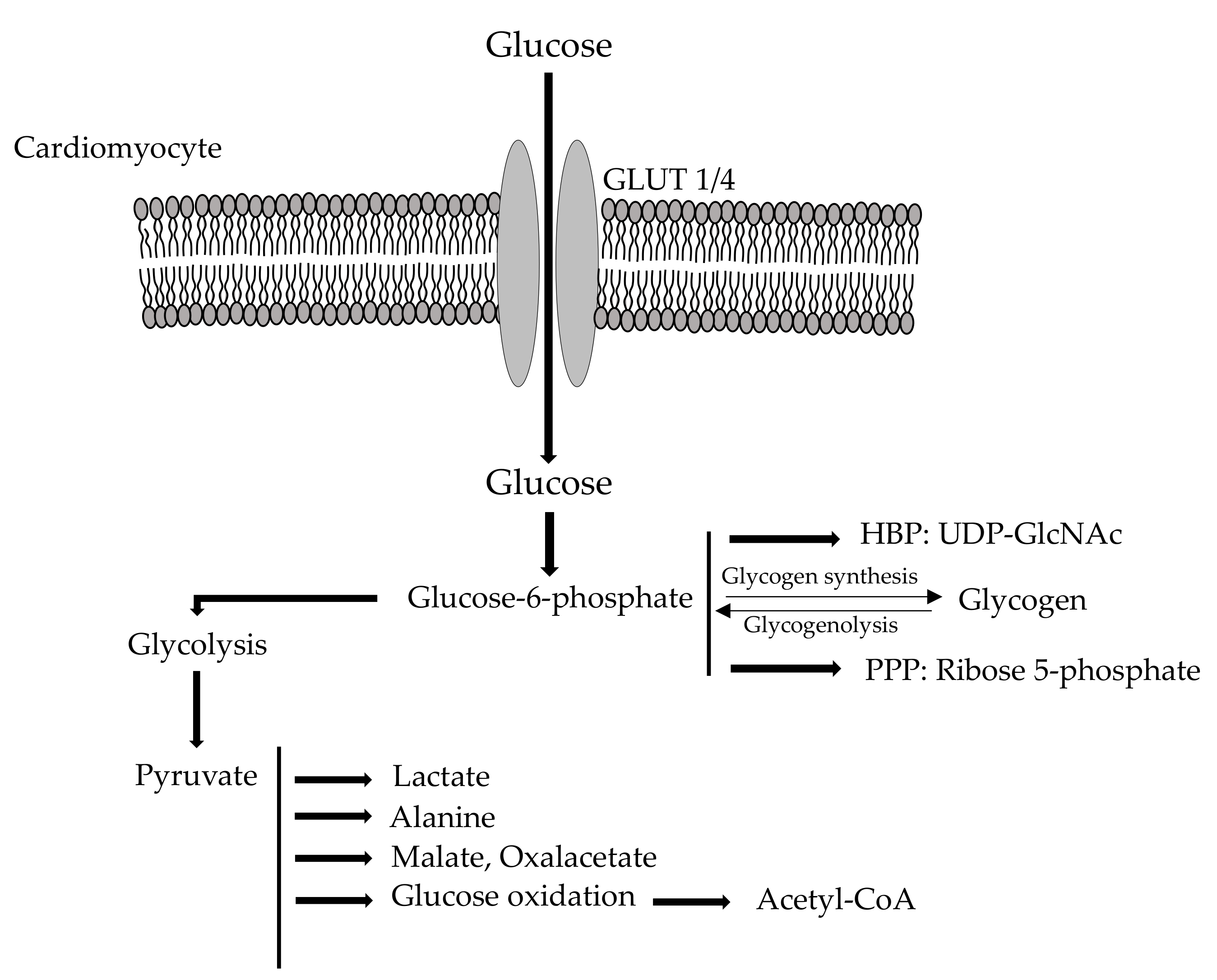
2.1. Cardio Metabolism of Glucose
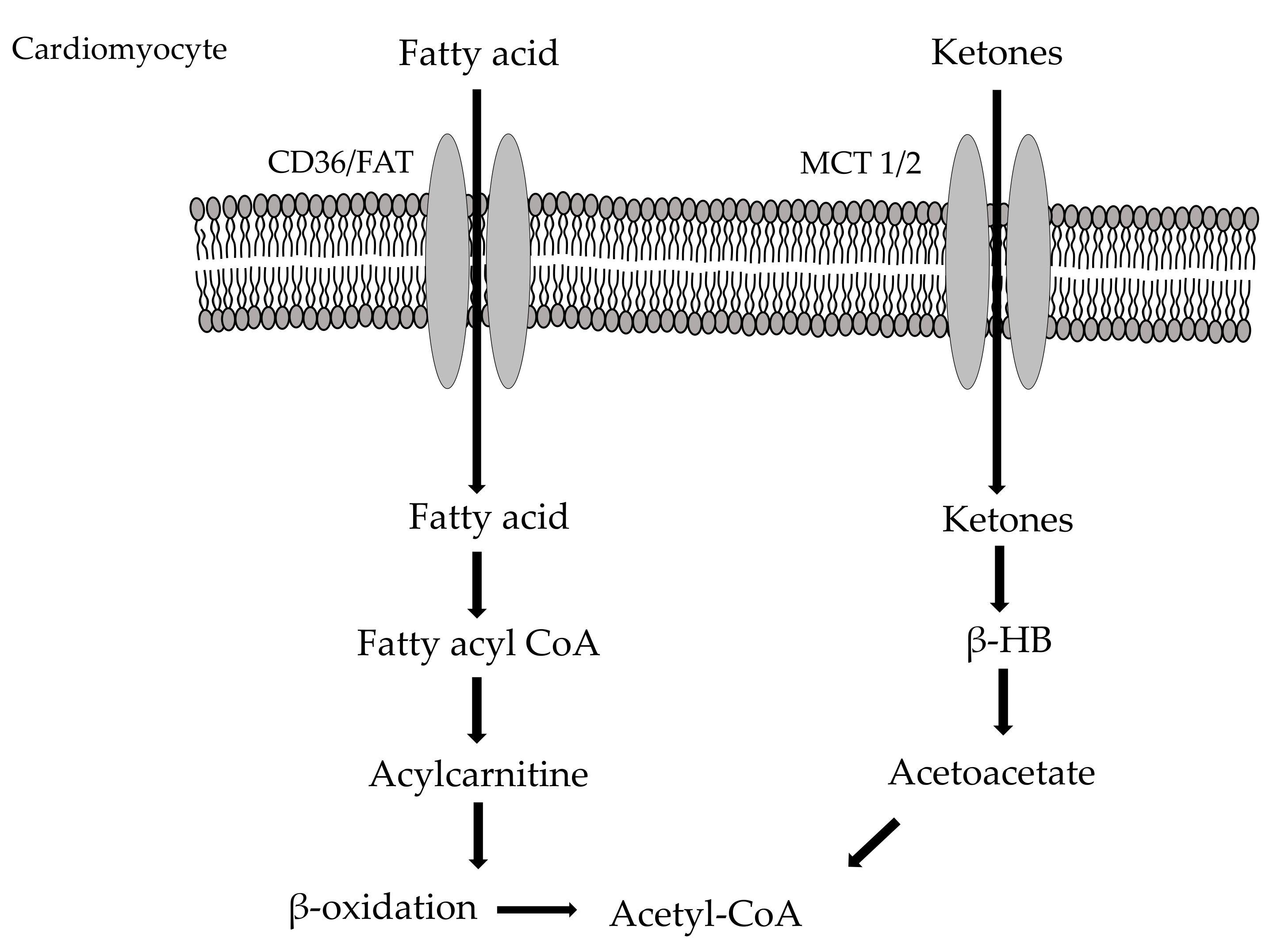
2.2. Myocardial Fatty Acid Metabolism
2.3. Cardio Metabolism of Ketone Bodies/Amino Acids
3. Cardiac Metabolic Impairment in Acute and Chronic Cardiac Diseases
3.1. Metabolic Changes Occurring during Acute Ischemia
3.2. Cardiometabolism in Chronic Ischemic Heart Disease
3.3. Metabolic Changes Occurring during Reperfusion
3.4. Cardiometabolic Profile in Inherited Cardiomyopathies
3.4.1. Fatty Acid Oxidation Disorders (FAODs)
3.4.2. Glycogen Storage Diseases (GSDs)
3.4.3. Lysosomal Storage Disorders (LSDs)
3.4.4. Mitochondrial Disorders
4. Cardiometabolic Adaptations in Heart Failure and Chronic Cardiac Diseases
4.1. The Shift towards Glucose Utilization in the Failing Heart
4.1.1. Is the Induction of Glucose Utilization a Double-Edged Sword in Heart Failure?
4.1.2. Significance of Glucose Utilization during Heart Failure
4.2. Impact of Glycogen Metabolism during Heart Failure
4.3. Role of Free Fatty Acid Metabolism in the Failing Heart
4.4. The Red Skeletal Muscle as a Counterpart: Common and Different Metabolic Traits
5. Therapeutic Strategies in Cardiac Diseases
5.1. Metabolic Therapies in Heart Failure
5.2. Metabolic Therapies in Myocardial Infarction
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAC | acetyl-CoA carboxylase |
| ADP | adenosine diphosphate |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ANF | natriuretic factor |
| ATP | adenosine triphosphate |
| Ca2+ | calcium |
| CABG | Coronary artery bypass graft surgery |
| CACT | carnitine acylcarnitine translocate |
| CAD | coronary artery disease |
| CPD | carnitine-palmitoyl transferase |
| CPT-I | carnitine palmitoyl transferase I |
| CPT-II | carnitine acyl-CoA transferase II |
| CVDs | cardiovascular diseases |
| FAODs | fatty acid oxidation disorders |
| G6P | glucose 6-phosphate |
| G6PDH | glucose 6-phosphate dehydrogenase |
| GFAT | fructose 6-phosphate amidotransferase |
| GLP-1 | glucagon-like peptide-1 |
| GLUT1 | insulin-independent glucose transporter |
| GLUT4 | insulin-sensitive glucose transporter |
| GSDs | glycogen storage diseases |
| GSK | glycogen synthase kinase |
| H+ | protons |
| HBP | hexosamine biosynthetic pathway |
| HDL | high-density lipoproteins |
| HF | heart failure |
| h-FABP | heart-specific fatty acid-binding protein |
| I/R | ischemia/reperfusion |
| IEM | inborn errors of metabolism |
| IPC | ischemic preconditioning |
| K+ | potassium |
| LCHAD | long chain 3-hydroxy-acyl-CoA dehydrogenase |
| LSDs | lysosomal storage disorders |
| MCD | malonyl-CoA decarboxylase |
| MI | myocardial infarction |
| mTOR | mechanistic Target of Rapamycin |
| ONOO– | peroxynitrite |
| PCD | primary carnitine deficiencies |
| PCI | primary percutaneous coronary intervention |
| PFK-1 | phospho-fructokinase-1 |
| PGC-1α | peroxisome proliferator-activated receptor-γ coactivator 1α |
| PH | primary hyperoxaluria |
| PPARα | peroxisome proliferator-activated receptor α |
| PPP | pentose phosphate pathway |
| PTP | permeability transition pore |
| ROS | reactive oxygen species |
| T2DM | type 2 diabetes mellitus |
| TGF-β | transforming growth factor β |
| UCP3 | uncoupling protein 3 |
| UDP-GlcNAc | uridine diphosphate-N-acetylglucosamine |
| VLCAD | very long-chain acyl-CoA dehydrogenase |
| WHO | World Health Organization |
References
- Taegtmeyer, H.; Lam, T.; Davogustto, G. Cardiac Metabolism in Perspective. Compr. Physiol. 2016, 6, 1675–1699. [Google Scholar] [PubMed]
- Kolwicz, S.C., Jr.; Olson, D.P.; Marney, L.C.; Garcia-Menendez, L.; Synovec, R.E.; Tian, R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 2012, 111, 728–738. [Google Scholar] [CrossRef]
- Gibb, A.A.; Hill, B.G. Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ. Res. 2018, 123, 107–128. [Google Scholar] [CrossRef]
- Geraets, C.N.W.; Veling, W.; Witlox, M.; Staring, A.B.P.; Matthijssen, S.J.M.A.; Cath, D. Virtual reality-based cognitive behavioural therapy for patients with generalized social anxiety disorder: A pilot study. Behav. Cogn. Psychother. 2019, 47, 745–750. [Google Scholar] [CrossRef]
- Stanley, W.C.; Morgan, E.E.; Huang, H.; McElfresh, T.A.; Sterk, J.P.; Okere, I.C.; Chandler, M.P.; Cheng, J.; Dyck, J.R.; Lopaschuk, G.D. Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation and reduces lactate production during demand-induced ischemia. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2304–H2309. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.E.; Krebs, H.A. The evolution of metabolic cycles. Nature 1981, 291, 381–382. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Taylor, C.S.; Taegtmeyer, H. Regulation of energy metabolism of the heart during acute increase in heart work. J. Biol. Chem. 1998, 273, 29530–29539. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Desvergne, B.; Dreyer, C.; Gavillet, M.; Laurini, R.N.; Wahli, W. PPAR expression and function during vertebrate development. Int. J. Dev. Biol. 2002, 46, 105–114. [Google Scholar] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1, regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef]
- Sugden, M.C.; Holness, M.J. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E855–E862. [Google Scholar] [CrossRef]
- Daniel, P.M.; Love, E.R.; Pratt, O.E. Factors affecting the supply of glucose to the heart of the rat, in vivo. J. Physiol. 1980, 309, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Manchester, J.; Kong, X.; Nerbonne, J.; Lowry, O.; Lawrence, J., Jr. Glucose transport and phosphorylation in single cardiac myocytes: Rate limiting steps in glucose metabolism. Am. J. Physiol. 1994, 266, E326–E333. [Google Scholar] [CrossRef]
- Olson, A.L.; Pessin, J.E. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu. Rev. Nutr. 1996, 16, 235–256. [Google Scholar] [CrossRef]
- Newsholme, E.A.; Crabtree, B. Theoretical principles in the approaches to the control of metabolic pathways and their application to glycolysis in muscle. J. Molec. Cell Cardiol. 1979, 11, 839–856. [Google Scholar] [CrossRef]
- Entman, M.L.; Kanike, K.; Goldstein, M.A.; Nelson, T.E.; Bornet, E.P.; Futch, T.W.; Schwartz, A. Association of glycogenolysis with cardiac sarcoplasmic reticulum. J. Biol. Chem. 1976, 251, 3140–3146. [Google Scholar]
- Chin, E.; Allen, D. Effects of reduced muscle glycogen concentrationon force, Ca2+ release and contractile protein function in intact mouse skeletal muscle. J. Physiol. 1997, 498, 17–29. [Google Scholar] [CrossRef]
- Johnson, M.; Everitt, B. The high concentration of glycogen in fetal cardiac muscle probably explainswhy the heart can maintain its contractile activity in the face of severe hypoxia. In Essential Reproduction, 3rd ed.; Blackwell Scientific Publishing: Oxford, UK, 1988; p. 275. [Google Scholar]
- Hue, L.; Rider, M.H. Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues. Biochem. J. 1987, 245, 313–324. [Google Scholar] [CrossRef]
- Kobayashi, K.; Neely, J. Control of maximum rates of glycolysis in rat cardiac muscle. Circ. Res. 1979, 44, 166–175. [Google Scholar] [CrossRef]
- Uyeda, K. Phosphofructokinase. Adv. Enzym. 1979, 48, 193–244. [Google Scholar]
- Rider, M.H.; Bertrand, L.; Vertommen, D.; Michels, P.A.; Rousseau, G.G.; Hue, L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Head-to-head with a bifunctional enzyme that controls glycolysis. Biochem. J. 2004, 381, 561–579. [Google Scholar] [CrossRef]
- Rovetto, M.; Lamberton, W.; Neely, J. Mechanisms of glycolytic inhibitionin ischemic rat heart. Circ. Res. 1975, 37, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Nuutila, P.; Koivisto, V.A.; Knuuti, J.; Ruotsalainen, U.; Teras, M.; Haaparanta, M.; Bergman, J.; Solin, O.; Voipio-Pulkki, L.M.; Wegelius, U. Glucose-free fatty acid cycle operates in human heart and skeletal muscle in vivo. J. Clin. Investig. 1992, 89, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, G.W.; Cohen, D.M.; Taegtmeyer, H. [5-3H] glucose overestimates glycolytic flux in isolated working rat heart: Role of the pentose phosphate pathway. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E502–E508. [Google Scholar] [CrossRef]
- Eggleston, L.V.; Krebs, H.A. Regulation of the pentose phosphate cycle. Biochem. J. 1974, 138, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, H.G. Regulation of and intervention into the oxidative pentose phosphate pathway and adenine nucleotide metabolism in the heart. Mol. Cell Biochem. 1996, 160, 101–109. [Google Scholar] [CrossRef]
- Dassanayaka, S.; Jones, S.P. O-GlcNAc and the cardiovascular system. Pharm. Ther. 2014, 142, 62–71. [Google Scholar] [CrossRef]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathwaymediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [CrossRef]
- Hu, Y.; Riesland, L.; Paterson, A.J.; Kudlow, J.E. Phosphorylation of mouse glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2) by cAMP-dependent protein kinase increases the enzyme activity. J. Biol. Chem. 2004, 279, 29988–29993. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Peterson, M.B.; Ragavan, V.V.; Ferguson, A.G.; Lesch, M. De novo alanine synthesis in isolated oxygen-deprived rabbit myocardium. J. Biol. Chem. 1977, 252, 5010–5018. [Google Scholar] [CrossRef]
- Peuhkurinen, K.J.; Hassinen, I.E. Pyruvate carboxylation as an anaplerotic mechanism in the isolated perfused rat heart. Biochem. J. 1982, 202, 67–76. [Google Scholar] [CrossRef]
- Russell, R.R.; Taegtmeyer, H. Pyruvate carboxylation prevents the decline in contractile function of rat hearts oxidizing acetoacetate. Am. J. Physiol. 1991, 261, H1756–H1762. [Google Scholar] [CrossRef] [PubMed]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef]
- Randle, P. Regulation of glycolysis and pyruvate oxidation in cardiac muscle. Circ. Res. 1976, 38, I8–I15. [Google Scholar]
- Randle, P.J.; Tubbs, P.K. Carbohydrate and fatty acid metabolism. In Handbook of Physiology: The Cardiovascular System; Berne, R., Sperelakis, V., Geiger, S., Eds.; American Physiological Society: Bethesda, MD, USA, 1979; pp. 805–844. [Google Scholar]
- Olson, M.S.; Dennis, S.C.; DeBuysere, M.S.; Padma, A. The regulation of pyruvate dehydrogenase in the isolated perfused rat heart. J. Biol. Chem. 1978, 253, 7369–7375. [Google Scholar] [CrossRef]
- Kerbey, A.L.; Randle, P.J.; Cooper, R.H.; Whitehouse, S.; Pask, H.T.; Denton, R.M. Regulation of pyruvate dehydrogenase in rat heart. Biochem. J. 1976, 154, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Bing, R.J. The metabolism of the heart. Trans. Am. Coll. Cardiol. 1955, 5, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J.; Wojtzak, A. Factors controlling the role of fatty acid beta oxidation in rat liver mitochondria. Biochem. Biophys. Acta 1972, 280, 515–530. [Google Scholar] [CrossRef]
- Binas, B.; Danneberg, H.; McWhir, J.; Mullins, L.; Clark, A.J. Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. 1999, 13, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E. Fatty acid transport: The roads taken. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E239–E246. [Google Scholar] [CrossRef]
- McGarry, J.D.; Mills, S.E.; Long, C.S.; Foster, D.W. Observations on the affinity for carnitine and malonyl-CoA sensitivity of carnitine palmitoyl transferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem. J. 1983, 214, 21–28. [Google Scholar] [CrossRef]
- Dyck, J.R.; Cheng, J.F.; Stanley, W.C.; Barr, R.; Chandler, M.P.; Brown, S.; Wallace, D.; Arrhenius, T.; Harmon, C.; Yang, G.; et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 2004, 94, e78–e84. [Google Scholar] [CrossRef]
- Essop, M.F.; Camp, H.S.; Choi, C.S.; Sharma, S.; Fryer, R.M.; Reinhart, G.A.; Guthrie, P.H.; Bentebibel, A.; Gu, Z.; Shulman, G.I.; et al. Reduced heart size and increased myocardial fuel substrate oxidation in ACC2 mutant mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H256–H265. [Google Scholar] [CrossRef]
- Young, M.E.; Goodwin, G.W.; Ying, J.; Guthrie, P.; Wilson, C.R.; Laws, F.A.; Taegtmeyer, H. Regulation of cardiac and skeletal muscle malonyl-CoA decarboxylase by fatty acids. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E471–E479. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Masoud, W.G.; Ussher, J.R.; Wang, W.; Jaswal, J.S.; Wagg, C.S.; Dyck, J.R.; Lygate, C.A.; Neubauer, S.; Clanachan, A.S.; Lopaschuk, G.D. Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc. Res. 2014, 101, 30–38. [Google Scholar] [CrossRef]
- Brandt, J.M.; Djouadi, F.; Kelly, D.P. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 1998, 273, 23786–23792. [Google Scholar] [CrossRef]
- van Bilsen, M.; van der Vusse, G.J.; Reneman, R.S. Transcriptional regulation of metabolic processes: Implications for cardiac metabolism. Pflug. Arch. 1998, 437, 2–14. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, K.A.; Vork, M.M.; de Vries, J.E.; Willemsen, P.H.; Glatz, J.F.; Reneman, R.S.; van der Vusse, G.J.; van Bilsen, M. Long-chain fatty acidinduced changes in gene expression in neonatal cardiac myocytes. J. Lipid Res. 2000, 41, 41–47. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; Harinstein, M.E.; Gheorghiade, M. More than bricks and mortar: Comments on protein and amino acid metabolism in the heart. Am. J. Cardiol. 2008, 101, 3E–7E. [Google Scholar] [CrossRef]
- Krebs, H.A. Some aspects of the regulation of fuel supply in omnivorous animals. Adv. Enzym. Regul. 1972, 10, 397–420. [Google Scholar] [CrossRef]
- Williamson, J.R.; Safer, B.; LaNoue, K.F.; Smith, C.M.; Walajtys, E. Mitochondrial-cytosolic interactions in cardiac tissue: Role of the malate-aspartate cycle in the removal of glycolytic NADH from the cytosol. Symp. Soc. Exp. Biol. 1973, 27, 241–281. [Google Scholar]
- Wentz, A.E.; d’Avignon, D.A.; Weber, M.L.; Cotter, D.G.; Doherty, J.M.; Kerns, R.; Nagarajan, R.; Reddy, N.; Sambandam, N.; Crawford, P.A. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J. Biol. Chem. 2010, 285, 24447–24456. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms Underlying Acute Protection from Cardiac Ischemia-Reperfusion Injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J. Reducing myocardial infarct size: Myth or reality. Heart Metab. 2016, 70, 2–3. [Google Scholar]
- Stanley, W.C. Changes in cardiac metabolism: A critical step from stable angina to ischaemic cardiomyopathy. Eur. Heart J. Suppl. 2001, 3, O2–O7. [Google Scholar] [CrossRef]
- Maengjo, K.; Rong, T. Targeting AMPK for cardiac protection: Opportunities and challenges. J. Mol. Cell Cardiol. 2011, 51, 548–553. [Google Scholar]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Docherty, J.C.; Rendell, J.C.; Clanachan, A.S.; Lopaschuk, G.D. High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J. Am. Coll. Cardiol. 2002, 20, 718–725. [Google Scholar] [CrossRef]
- Ferrari, R.; Ceconi, C.; Curello, S.; Cargnoni, A.; Condorelli, E.; Belloli, S.; Albertini, A.; Visioli, O. Metabolic changes during post-ischaemic reperfusion. J. Mol. Cell Cardiol. 1988, 20 (Suppl. 2), 119–133. [Google Scholar] [CrossRef]
- Brown, D.I.; Willis, M.S.; Berthiame, J.M. Influence of Ischemia-Reperfusion Injury on Cardiac Metabolism. In The Scientist’s Guide to Cardiac Metabolism; Academic Press: Cambridge, MA, USA, 2016; Chapter 11; pp. 155–167. [Google Scholar]
- Rezende, P.C.; Ribas, F.F.; Serrano, C.V.; Hueb, W. Clinical significance of chronic myocardial ischemia in coronary artery disease patients. J. Thorac. Dis. 2019, 11, 1005–1015. [Google Scholar] [CrossRef]
- Khand, A.; Fisher, M.; Jones, J.; Patel, B.; Perry, R.; Mitsudo, K. The collateral circulation of the heart in coronary total arterial occlusions in man: Systematic review of assessment and pathophysiology. Am. Heart J. 2013, 166, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Kloner, R.A. Stunned and Hibernating Myocardium: Where Are We Nearly 4 Decades Later? J. Am. Heart Assoc. 2020, 9, e015502. [Google Scholar] [CrossRef]
- Depre, C.; Vatner, S.F. Cardioprotection in stunned and hibernating myocardium. Heart Fail. Rev. 2007, 12, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Ross, J. Myocardial perfusion-contraction matching. Circulation 1991, 83, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Peppas, A.; Hong, S.K.; Yang, G.; Huang, Y.; Diaz, G.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F. Persistent Stunning Induces Myocardial Hibernation and Protection. Circ. Res. 2003, 92, 1233–1239. [Google Scholar] [CrossRef]
- Cross, H.R.; Opie, L.H.; Radda, G.K.; Clarke, K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ. Res. 1996, 78, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Pasqua, T.; Boukhzar, L.; Anouar, Y.; Angelone, T. Progress in the emerging role of selenoproteins in cardiovascular disease: Focus on endoplasmic reticulum-resident selenoproteins. Cell Mol. Life Sci. 2019, 76, 3969–3985. [Google Scholar] [CrossRef] [PubMed]
- Piper, H.M.; Garcia-Dorado, D.; Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 1998, 38, 291–300. [Google Scholar] [CrossRef]
- di Lisa, F.; Bernardi, P. Mitochondria and ischemia–reperfusion injury of the heart: Fixing a hole. J. Mol. Cell Cardiol. 2003, 35, 339–341. [Google Scholar]
- Keeley, E.C.; Boura, J.A.; Grines, C.L. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: A quantitative review of 23 randomised trials. Lancet 2003, 361, 13–20. [Google Scholar] [CrossRef]
- Bolli, R. Myocardial stunning in man. Circulation 1992, 86, 1671–1691. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Belke, D.D.; Gamble, J.; Itoi, T.; Schönekess, B.O. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim. Biophys. Acta 1994, 1213, 263–276. [Google Scholar] [CrossRef]
- di Lisa, F.; Kaludercic, N.; Carpi, A.; Menabò, R.; Giorgio, M. Mitochondria and vascular pathology. Pharm. Rep. 2009, 61, 123–130. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [PubMed]
- Kvietys, P.R.; Granger, D.N. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic. Biol. Med. 2012, 52, 556–592. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.; Fanton, Z.; Tome, M.; Behr, E.R. Inherited cardiomyopathies. BMJ 2019, 365, l1570. [Google Scholar] [CrossRef]
- Sacchetto, C.; Sequeira, V.; Bertero, E.; Dudek, J.; Maack, C.; Calore, M. Metabolic Alterations in Inherited Cardiomyopathies. J. Clin. Med. 2019, 8, 2195. [Google Scholar] [CrossRef]
- Guertl, B.; Noehammer, C.; Hoefler, G. Metabolic cardiomyopathies. Int. J. Exp. Pathol. 2000, 81, 349–372. [Google Scholar] [CrossRef]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Tang, N.L.S.; Ganapathy, V.; Wu, X. Mutations of OCTN2, an organic cation/carnitine transporter, lead to a deficient cellular carnitine uptake in primary carnitine deficiency. Hum. Mol. Genet. 1999, 8, 655–660. [Google Scholar] [CrossRef]
- Burwinkel, B.; Scott, J.W.; Buhrer, C. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2 subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am. J. Hum. Genet. 2005, 76, 1034–1049. [Google Scholar] [CrossRef] [PubMed]
- Tein, I.; Devivo, D.C.; Bierman, F. Impaired skin fibroblast uptake in primary systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy. Pediatr. Res. 1990, 28, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.A.; Deleeuw, S.; Coates, P.M. Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann. Neurol. 1991, 30, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.S. Dilated cardiomyopathy caused by plasma membrane carnitnine transport defect. J. Inherit. Metab. Dis. 1998, 21, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Waber, L.; Valle, D.; Neill, C.; Dimauro, S.; Shug, A. Carnitine deficiency presenting as familial cardiomyopathy: A treatable defect in carnitine transport. J. Pediatr. 1982, 101, 700–705. [Google Scholar] [CrossRef]
- Garavaglia, B.; Uziel, G.; Dworzak, F.; Carrara, F.; Didonato, S. Primary carnitine deficiency: Heterozygote and intrafamilial phenotypic variation. Neurology 1991, 41, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Hug, G.; Bove, K.E.; Soukup, S. Lethal neonatal multiorgan deficiency of carnitine palmitoyltransferase II. N. Engl. J. Med. 1991, 325, 1862–1864. [Google Scholar] [CrossRef]
- North, K.N.; Hoppel, C.L.; Degirolami, U. Lethal neonatal deficiency of carnitine palmitoyltransferase II associated with dysgenesis of the brain and kidneys. J. Pediatr. 1995, 127, 414–420. [Google Scholar] [CrossRef]
- Taroni, F.; Verderio, E.; Fiorucci, S. Molecular characterization of inherited carnitine palmitoyltransferase II deficiency. Proc. Natl. Acad. Sci. USA 1992, 89, 8429–8433. [Google Scholar] [CrossRef]
- Strauss, A.W.; Powell, C.K.; Hale, D.E. Molecular basis of human mitochondrial very-long-chain acyl-CoA dehydrogenase deficiency causing cardiomyopathy and sudden death in childhood. Proc. Natl. Acad. Sci. USA 1995, 92, 10496–10500. [Google Scholar] [CrossRef]
- De Lonlay-Debeney, P.; Fournet, J.C.; Bonnet, D. Fatty acid b-oxidation deficiency masquerading as fulminant myocarditis. Int. J. Cardiol. 1998, 65, 287–289. [Google Scholar] [CrossRef]
- Parini, R.; Menni, F.; Garavaglia, B. Acute, severe cardiomyopathy as main symptom of late-onset very longchain acyl-coenzyme A dehydrogenase deficiency. Eur. J. Pediatr. 1998, 157, 992–995. [Google Scholar] [CrossRef]
- Tyni, T.; Palotie, A.; Viikina, L. Long-chain 3- hydroxyl-coenzyme A dehydrogenase deficiency with the G1528C mutation: Clinical presentation of thirteen patients. J. Pediatr. 1997, 130, 67–76. [Google Scholar] [CrossRef]
- Bonnet, D.; de Martin, D.; Lonlay, P. Arrhythmias and conduction defects as presenting symptoms of fatty acids oxidaton disorders in children. Circulation 1999, 100, 2248–2253. [Google Scholar] [CrossRef]
- Duran, M.; Wanders, R.J.A.; Dejager, J.P. 3-hydroxydicarboxylic aciduria due to long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency associated with sudden neonatal death: Protective effect of medium-chain triglycerid treatment. Eur. J. Pediatr. 1991, 150, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Bartlett, K.; Land, J. Long-chain 3- hydroxyl-CoA dehydrogenase deficiency. Pediatr. Res. 1991, 29, 406–411. [Google Scholar] [CrossRef][Green Version]
- Dasm, A.M.; Fingerhut, R.; Wanders, R.J.A.; Ullrich, K. Secondary respiratory chain defect in a boy with long-chain 3-hydroxylacyl-CoA dehydrogenase deficiency: Possible diagnostic pitfalls. Eur. J. Pediatr. 2000, 159, 243–246. [Google Scholar]
- Wicks, E.C.; Elliott, P.M. Genetics and metabolic cardiomyopathies. Herz 2012, 37, 598–610. [Google Scholar] [CrossRef]
- Kollberg, G.; Tulinius, M.; Gilljam, T.; Ostman-Smith, I.; Forsander, G.; Jotorp, P.; Oldfors, A.; Holme, E. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease. N. Engl. J. Med. 2007, 357, 1507–1514. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- Akhtar, M.M.; Elliot, P.M. Anderson-Fabry disease in heart failure. Biophys. Rev. 2018, 10, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; O’Mahony, C.; Hughes, D. Clinical and genetic predictors of major cardiac events in patients with Anderson-Fabry disease. Heart 2015, 101, 961–966. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- George, R. Severe valvular and aortic arch calcification in a patient with Gaucher’s disease homozygous for the D409H mutation. Clin. Genet. 2001, 59, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Levran, O.; Pereira, L.V.; Desnick, R.J. Structural organization and complete nucleotide sequence of the gene encoding human acid sphingomyelinase (SMPD1). Genomics 1992, 12, 197–205. [Google Scholar] [CrossRef]
- Westwood, M. Endocardial fibroelastosis and NiemannPick disease. Br. Heart J. 1977, 39, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of mucopolysaccharidoses, an update. Int. J. Mol. Sci. 2020, 21, 2515. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. J. Am. Coll. Cardiol. 2010, 55, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Kohlschutter, A.; Sieg, K.; Schulte, F.J.; Hayek, H.W.; Goebel, H.H. Infantile cardiomyopathy and neuromyopathy with, -galactosidase deficiency. Eur. J. Pediatr. 1982, 139, 75–81. [Google Scholar] [CrossRef]
- Merritt, J.L., 2nd; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6, 473. [Google Scholar] [CrossRef]
- Nezu, J.; Tamai, I.; Oku, A.; Ohashi, R.; Yabuuchi, H.; Hashimoto, N.; Nikaido, H.; Sai, Y.; Koizumi, A.; Shoji, Y.; et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat. Genet. 1999, 21, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ye, J.; Ganapathy, V.; Longo, N. Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc. Natl. Acad. Sci. USA 1999, 96, 2356–2360. [Google Scholar] [CrossRef]
- Tein, I. Carnitine transport: Pathophysiology and metabolism of known molecular defects. J. Inherit. Metab. Dis. 2003, 26, 147–169. [Google Scholar] [CrossRef]
- Mathur, A.; Sims, H.F.; Gopalakrishnan, D.; Gibson, B.; Rinaldo, P.; Vockley, J.; Hug, G.; Strauss, A.W. Molecular Heterogeneity in Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency Causing Pediatric Cardiomyopathy and Sudden Death. Circulation 1999, 99, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Brooks, E.D.; Koeberl, D.D. Preclinical Development of New Therapy for Glycogen Storage Diseases. Curr. Gene Ther. 2015, 15, 338–347. [Google Scholar] [CrossRef]
- Regalado, J.J.; Rodriguez, M.M.; Ferrer, P.L. Infantile hypertrophic cardiomyopathy of glycogenosis type IX: Isolated cardiac phosphorylase kinase deficiency. Pediatr. Cardiol. 1999, 20, 304–307. [Google Scholar] [CrossRef] [PubMed]
- van der Ploeg, A.T.; Reuser, A.J.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- D’souza, R.S.; Levandowski, C.; Slavov, D.; Graw, S.L.; Allen, L.A.; Adler, E.; Mestroni, L.; Taylor, M.R.G. Danon disease: Clinical features, evaluation, and management. Circ. Heart Fail. 2014, 7, 843–849. [Google Scholar] [CrossRef]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lüllmann-Rauch, R.; Janssen, P.M.L.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Benson, D.W.; Perez-Atayde, A.R.; McKenna, W.J.; Sparks, E.A.; Kanter, R.J.; McGarry, K.; Seidman, J.G.; Seidman, C.E. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J. Clin. Investig. 2002, 109, 357–362. [Google Scholar] [CrossRef]
- Blair, E.; Redwood, C.; Ashrafian, H.; Oliveira, M.; Broxholme, J.; Kerr, B.; Salmon, A.; Ostman-Smith, I.; Watkins, H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet. 2001, 10, 1215–1220. [Google Scholar] [CrossRef]
- Luptak, I.; Shen, M.; He, H.; Hirshman, M.F.; Musi, N.; Goodyear, L.J.; Yan, J.; Wakimoto, H.; Morita, H.; Arad, M.; et al. Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J. Clin. Investig. 2007, 117, 1432–1439. [Google Scholar] [CrossRef]
- Wolf, C.M.; Arad, M.; Ahmad, F.; Sanbe, A.; Bernstein, S.A.; Toka, O.; Konno, T.; Morley, G.; Robbins, J.; Seidman, J.G.; et al. Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations. Circulation 2008, 117, 144–154. [Google Scholar] [CrossRef]
- Xu, Y.; Gray, A.; Hardie, D.G.; Uzun, A.; Shaw, S.; Padbury, J.; Phornphutkul, C.; Tseng, Y.T. A novel, de novo mutation in the PRKAG2 gene: Infantile-onset phenotype and the signaling pathway involved. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H283–H292. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Bertrand, L.; Beauloye, C.R.; Andreadou, I.; Ruiz-Meana, M.; Jespersen, N.R.; Kula-Alwar, D.; Prag, H.A.; Botker, E.H.; Dambrova, M.; et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J. Cell Mol. Med. 2020, 24, 5937–5954. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; et al. American Heart Association Statistics Committee; Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2016 Update: A Report from the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar]
- Lee, L.; Horowitz, J.; Frenneaux, M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur. Heart J. 2004, 25, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H. Metabolic Management of Acute Myocardial Infarction Comes to the Fore and Extends Beyond Control of Hyperglycemia. Circulation 2008, 117, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Murry, C.E. Hallmarks of cardiac regeneration. Nat. Rev. Cardiol. 2018, 15, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Ravingerová, T.; Kindernay, L.; Barteková, M.; Ferko, M.; Adameová, A.; Zohdi, V.; Bernátová, I.; Ferenczyová, K.; Lazou, A. The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection. Int. J. Mol. Sci. 2020, 21, 7889. [Google Scholar] [CrossRef] [PubMed]
- Mayr, M.; Yusuf, S.; Weir, G.; Chung, Y.L.; Mayr, U.; Yin, X.; Ladroue, C.; Madhu, B.; Roberts, N.; de Souza, A.; et al. Combined metabolomic and proteomic analysis of human atrial fibrillation. J. Am. Coll. Cardiol. 2008, 5, 585–594. [Google Scholar] [CrossRef]
- van der Velden, J.; Tocchetti, C.G.; Varricchi, G.; Bianco, A.; Sequeira, V.; Hilfiker-Kleiner, D.; Hamdani, N.; Leite-Moreira, A.D.; Mayr, M.; Falcão-Pires, I.; et al. Metabolic changes in hypertrophic cardiomyopathies: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [PubMed]
- Verdonschot, J.A.J.; Hazebroek, M.R.; Ware, J.S.; Prasad, S.K.; Heymans, S.R.B. Role of Targeted Therapy in Dilated Cardiomyopathy: The Challenging Road Toward a Personalized Approach. JAHA 2019, 8, e012514. [Google Scholar] [CrossRef]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive Cardiomyopathy- Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 819–837. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Rajamannan, N.M.; Evans, F.J.; Aikawa, E.; Grande-Allen, K.J.; Demer, L.L.; Heistad, D.D.; Simmons, C.A.; Masters, K.S.; Mathieu, P.; O’Brien, K.D.; et al. Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011, 18, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017, a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Groenewegen, A.; Rutten, F.H.; Mosterd, M.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Lorenzoni, G.; Azzolina, D.; Lanera, C.; Brianti, G.; Gregori, D.; Vanuzzo, D.; Baldi, I. Time trends in first hospitalization for heart failure in a community-based population. Int J. Cardiol 2017, 243, 385–388. [Google Scholar] [CrossRef]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiov. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the fetal gene program A suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic gene expression in fetal and failing human heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Nascimben, L.; Ingwall, J.S.; Lorell, B.H.; Pinz, I.; Schultz, V.; Tornheim, K.; Tian, R. Mechanisms for Increased Glycolysis in the Hypertrophied Rat Heart. Hypertension 2004, 44, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Jain, M.; Cui, L.; D’Agostino, J.; Aiello, F.; Luptak, I.; Ngoy, S.; Mortensen, R.M.; Tian, R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002, 106, 2125–2131. [Google Scholar] [CrossRef]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar]
- Domenighetti, A.A.; Danes, V.R.; Curl, C.L.; Favaloro, J.M.; Proietto, J.; Delbridge, L.M.D. Targeted GLUT-4 deficiency in the heart induces cardiomyocyte hypertrophy and impaired contractility linked with Ca (2+) and proton flux dysregulation. J. Mol. Cell Cardiol. 2010, 48, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Abel, E.D. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 2001, 103, 2961–2966. [Google Scholar] [CrossRef]
- Wambolt, R.B.; Henning, S.L.; English, D.R.; Dyachkova, Y.; Lopaschuk, G.D.; Allard, M.F. Glucose utilization and glycogen turnover are accelerated in hypertrophied rat hearts during severe low-flow ischemia. J. Mol. Cell Cardiol. 1999, 31, 493–502. [Google Scholar] [CrossRef]
- McCommis, K.S.; Douglas, D.L.; Krenz, M.; Baines, C.P. Cardiac-specific Hexokinase 2 Overexpression Attenuates Hypertrophy by Increasing Pentose Phosphate Pathway Flux. J. Am. Heart Assoc. 2013, 2, e000355. [Google Scholar] [CrossRef]
- Wu, R.; Wyatt, E.; Chawla, K.; Tran, M.; Ghanefar, M.; Laakso, M.; Epting, C.L.; Ardehali, H. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol. Med. 2012, 4, 633–646. [Google Scholar] [CrossRef]
- Liang, Q.; Donthi, R.V.; Kralik, P.M.; Epstein, P.N. Elevated hexokinase increases cardiac glycolysis in transgenic mice. Cardiovasc. Res. 2002, 53, 423–430. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Tian, R. Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef]
- Gupte, S.A.; Levine, R.J.; Gupte, R.S.; Young, M.E.; Lionetti, V.; Labinskyy, V.; Floyd, B.C.; Ojaimi, C.; Bellomo, M.; Wolin, M.S.; et al. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J. Mol. Cell Cardiol. 2006, 41, 340–349. [Google Scholar] [CrossRef]
- Watson, L.J.; Facundo, H.T.; Ngoh, G.A.; Ameen, M.; Brainard, R.E.; Lemma, K.M.; Long, B.W.; Prabhu, S.D.; Xuan, Y.T.; Jones, S.P. O-linked β-N-acetylglucosamine transferase is indispensable in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 17797–17802. [Google Scholar] [CrossRef]
- Barger, P.M.; Kelly, D.P. PPAR signaling in the control of cardiac energy metabolism. Trends. Cardiovasc. Med. 2000, 10, 238–245. [Google Scholar] [CrossRef]
- Memon, R.A.; Tecott, L.H.; Nonogaki, K.; Beigneux, A.; Moser, A.H.; Grunfeld, C.; Feingold, K.R. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: Troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 2000, 141, 4021–4031. [Google Scholar] [PubMed]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kundu, B.K.; Wu, H.C.; Hashmi, S.S.; Guthrie, P.; Locke, L.W.; Roy, R.J.; Matherne, G.P.; Berr, S.S.; Terwelp, M.; et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J. Am. Heart Assoc. 2013, 2, e004796. [Google Scholar] [CrossRef]
- Hamirani, Y.S.; Kundu, B.K.; Zhong, M.; McBride, A.; Li, Y.; Davogustto, G.E.; Taegtmeyer, H.; Bourque, J.M. Noninvasive Detection of Early Metabolic Left Ventricular Remodeling in Systemic Hypertension. Cardiology 2016, 133, 157–162. [Google Scholar] [CrossRef]
- Chu, T.F.; Rupnick, M.A.; Kerkela, R.; Dallabrida, S.M.; Zurakowski, D.; Nguyen, L.; Woulfe, K.; Pravda, E.; Cassiola, F.; Desai, J.; et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007, 370, 2011–2019. [Google Scholar] [CrossRef]
- Rees, M.L.; Subramaniam, J.; Li, Y.; Hamilton, D.J.; Frazier, O.H.; Taegtmeyer, H. A PKM2 signature in the failing heart. Biochem. Biophys. Res. Commun. 2015, 459, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Pascual, F.; Coleman, R.A. Fuel availability and fate in cardiac metabolism: A tale of two substrates. Biochim. Et Biophys. Acta 2016, 1861, 1425–1433. [Google Scholar] [CrossRef]
- Pereira, R.O.; Wende, A.R.; Olsen, C.; Soto, J.; Rawlings, T.; Zhu, Y.; Anderson, S.M.; Abel, E.D. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J. Am. Heart Assoc. 2013, 2, e000301. [Google Scholar] [CrossRef]
- Yan, J.; Young, M.E.; Cui, L.; Lopaschuk, G.D.; Liao, R.; Tian, R. Increased Glucose Uptake and Oxidation in Mouse Hearts Prevent High Fatty Acid Oxidation but Cause Cardiac Dysfunction in Diet-Induced Obesity. Circulation 2009, 119, 2818–2828. [Google Scholar] [CrossRef]
- Wende, A.R.; Schell, J.C.; Ha, C.M.; Pepin, M.E.; Khalimonchuk, O.; Schwertz, H.; Pereira, R.O.; Brahma, M.K.; Tuinei, J.; Contreras-Ferrat, A.; et al. Maintaining Myocardial Glucose Utilization in Diabetic Cardiomyopathy Accelerates Mitochondrial Dysfunction. Diabetes 2020, 69, 2094–2111. [Google Scholar] [CrossRef]
- Ardehali, H.; Sabbah, H.N.; Burke, M.A.; Sarma, S.; Liu, P.P.; Cleland, J.G.; Maggioni, A.; Fonarow, G.C.; Abel, E.D.; Campia, U.; et al. Targeting myocardial substrate metabolism in heart failure: Potential for new therapies. Eur. J. Heart Fail. 2012, 14, 120–129. [Google Scholar] [CrossRef]
- Pederson, B.A.; Chen, H.; Schroeder, J.M.; Shou, W.; DePaoli-Roach, A.A.; Roach, P.J. Abnormal Cardiac Development in the Absence of Heart Glycogen. Mol. Cell Biol. 2004, 24, 7179–7187. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Kassiotis, C.; Razeghi, P.; Taegtmeyer, H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail. Rev. 2007, 12, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Vanoverschelde, J.L.J.; Taegtmeyer, H. Glucose for the heart. Circulation 1999, 99, 578–588. [Google Scholar] [CrossRef]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation—A novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta 2011, 1813, 1333–1350. [Google Scholar] [CrossRef]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid β-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Huber, K.; Petzold, J.; Rehfeldt, C.; Ender, K.; Fiedler, I. Muscle energy metabolism: Structural and functional features in different types of porcine striated muscles. J. Muscle Res. Cell Motil. 2007, 28, 249–258. [Google Scholar] [CrossRef]
- Morash, A.J.; Kotwica, A.O.; Murray, A.J. Tissue-specific changes in fatty acid oxidation in hypoxic heart and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R534–R541. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef] [PubMed]
- Jeukendrup, A.E. Regulation of fat metabolism in skeletal muscle. Ann. N. Y. Acad. Sci. 2002, 967, 217–235. [Google Scholar] [CrossRef]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [PubMed]
- Kjøbsted, R.; Hingst, J.R.; Fentz, J.; Foretz, M.; Sanz, M.N.; Pehmøller, C.; Shum, M.; Marette, A.; Mounier, R.; Treebak, J.T.; et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018, 32, 1741–1777. [Google Scholar] [CrossRef] [PubMed]
- Petrick, H.L.; Brunetta, H.S.; Pignanelli, C.; Nunes, E.A.; van Loon, L.J.C.; Burr, J.F.; Holloway, G.P. In vitro ketone-supported mitochondrial respiration is minimal when other substrates are readily available in cardiac and skeletal muscle. J. Physiol. 2020, 598, 4869–4885. [Google Scholar] [CrossRef] [PubMed]
- Lunde, P.K.; Sjaastad, I.; Schiøtz-Thorud, H.M.; Sejersted, O.M. Skeletal muscle disorders in heart failure. Acta Physiol. Scand. 2001, 171, 277–294. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.; Lechêne, P.; Fortin, D.; N’Guessan, B.; Belmadani, S.; Bigard, X.; Veksler, V.; Ventura-Clapier, R. Cardiac and skeletal muscle energy metabolism in heart failure: Beneficial effects of voluntary activity. Cardiovasc. Res. 2002, 56, 260–268. [Google Scholar] [CrossRef]
- Takada, S.; Sabe, H.; Kinugawa, S. Abnormalities of Skeletal Muscle, Adipocyte Tissue, and Lipid Metabolism in Heart Failure: Practical Therapeutic Targets. Front. Cardiovasc. Med. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H. Heart Physiology: From Cell to Circulation, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004. [Google Scholar]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Tuunanen, H.; Engblom, E.; Naum, A.; Någren, K.; Hesse, B.; Airaksinen, K.E.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Opie, L.H.; et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 2006, 114, 2130–2137. [Google Scholar] [CrossRef]
- Halbirk, M.; Nørrelund, H.; Møller, N.; Schmitz, O.; Gøtzsche, L.; Nielsen, R.; Nielsen-Kudsk, J.E.; Nielsen, S.S.; Nielsen, T.T.; Eiskjær, H.; et al. Suppression of circulating free fatty acids with acipimox in chronic heart failure patients changes whole body metabolism but does not affect cardiac function. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1220–H1225. [Google Scholar] [CrossRef] [PubMed]
- Zarain-Herzberg, A.; Rupp, H.; Elimban, V.; Dhalla, N.S. Modification of sarcoplasmic reticulum gene expression in pressure overload cardiac hypertrophy by etomoxir. FASEB J. 1996, 10, 1303–1309. [Google Scholar] [CrossRef]
- Turcani, M.; Rupp, H. Etomoxir improves left ventricular performance of pressure-overloaded rat heart. Circulation 1997, 96, 3681–3686. [Google Scholar] [CrossRef]
- Schmidt-Schweda, S.; Holubarsch, C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin. Sci. 2000, 99, 27–35. [Google Scholar] [CrossRef]
- Lee, L.; Campbell, R.; Scheuermann-Freestone, M.; Taylor, R.; Gunaruwan, P.; Williams, L.; Ashrafian, H.; Horowitz, J.; Fraser, A.G.; Clarke, K.; et al. Metabolic modulation with perhexiline in chronic heart failure: A randomized, controlled trial of short-term use of a novel treatment. Circulation 2005, 112, 3280–3288. [Google Scholar] [CrossRef]
- Abozguia, K.; Elliott, P.; McKenna, W.; Phan, T.T.; Nallur-Shivu, G.; Ahmed, I.; Maher, A.R.; Kaur, K.; Taylor, J.; Henning, A.; et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 2010, 122, 1562–1569. [Google Scholar] [CrossRef]
- Lionetti, V.; Linke, A.; Chandler, M.P.; Young, M.E.; Penn, M.S.; Gupte, S.; D’Agostino, C.; Hintze, T.H.; Stanley, W.C.; Recchia, F.A. Carnitine palmitoyl transferase-I inhibition prevents ventricular remodeling and delays decompensation in pacing-induced heart failure. Cardiovasc. Res. 2005, 66, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Churchill, E.N.; Murriel, C.L.; Chen, C.H.; Mochly-Rosen, D.; Szweda, L.I. Reperfusion-induced translocation of deltaPKC to cardiac mitochondria prevents pyruvate dehydrogenase reactivation. Circ. Res. 2005, 97, 78–85. [Google Scholar] [CrossRef]
- Piao, L.; Fang, Y.H.; Cadete, V.J.; Wietholt, C.; Urboniene, D.; Toth, P.T.; Marsboom, G.; Zhang, H.J.; Haber, I.; Rehman, J.; et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J. Mol. Med. 2010, 88, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Atherton, H.J.; Dodd, M.S.; Heather, L.C.; Schroeder, M.A.; Griffin, J.L.; Radda, G.K.; Clarke, K.; Tyler, D.J. Role of pyruvate dehydrogenase inhibition in the development of hypertrophy in the hyperthyroid rat heart: A combined magnetic resonance imaging and hyperpolarized magnetic resonance spectroscopy study. Circulation 2011, 123, 2552–2561. [Google Scholar] [CrossRef]
- Wargovich, T.J.; MacDonald, R.G.; Hill, J.A.; Feldman, R.L.; Stacpoole, P.W.; Pepine, C.J. Myocardial metabolic and hemodynamic effects of dichloroacetate in coronary artery disease. Am. J. Cardiol. 1988, 61, 65–70. [Google Scholar] [CrossRef]
- Spruijt, L.; Naviaux, R.K.; McGowan, K.A.; Nyhan, W.L.; Sheean, G.; Haas, R.H.; Barshop, B.A. Nerve conduction changes in patients with mitochondrial diseases treated with dichloroacetate. Muscle Nerve 2001, 24, 916–924. [Google Scholar] [CrossRef]
- Oishi, K.; Yoshioka, M.; Ozawa, R.; Yamamoto, T.; Oya, Y.; Ogawa, M.; Kawai, M. Dichloroacetate treatment for adult patients with mitochondrial disease. Rinsho Shinkeigaku 2003, 43, 154–161. [Google Scholar] [PubMed]
- Ingelsson, E.; Sundström, J.; Arnlöv, J.; Zethelius, B.; Lind, L. Insulin resistance and risk of congestive heart failure. JAMA 2005, 294, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Sidell, R.J.; Cole, M.A.; Draper, N.J.; Desrois, M.; Buckingham, R.E.; Clarke, K. Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the zucker Fatty rat heart. Diabetes 2002, 51, 1110–1117. [Google Scholar] [CrossRef]
- Zhu, P.; Lu, L.; Xu, Y.; Schwartz, G.G. Troglitazone improves recovery of left ventricular function after regional ischemia in pigs. Circulation 2000, 101, 1165–1171. [Google Scholar] [CrossRef]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- Di Franco, A.; Cantini, G.; Tani, A.; Coppini, R.; Zecchi-Orlandini, S.; Raimondi, L.; Luconi, M.; Mannucci, E. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 2017, 243, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; Wüst, R.C.; Fiolet, J.W.; Stienen, G.J.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. DAPA-HF Trial Committees and Investigators. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. EMPA-REG OUTCOME Investigators. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. CANVAS Program Collaborative Group. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. DECLARE–TIMI 58 Investigators. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. CREDENCE Trial Investigators. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Eurich, D.T.; Weir, D.L.; Majumdar, S.R.; Tsuyuki, R.T.; Johnson, J.A.; Tjosvold, L.; Vanderloo, S.E.; McAlister, F.A. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: Systematic review of observational studies involving 34,000 patients. Circ. Heart Fail. 2013, 6, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Doney, A.S.; AlZadjali, M.A.; Ogston, S.A.; Petrie, J.R.; Morris, A.D.; Struthers, A.D.; Wong, A.K.; Lang, C.C. Effect of Metformin on mortality in patients with heart failure and type 2 diabetes mellitus. Am. J. Cardiol. 2010, 106, 1006–1010. [Google Scholar] [CrossRef]
- Aguilar, D.; Chan, W.; Bozkurt, B.; Ramasubbu, K.; Deswal, A. Metformin use and mortality in ambulatory patients with diabetes and heart failure. Circ. Heart Fail. 2011, 4, 53–58. [Google Scholar] [CrossRef]
- Romero, S.P.; Andrey, J.L.; Garcia-Egido, A.; Escobar, M.A.; Perez, V.; Corzo, R.; Garcia-Domiguez, G.J.; Gomez, F. Metformin therapy and prognosis of patients with heart failure and new-onset diabetes mellitus. A propensity-matched study in the community. Int. J. Cardiol. 2013, 166, 404–412. [Google Scholar] [CrossRef]
- Wong, A.K.; Symon, R.; AlZadjali, M.A.; Ang, D.S.; Ogston, S.; Choy, A.; Petrie, J.R.; Struthers, A.D.; Lang, C.C. The effect of metformin on insulin resistance and exercise parameters in patients with heart failure. Eur. J. Heart Fail. 2012, 14, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Cadeddu, C.; Nocco, S.; Cugusi, L.; Deidda, M.; Bina, A.; Orru, F.; Bandinu, S.; Cossu, E.; Baroni, M.G.; Mercuro, G. Effects of metformin and exercise training, alone or in association, on cardio-pulmonary performance and quality of life in insulin resistance patients. Cardiovasc. Diabetol. 2014, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Ginion, A.; Beauloye, C.; Hebert, A.D.; Guigas, B.; Hue, L.; Vanoverschelde, J.L. AMPK activation restores the stimulation of glucose uptake in an in vitro model of insulin-resistant cardiomyocytes via the activation of protein kinase B. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H239–H250. [Google Scholar] [CrossRef]
- Wang, X.F.; Zhang, J.Y.; Li, L.; Zhao, X.Y.; Tao, H.L.; Zhang, L. Metformin improves cardiac function in rats via activation of AMP-activated protein kinase. Clin. Exp. Pharm. Physiol. 2011, 38, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Cittadini, A.; Napoli, R.; Monti, M.G.; Rea, D.; Longobardi, S.; Netti, P.A.; Walser, M.; Samà, M.; Aimaretti, G.; Isgaard, J.; et al. Metformin prevents the development of chronic heart failure in the SHHF rat model. Diabetes 2012, 61, 944–953. [Google Scholar] [CrossRef]
- Nikolaidis, L.A.; Mankad, S.; Sokos, G.G.; Miske, G.; Shah, A.; Elahi, D.; Shannon, R.P. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004, 109, 962–965. [Google Scholar] [CrossRef]
- Garber, A.J. Long-acting glucagon-like peptide 1 receptor agonists: A review of their efficacy and tolerability. Diabetes Care 2011, 34 (Suppl. 2), S279–S284. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. SUSTAIN-6 Investigators. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. REWIND Investigators. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Opie, L.L.; Lopaschuk, G.D. Fuels, aerobic and anaerobic metabolism. In Heart Physiology: From Cell to Circulation; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004; p. 306. [Google Scholar]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef]
- Clanachan, A.S. Contribution of protons to post-ischemic Na+ and Ca2+ overload and left ventricular mechanical dysfunction. J. Cardiovasc. Electrophysiol. 2006, 17, S141–S148. [Google Scholar] [CrossRef] [PubMed]
- Dalgas, C.; Povlsen, J.A.; Løfgren, B.; Erichsen, S.B.; Bøtker, H.E. Effects of FA on cardioprotection by pre-ischaemic inhibition of the malate–aspartate shuttle. Clin. Exp. Pharm. Physiol. 2012, 39, 878–885. [Google Scholar] [CrossRef]
- Abozguia, K.; Clarke, K.; Lee, L.; Frenneaux, M. Modification of myocardial substrate use as a therapy for heart failure. Nat. Clin. Pr. Cardiovasc. Med. 2006, 3, 490–498. [Google Scholar] [CrossRef]
- Kudo, N.; Barr, A.J.; Barr, R.L.; Desai, S.; Lopaschuk, G.D. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 1995, 270, 17513–17520. [Google Scholar] [CrossRef] [PubMed]
- Heather, L.C.; Clarke, K. Metabolism, hypoxia and the diabetic heart. J. Mol. Cell Cardiol. 2011, 50, 598–605. [Google Scholar] [CrossRef]
- Terrand, J.; Papageorgiou, I.; Rosenblatt-Velin, N.; Lerch, R. Calcium mediated activation of pyruvate dehydrogenase in severely injured postischemic myocardium. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H722–H730. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Barr, R.L.; Wolff, A.A.; Lopaschuk, G.D. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 1996, 93, 135–142. [Google Scholar] [CrossRef]
- Mansor, L.S.; Sousa-Fialho, M.D.L.; Yea, G.; Coumans, W.A.; West, J.A.; Kerr, M.; Carr, C.A.; Luiken, J.J.F.P.; Glatz, J.F.C.; Evans, R.D.; et al. Affiliations expand Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc. Res. 2017, 113, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef]
- Fang, Y.H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. 2012, 90, 31–43. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Barr, R.; Thomas, P.D.; Dyck, J.R. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ. Res. 2003, 93, e33–e37. [Google Scholar] [CrossRef] [PubMed]
- Marzilli, M.; Vinereanu, D.; Lopaschuk, G.; Chen, Y.; Dalal, J.J.; Danchin, N.; Etriby, E.; Ferrari, R.; Gowdak, L.H.; Lopatin, Y.; et al. Trimetazidine in cardiovascular medicine. Int. J. Cardiol. 2019, 293, 39–44. [Google Scholar] [CrossRef]
- Lopatin, Y.M.; Rosano, G.M.; Fragasso, G.; Lopaschuk, G.D.; Seferovic, P.M.; Gowdak, L.H.; Vinereanu, D.; Hamid, M.A.; Jourdain, P.; Ponikowski, P. Rationale and benefits of trimetazidine by acting on cardiac metabolism in heart failure. Int. J. Cardiol. 2016, 203, 909–915. [Google Scholar] [CrossRef]
- McVeigh, J.J.; Lopaschuk, G.D. Dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. Am. J. Physiol. Heart Circ. Physiol. 1990, 259, H1079–H1085. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, E.D.; White, L.T. Pyruvate dehydrogenase influences postischemic heart function. Circulation 1995, 91, 2071–2079. [Google Scholar] [CrossRef]
- Liu, B.; El-Alaoui-Talibi, Z.; Clanachan, A.S.; Schulz, R.; Lopaschuk, G.D. Uncoupling of contractile function from mitochondrial TCA cycle activity and MVO2 during reperfusion of ischemic hearts. Am. J. Physiol. Heart Circ. Physiol. 1996, 39, H72–H80. [Google Scholar] [CrossRef]
- Kingsley, P.B.; Sako, E.Y.; Yang, M.Q.; Zimmer, S.D.; Ugurbil, K.; Foker, J.E.; From, A.H. Ischemic contracture begins when anaerobic glycolysis stops: A 31P-NMR study of isolated rat hearts. Am. J. Physiol. 1991, 261, H469–H478. [Google Scholar] [CrossRef]
- Vanoverschelde, J.J.; Janier, M.F.; Bakke, J.E.; Marshall, D.R.; Bergmann, S.R. Rate of glycolysis during ischemia determines extent of ischemic injury and functional recovery after reperfusion. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H1785–H1794. [Google Scholar] [CrossRef] [PubMed]
- Beltran, C.; Pardo, R.; Bou-Teen, D.; Ruiz-Meana, M.; Villena, J.A.; Ferreira-González, I.; Barba, I. Enhancing Glycolysis Protects against Ischemia-Reperfusion Injury by Reducing ROS Production. Metabolites 2020, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.S.; Miller, E.J.; Wright, T.M.; Li, J.; Qi, D.; Atsina, K.; Zaha, V.; Sakamoto, K.; Young, L.H. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J. Mol. Cell Cardiol. 2011, 51, 24–32. [Google Scholar] [CrossRef]
- Timmermans, A.D.; Balteau, M.; Gelinas, R.; Renguet, E.; Ginion, A.; de Meester, C.; Sakamoto, K.; Balligand, J.L.; Bontemps, F.; Vanoverschelde, J.L.; et al. A-769662 potentiates the effect of other AMPK-activated protein kinase activators on cardiac glucose uptake. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1619–H1630. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.M.; Yao, H.; McBurney, M.W.; Gu, W.; Guarente, L.; Rahman, I.; Brookes, P.S. SIRT1-mediated acute cardioprotection. Am. J. Physiol. Circ. Physiol. 2015, 301, H1506–H1512. [Google Scholar] [CrossRef] [PubMed]
- Barron, J.T.; Gu, L.; Parrillo, J.E. Malate-aspartate shuttle, cytoplasmic NADH redox potential, and energetics in vascular smooth muscle. J. Mol. Cell Cardiol. 1998, 30, 1571–1579. [Google Scholar] [CrossRef]
- Bunger, R.; Glanert, S.; Sommer, O.; Gerlach, E. Inhibition by (aminooxy) acetate of the malate-aspartate cycle in the isolated working guinea pig heart. Hoppe Seylers Z Physiol. Chem. 1980, 361, 907–914. [Google Scholar] [CrossRef]
- Støttrup, N.B.; Løfgren, B.; Birkler, R.D.; Nielsen, J.M.; Wang, L.; Caldarone, C.A.; Kristiansen, S.B.; Contractor, H.; Johannsen, M.; Bøtker, H.E.; et al. Inhibition of the malate–aspartate shuttle by pre-ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc. Res. 2010, 88, 257–266. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Priestman, D.A.; Mistry, S.C.; Halsall, A. Glucose fatty acid interactions and the regulation of glucose disposal. J. Cell Biochem. 1994, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, J.; Hu, H.; Lu, S.; Lu, Q.; Quan, N.; Rousselle, T.; Patel, M.S.; Li, J. Dichloroacetate Ameliorates Cardiac Dysfunction Caused by Ischemic Insults Through AMPK Signal Pathway-Not Only Shifts Metabolism. Toxicol. Sci. 2019, 167, 604–617. [Google Scholar] [CrossRef]
- Ussher, J.R.; Wang, W.; Gandhi, M.; Keung, W.; Samokhvalov, V.; Oka, T.; Wagg, C.S.; Jaswal, J.S.; Harris, R.A.; Clanachan, A.S.; et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc. Res. 2012, 94, 359–369. [Google Scholar] [CrossRef]
- Heywood, S.E.; Richart, A.L.; Henstridge, D.C.; Alt, K.; Kiriazis, H.; Zammit, C.; Carey, A.L.; Kammoun, H.L.; Delbridge, L.M.; Reddy, M.; et al. High-density lipoprotein delivered after myocardial infarction increases cardiac glucose uptake and function in mice. Sci. Transl. Med. 2017, 9, eaam6084. [Google Scholar] [CrossRef]
- Pham, V.; Zhang, W.; Chen, V.; Whitney, T.; Yao, J.; Froese, D.; Friesen, A.D.; Diakur, J.M.; Haque, W. Design and synthesis of novel pyridoxine 5′-phosphonates as potential antiischemic agents. J. Med. Chem. 2003, 46, 3680–3687. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Aravindhan, K.; Alsaid, H.; Chendrimada, T.; Szapacs, M.; Citerone, D.R.; Harpel, M.R.; Willette, R.N.; Lepore, J.L.; Jucker, B.M. Albiglutide, a long lasting glucagon-like peptide-1 analog, protects the rat heart against ischemia/reperfusion injury: Evidence for improving cardiac metabolic efficiency. PLoS ONE 2011, 6, e23570. [Google Scholar] [CrossRef]
- Yue, T.L.; Bao, W.; Gu, J.L.; Cui, J.; Tao, L.; Ma, X.L.; Ohlstein, E.H.; Jucker, B.M. Rosiglitazone treatment in Zucker diabetic fatty rats Is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes 2005, 54, 554–562. [Google Scholar] [CrossRef]
- de Micheli, A.; Medrano, G.A.; Villarreal, A.; Sodi-Pallares, D. Protective effect of glucose-insulin-potassium solutions in myocardial damage caused by emetine. Arch. Inst. Cardiol. Mex. 1975, 45, 469–486. [Google Scholar] [PubMed]
- Oates, A.; Nubani, R.; Smiley, J.; Kistler, L.; Hughey, S.; Theiss, P.; Perez-Tamayo, R.A.; Eiferman, D.; Lonchyna, V.; Higgins, R.S. Myocardial protection of insulin and potassium in a porcine ischemia-reperfusion model. Surgery 2009, 146, 23–30. [Google Scholar] [CrossRef]
- Eiferman, D.; Perez-Tamayo, R.A.; Abe, K.; Okum, E.; Higgins, R. Real-time monitoring of cardiac metabolism using biosensors shows myocardial protection during ischemia-reperfusion injury with glucose-insulin-potassium administration. Surgery 2007, 142, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhang, H.F.; Yu, L.; Zhang, Q.J.; Li, J.; Huo, J.H.; Li, X.; Guo, W.Y.; Wang, H.C.; Gao, F. Vasculoprotective effect of insulin in the ischemic/reperfused canine heart: Role of Akt-stimulated NO production. Cardiovasc. Res. 2006, 69, 57–65. [Google Scholar] [CrossRef]
- Selker, H.P.; Beshansky, J.R.; Griffith, J.L.; D’Agostino, R.B.; Massaro, J.M.; Udelson, J.E.; Rashba, E.J.; Ruthazer, R.; Sheehan, P.R.; Desvigne-Nickens, P.; et al. Study design for the Immediate Myocardial Metabolic Enhancement During Initial Assessment and Treatment in Emergency Care (IMMEDIATE) Trial: A double-blind randomized controlled trial of intravenous glucose, insulin, and potassium for acute coronary syndromes in emergency medical services. Am. Heart J. 2012, 163, 315–322. [Google Scholar]
- Selker, H.P.; Udelson, J.E.; Massaro, J.M.; Ruthazer, R.; D’Agostino, R.B.; Griffith, J.L.; Sheehan, P.R.; Desvigne-Nickens, P.; Rosenberg, Y.; Tian, X.; et al. One-year outcomes of out-of-hospital administration of intravenous glucose, insulin, and potassium (GIK) in patients with suspected acute coronary syndromes (from the IMMEDIATE [Immediate Myocardial Metabolic Enhancement During Initial Assessment and Treatment in Emergency Care] Trial). Am. J. Cardiol. 2014, 113, 1599–1605. [Google Scholar] [PubMed]
- Licker, M.; Reynaud, T.; Garofano, N.; Sologashvili, T.; Diaper, J.; Ellenberger, C. Pretreatment with glucose-insulin-potassium improves ventricular performances after coronary artery bypass surgery: A randomized controlled trial. J. Clin. Monit. Comput. 2020, 34, 29–40. [Google Scholar] [CrossRef]
- Wu, R.; Smeele, K.M.; Wyatt, E.; Ichikawa, Y.; Eerbeek, O.; Sun, L.; Chawla, K.; Hollmann, M.W.; Nagpal, V.; Heikkinen, S.; et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodelling after ischemia-reperfusion injury. Circ. Res. 2011, 108, 60–69. [Google Scholar] [CrossRef]
- Fernandez-Sanz, C.; Ruiz-Meana, M.; Castellano, J.; Miro-Casas, E.; Nuñez, E.; Inserte, J.; Vázquez, J.; Garcia-Dorado, D. Altered FoF1 ATPase synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb. Haemost. 2015, 113, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Pasqua, T.; Cerra, M.C.; Angelone, T. Cardiac Damage in Anthracyclines Therapy: Focus on Oxidative Stress and Inflammation. Antioxid. Redox Signal. 2020, 20, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Abdurrachim, D.; Luiken, J.J.; Nicolay, K.; Glatz, J.F.C.; Prompers, J.J.; Nabben, M. Good and bad consequences of altered fatty acid metabolism in heart failure: Evidence from mouse models. Cardiovasc. Res. 2015, 106, 194–205. [Google Scholar] [CrossRef]
- Fillmore, N.; Lopaschuk, G.D. Malonyl CoA: A promising target for the treatment of cardiac disease. IUBMB Life 2014, 66, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Liepinsh, E.; Makrecka-Kuka, M.; Kuka, J.; Vilskersts, R.; Makarova, E.; Cirule, H.; Loza, E.; Lola, D.; Grinberga, S.; Pugovics, O.; et al. Inhibition of L-carnitine biosynthesis and transport by methyl-gamma-butyrobetaine decreases fatty acid oxidation and protects against myocardial infarction. Br. J. Pharm. 2015, 172, 1319–1332. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genetic Metabolic Cardiomyopathies |
Cardiac Manifestations |
Cardiometabolic Changes | References |
|---|---|---|---|
| Fatty acid oxidation disorders | |||
| Carnitine deficiency | Dilated cardiomyopathy, cardiac arrest | Defective carnitine biosynthesis ↓ Fatty acid oxidation Lipid accumulation | [86,87,88,89,90,91,92,93,94,95] |
| Carnitine palmitoyltransferase II deficiency | Pansystolic murmur, septal hypertrophy, cardiac arrhythmias | Impaired mitochondrial acyl–CoA transport and fatty acid oxidation Cardiac lipidosis | [86,87,88,96,97,98] |
| Very-long-chain acyl-CoA dehydrogenase deficiency | Cardiac hypertrophy | ↓ Fatty acid oxidation Cardiac lipidosis | [86,87,99,100,101] |
| Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency | Cardiac hypertrophy, cardiac arrhythmias | ↓ Fatty acid oxidation Cardiac lipidosis | [86,87,102,103,104,105,106] |
| Mitochondrial trifunctional protein deficiency | Cardiac arrhythmias, conduction disorder, cardiorespiratory arrest | ↓ Fatty acid oxidation Cardiac lipidosis | [86,87,107] |
| Glycogen storage diseases | |||
| Glycogen storage diseases types II, III, IV and VI | Restrictive or dilated cardiomyopathies, conduction disorder | Glycogen accumulation Impaired autophagy and expression of mitochondrial genes | [86,107,108] |
| Lysosomal storage disorders | |||
| Anderson–Fabry disease | Ventricular hypertrophy, valvular abnormalities, conduction disorder, cardiac arrhythmias | Globotriaosylceramide accumulation ↓ Activity of respiratory chain enzymes ↑ Oxidative stress ↑ Release of proinflammatory cytokines | [86,87,107,109,110,111] |
| Gaucher disease | Heart valve diseases | Glucosylceramide accumulation ↓ Activity of respiratory chain enzymes ↑ Oxidative stress ↑ Release of proinflammatory cytokines | [87,107,112,113] |
| Niemann–Pick disease | Endocardial fibroelastosis | Sphingomyelin accumulation ↓ Activity of respiratory chain enzymes ↑ Oxidative stress ↑ Release of proinflammatory cytokines | [87,107,114,115] |
| GM1 gangliosidosis | Heart failure | GM1-ganglioside accumulation ↓ Activity of respiratory chain enzymes ↑ Oxidative stress ↑ Release of proinflammatory cytokines | [87,107,109,110,112,114,116,117,118] |
| Mitochondrial disorders | |||
| Friedreich ataxia | Cardiac hypertrophy, heart failure | ↑ Oxidative stress Impaired mitochondrial respiratory function and iron metabolism | [86,107] |
| Barth syndrome | Dilated cardiomyopathy | ↓ Electron transport chain activity ↓ Oxygen consumption ↑ Oxidative stress | [86,107] |
| Acute and Chronic Cardiac Diseases | Cardiometabolic Alterations | References |
|---|---|---|
| Ischemic heart disease | ↓ Mitochondrial oxidative metabolism ↑ Utilization of glucose ↑ Rates of free fatty acid oxidation Accumulation of lactate and protons Reduction in intracellular pH | [138,139] |
| Heart failure | ↑ Uptake of glucose and free fatty acid ↓ Uptake and oxidation of glucose and free fatty acid in mitochondria Cytosolic accumulation of metabolic intermediates Lipotoxicity and glucotoxicity ↑ Reliance on ketone bodies | [140] |
| Arrhythmias | Abnormalities in Ca2+, K+, Na+ homeostasis Oxidative stress | [141] |
| Atrial fibrillation | ↑ β-hydroxybutyrate generation, ketogenic amino acids (tyrosine and glycine) and 3-oxoacid-CoA transferase Mitochondrial dysfunction Oxidative stress | [142] |
| Hypertrophic cardiomyopathy | ↓ Free fatty acid oxidation ↑ Ketone bodies and glucose oxidation | [143,144] |
| Dilated cardiomyopathy | ↓ Oxidative metabolism in cardiomyocytes ↑ Anaerobic glycolysis ↑ Acylcarnitine and ketone bodies | [145] |
| Restrictive cardiomyopathy | Glycogen accumulation Amyloid deposits Iron overload Glycosphingolipids accumulation | [146] |
| Diabetic cardiomyopathy | ↑ Free fatty acid release and myocyte sarcolemmal free fatty acid transporters. Lipotoxicity ↑ Triacylglycerols Impaired mitochondrial Ca2+ handling ↑ Oxidative stress | [147] |
| Valvular heart disease | ↑ Lipid deposition and oxidized low-density lipoprotein formation ↑ Inflammation-associated factors ↑ Superoxide and hydrogen peroxide levels | [148] |
| Cardiac Diseases | Pharmacological Intervention | Mechanism of Action | References |
|---|---|---|---|
| Heart Failure | Acipimox | Lipolysis inhibitor ↑ Glucose utilization | [191,192] |
| Etomoxir, Perhexiline, and Oxfenicine | CPT-I inhibitors ↓ Fatty acid oxidation ↑ Glucose utilization | [2,193,194,195,196,197,198] | |
| Dichloroacetate | PDK inhibitor ↑ Glucose oxidation | [199,200,201,202] | |
| Thiazolidinediones | PPARγ agonists ↑ Glucose utilization | [206,207] | |
| SGLT2 inhibitors | ↓ Na+/H+ exchanger activity ↑ Antioxidant ability of mitochondria | [208,209,210,211,212] | |
| Metformin | AMPK activator ↓ Oxidative stress and inflammation Improved endothelial function ↑ Glucose uptake | [218,219,220,221,222,223,224,225,226,227] | |
| GLP-1RAs | GLP-1 Receptor Analogues ↑ Glucose utilization | [229,230,231,232] | |
| Myocardial infarction | AMPK activators | ↑ Glycolytic pathway | [237,238,239] |
| Aminooxyacetate and similar | Malate-aspartate shuttle inhibitors ↓ ROS production and oxidative stress | [240,241,242,243] | |
| Trimetazidine | 3-KAT inhibitor ↓ Fatty acid oxidation ↑ Glucose utilization | [244,245,246,247] | |
| Dichloroacetate | PDK inhibitor ↑ Glucose oxidation | [248,249,250,251,252,253,254] | |
| GLP-1RAs | GLP-1 Receptor Analogues ↑ Glucose utilization | [255,256] | |
| GIK infusion | ↑ Glucose metabolism ↓ Fatty acid utilization | [257,258,259,260] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasqua, T.; Rocca, C.; Giglio, A.; Angelone, T. Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications. J. Clin. Med. 2021, 10, 721. https://doi.org/10.3390/jcm10040721
Pasqua T, Rocca C, Giglio A, Angelone T. Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications. Journal of Clinical Medicine. 2021; 10(4):721. https://doi.org/10.3390/jcm10040721
Chicago/Turabian StylePasqua, Teresa, Carmine Rocca, Anita Giglio, and Tommaso Angelone. 2021. "Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications" Journal of Clinical Medicine 10, no. 4: 721. https://doi.org/10.3390/jcm10040721
APA StylePasqua, T., Rocca, C., Giglio, A., & Angelone, T. (2021). Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications. Journal of Clinical Medicine, 10(4), 721. https://doi.org/10.3390/jcm10040721