
Recent Advances on the Role and Therapeutic Potential of Regulatory T Cells in Atherosclerosis


Abstract
:1. Introduction
2. Role of the Immune System in Atherosclerotic Disease
3. Protective Role of Tregs in Atherosclerotic Disease
3.1. Protective Role of Tregs in Experimental Atherosclerosis
3.2. Possible Protective Role of Tregs in Human Atherosclerosis
3.3. Protective Role of Tregs in AAA
3.4. Mechanisms of Treg-Mediated Atheroprotection
3.5. Treg Immune Responses under Hypercholesterolemia
3.6. Stability, Plasticity, and Antigen-Specificity of Tregs in Atherosclerosis
4. Therapeutic Approaches for Atherosclerotic Disease by Promoting Regulatory Immune Responses
4.1. Vaccination Strategies
4.2. Modulation of DC Functions
4.3. Modulation of Intestinal Immunity
4.4. Treatment with Antibodies and Cytokines
4.5. Ultraviolet B (UVB)-Based Phototherapy
4.6. Approaches to Regress Atherosclerosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-macrophages and T cells in atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Lutgens, E.; Atzler, D.; Doring, Y.; Duchene, J.; Steffens, S.; Weber, C. Immunotherapy for cardiovascular disease. Eur. Heart J. 2019, 40, 3937–3946. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T cells and human disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Yamashita, T.; Kasahara, K.; Takeda, M.; Hirata, K. Regulatory T cells and tolerogenic dendritic cells as critical immune modulators in atherogenesis. Curr. Pharm. Des. 2015, 21, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Lavillegrand, J.R.; Tedgui, A. Regulatory T cell-enhancing therapies to treat atherosclerosis. Cells 2021, 10, 723. [Google Scholar]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef]
- Ishibashi, S.; Brown, M.S.; Goldstein, J.L.; Gerard, R.D.; Hammer, R.E.; Herz, J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Investig. 1993, 92, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Ait-Oufella, H.; Sage, A.P.; Mallat, Z.; Tedgui, A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ. Res. 2014, 114, 1640–1660. [Google Scholar] [CrossRef]
- Paulsson, G.; Zhou, X.; Tornquist, E.; Hansson, G.K. Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.; Hansson, G.K. Vaccination strategies and immune modulation of atherosclerosis. Circ. Res. 2020, 126, 1281–1296. [Google Scholar]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.J.; Cochain, C.; Vafadarnejad, E.; Saliba, A.E.; et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ. Res. 2018, 122, 1675–1688. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Pablo, A.M.; Jiang, X.; Wang, N.; Tall, A.R.; Schindler, C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Investig. 1997, 99, 2752–2761. [Google Scholar] [CrossRef]
- Whitman, S.C.; Ravisankar, P.; Elam, H.; Daugherty, A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. Am. J. Pathol. 2000, 157, 1819–1824. [Google Scholar] [CrossRef]
- Buono, C.; Binder, C.J.; Stavrakis, G.; Witztum, J.L.; Glimcher, L.H.; Lichtman, A.H. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc. Natl. Acad. Sci. USA 2005, 102, 1596–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frostegard, J.; Ulfgren, A.K.; Nyberg, P.; Hedin, U.; Swedenborg, J.; Andersson, U.; Hansson, G.K. Cytokine expression in advanced human atherosclerotic plaques: Dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 1999, 145, 33–43. [Google Scholar] [CrossRef]
- King, V.L.; Szilvassy, S.J.; Daugherty, A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor−/− mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 456–461. [Google Scholar] [CrossRef] [Green Version]
- King, V.L.; Cassis, L.A.; Daugherty, A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am. J. Pathol. 2007, 171, 2040–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littman, D.R.; Rudensky, A.Y. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010, 140, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Erbel, C.; Chen, L.; Bea, F.; Wangler, S.; Celik, S.; Lasitschka, F.; Wang, Y.; Bockler, D.; Katus, H.A.; Dengler, T.J. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J. Immunol. 2009, 183, 8167–8175. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.; Prasad, K.M.; Butcher, M.; Dobrian, A.; Kolls, J.K.; Ley, K.; Galkina, E. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation 2010, 121, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Danzaki, K.; Matsui, Y.; Ikesue, M.; Ohta, D.; Ito, K.; Kanayama, M.; Kurotaki, D.; Morimoto, J.; Iwakura, Y.; Yagita, H.; et al. Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Kyaw, T.; Winship, A.; Tay, C.; Kanellakis, P.; Hosseini, H.; Cao, A.; Li, P.; Tipping, P.; Bobik, A.; Toh, B.H. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in ApoE-deficient mice. Circulation 2013, 127, 1028–1039. [Google Scholar] [CrossRef] [Green Version]
- van Duijn, J.; Kritikou, E.; Benne, N.; van der Heijden, T.; van Puijvelde, G.H.; Kroner, M.J.; Schaftenaar, F.H.; Foks, A.C.; Wezel, A.; Smeets, H.; et al. CD8+ T-cells contribute to lesion stabilization in advanced atherosclerosis by limiting macrophage content and CD4+ T-cell responses. Cardiovasc. Res. 2019, 115, 729–738. [Google Scholar] [CrossRef]
- Ley, K.; Gerdes, N.; Winkels, H. ATVB distinguished scientist award: How costimulatory and coinhibitory pathways shape atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 764–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, K.; Yamaguchi, T.; Sakaguchi, S. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. 2011, 32, 428–433. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sasaki, N.; Yamashita, T.; Emoto, T.; Kasahara, K.; Mizoguchi, T.; Hayashi, T.; Yodoi, K.; Kitano, N.; Saito, T.; et al. Overexpression of cytotoxic T-lymphocyte-associated antigen-4 prevents atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Amin, H.Z.; Sasaki, N.; Yamashita, T.; Mizoguchi, T.; Hayashi, T.; Emoto, T.; Matsumoto, T.; Yoshida, N.; Tabata, T.; Horibe, S.; et al. CTLA-4 protects against angiotensin II-induced abdominal aortic aneurysm formation in mice. Sci. Rep. 2019, 9, 8065. [Google Scholar] [CrossRef] [PubMed]
- Amin, H.Z.; Sasaki, N.; Hirata, K.I.; Rikitake, Y. Cytotoxic T lymphocyte-associated antigen-4 protects against angiotensin II-induced kidney injury in mice. Circ. Rep. 2020, 2, 339–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsley, P.S.; Nadler, S.G. The clinical utility of inhibiting CD28-mediated costimulation. Immunol. Rev. 2009, 229, 307–321. [Google Scholar] [CrossRef]
- Ma, K.; Lv, S.; Liu, B.; Liu, Z.; Luo, Y.; Kong, W.; Xu, Q.; Feng, J.; Wang, X. CTLA4-IgG ameliorates homocysteine-accelerated atherosclerosis by inhibiting T-cell overactivation in apoE−/− mice. Cardiovasc. Res. 2013, 97, 349–359. [Google Scholar] [CrossRef]
- Gotsman, I.; Grabie, N.; Dacosta, R.; Sukhova, G.; Sharpe, A.; Lichtman, A.H. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J. Clin. Investig. 2007, 117, 2974–2982. [Google Scholar] [CrossRef]
- Bu, D.X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoInt blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association between immune checkpoInt. inhibitors with cardiovascular events and atherosclerotic plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; Paeper, B.; Clark, L.B.; Yasayko, S.A.; Wilkinson, J.E.; Galas, D.; Ziegler, S.F.; Ramsdell, F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001, 27, 68–73. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Shevach, E.M.; Thornton, A.M. tTregs, pTregs, and iTregs: Similarities and differences. Immunol. Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.K.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Planer, D.; Luboshits, G.; Afek, A.; Metzger, S.; Chajek-Shaul, T.; Keren, G.; George, J. Role of naturally occurring CD4+ CD25+ regulatory T cells in experimental atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 893–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkels, H.; Meiler, S.; Lievens, D.; Engel, D.; Spitz, C.; Burger, C.; Beckers, L.; Dandl, A.; Reim, S.; Ahmadsei, M.; et al. CD27 co-stimulation increases the abundance of regulatory T cells and reduces atherosclerosis in hyperlipidaemic mice. Eur. Heart J. 2017, 38, 3590–3599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahl, K.; Loddenkemper, C.; Drouin, C.; Freyer, J.; Arnason, J.; Eberl, G.; Hamann, A.; Wagner, H.; Huehn, J.; Sparwasser, T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 2007, 204, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, R.; Gerdes, N.; Badeau, R.M.; Gistera, A.; Strodthoff, D.; Ketelhuth, D.F.; Lundberg, A.M.; Rudling, M.; Nilsson, S.K.; Olivecrona, G.; et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Investig. 2013, 123, 1323–1334. [Google Scholar] [CrossRef]
- De Boer, O.J.; van der Meer, J.J.; Teeling, P.; van der Loos, C.M.; van der Wal, A.C. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS ONE 2007, 2, e779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Ord, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef]
- Mor, A.; Luboshits, G.; Planer, D.; Keren, G.; George, J. Altered status of CD4(+)CD25(+) regulatory T cells in patients with acute coronary syndromes. Eur. Heart J. 2006, 27, 2530–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammirati, E.; Cianflone, D.; Banfi, M.; Vecchio, V.; Palini, A.; De Metrio, M.; Marenzi, G.; Panciroli, C.; Tumminello, G.; Anzuini, A.; et al. Circulating CD4+CD25hiCD127lo regulatory T-Cell levels do not reflect the extent or severity of carotid and coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1832–1841. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Schwartzenberg, S.; Medvedovsky, D.; Jonas, M.; Charach, G.; Afek, A.; Shamiss, A. Regulatory T cells and IL-10 levels are reduced in patients with vulnerable coronary plaques. Atherosclerosis 2012, 222, 519–523. [Google Scholar] [CrossRef]
- Zhang, W.C.; Wang, J.; Shu, Y.W.; Tang, T.T.; Zhu, Z.F.; Xia, N.; Nie, S.F.; Liu, J.; Zhou, S.F.; Li, J.J.; et al. Impaired thymic export and increased apoptosis account for regulatory T cell defects in patients with non-ST segment elevation acute coronary syndrome. J. Biol. Chem. 2012, 287, 34157–34166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potekhina, A.V.; Pylaeva, E.; Provatorov, S.; Ruleva, N.; Masenko, V.; Noeva, E.; Krasnikova, T.; Arefieva, T. Treg/Th17 balance in stable CAD patients with different stages of coronary atherosclerosis. Atherosclerosis 2015, 238, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Wigren, M.; Bjorkbacka, H.; Andersson, L.; Ljungcrantz, I.; Fredrikson, G.N.; Persson, M.; Bryngelsson, C.; Hedblad, B.; Nilsson, J. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2000–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, R.; Brokopp, C.E.; Grives, A.; Courtier, A.; Jaguszewski, M.; Pasqual, N.; Vlaskou Badra, E.; Lewandowski, A.; Gaemperli, O.; Hoerstrup, S.P.; et al. Clonal restriction and predominance of regulatory T cells in coronary thrombi of patients with acute coronary syndromes. Eur. Heart J. 2015, 36, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [Green Version]
- Emoto, T.; Sasaki, N.; Yamashita, T.; Kasahara, K.; Yodoi, K.; Sasaki, Y.; Matsumoto, T.; Mizoguchi, T.; Hirata, K. Regulatory/effector T-cell ratio is reduced in coronary artery disease. Circ. J. 2014, 78, 2935–2941. [Google Scholar] [CrossRef] [Green Version]
- Joly, A.L.; Seitz, C.; Liu, S.; Kuznetsov, N.V.; Gertow, K.; Westerberg, L.S.; Paulsson-Berne, G.; Hansson, G.K.; Andersson, J. Alternative splicing of FOXP3 controls regulatory T cell effector functions and is associated with human atherosclerotic plaque stability. Circ. Res. 2018, 122, 1385–1394. [Google Scholar] [CrossRef]
- Dale, M.A.; Ruhlman, M.K.; Baxter, B.T. Inflammatory cell phenotypes in AAAs: Their role and potential as targets for therapy. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1746–1755. [Google Scholar] [CrossRef] [Green Version]
- Ait-Oufella, H.; Wang, Y.; Herbin, O.; Bourcier, S.; Potteaux, S.; Joffre, J.; Loyer, X.; Ponnuswamy, P.; Esposito, B.; Dalloz, M.; et al. Natural regulatory T cells limit angiotensin II-induced aneurysm formation and rupture in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2374–2379. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Yang, J.; Zhang, K.; An, G.; Kong, J.; Jiang, F.; Zhang, Y.; Zhang, C. Regulatory T cells prevent angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E knockout mice. Hypertension 2014, 64, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Yodoi, K.; Yamashita, T.; Sasaki, N.; Kasahara, K.; Emoto, T.; Matsumoto, T.; Kita, T.; Sasaki, Y.; Mizoguchi, T.; Sparwasser, T.; et al. Foxp3+ regulatory T cells play a protective role in angiotensin II-induced aortic aneurysm formation in mice. Hypertension 2015, 65, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Zhang, J.; Wang, Y.; Wang, S.; Bockler, D.; Duan, Z.; Xin, S. Deficient CD4+CD25+ T regulatory cell function in patients with abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1825–1831. [Google Scholar] [CrossRef] [Green Version]
- Ait-Oufella, H.; Taleb, S.; Mallat, Z.; Tedgui, A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 969–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinderski, L.J.; Fischbein, M.P.; Subbanagounder, G.; Fishbein, M.C.; Kubo, N.; Cheroutre, H.; Curtiss, L.K.; Berliner, J.A.; Boisvert, W.A. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circ. Res. 2002, 90, 1064–1071. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.K.; Rudling, M.; Zhou, X.; Gorelik, L.; Flavell, R.A.; Hansson, G.K. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J. Clin. Investig. 2003, 112, 1342–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proto, J.D.; Doran, A.C.; Gusarova, G.; Yurdagul, A., Jr.; Sozen, E.; Subramanian, M.; Islam, M.N.; Rymond, C.C.; Du, J.; Hook, J.; et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity 2018, 49, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maganto-Garcia, E.; Tarrio, M.L.; Grabie, N.; Bu, D.X.; Lichtman, A.H. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation 2011, 124, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailer, R.K.W.; Gistera, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia enhances T cell receptor signaling and increases the regulatory T cell population. Sci. Rep. 2017, 7, 15655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailer, R.K.W.; Gistera, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia induces differentiation of regulatory T cells in the liver. Circ. Res. 2017, 120, 1740–1753. [Google Scholar] [CrossRef]
- Amersfoort, J.; Schaftenaar, F.H.; Douna, H.; van Santbrink, P.J.; van Puijvelde, G.H.M.; Slutter, B.; Foks, A.C.; Harms, A.; Moreno-Gordaliza, E.; Wang, Y.; et al. Diet-induced dyslipidemia induces metabolic and migratory adaptations in regulatory T cells. Cardiovasc. Res. 2021, 117, 1309–1324. [Google Scholar] [CrossRef] [PubMed]
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as therapeutic targets in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 583–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonacina, F.; Martini, E.; Svecla, M.; Nour, J.; Cremonesi, M.; Beretta, G.; Moregola, A.; Pellegatta, F.; Zampoleri, V.; Catapano, A.L.; et al. Adoptive transfer of CX3CR1 transduced-T regulatory cells improves homing to the atherosclerotic plaques and dampens atherosclerosis progression. Cardiovasc. Res. 2021, 117, 2069–2082. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Vignali, D.A.; Rudensky, A.Y.; Niec, R.E.; Waldmann, H. The plasticity and stability of regulatory T cells. Nat. Rev. Immunol. 2013, 13, 461–467. [Google Scholar] [CrossRef]
- Ali, A.J.; Makings, J.; Ley, K. Regulatory T cell stability and plasticity in atherosclerosis. Cells 2020, 9, 2665. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Filipowicz, A.R.; Waseem, T.C.; McGary, C.M.; Crow, K.J.; Magilnick, N.; Boldin, M.; Lundberg, P.S.; Galkina, E.V. Atherosclerosis-driven treg plasticity results in formation of a dysfunctional subset of plastic IFNgamma+ Th1/tregs. Circ. Res. 2016, 119, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; McArdle, S.; Gholami, A.; Kimura, T.; Wolf, D.; Gerhardt, T.; Miller, J.; Weber, C.; Ley, K. CCR5+T-bet+FoxP3+ effector CD4 T cells drive atherosclerosis. Circ. Res. 2016, 118, 1540–1552. [Google Scholar] [CrossRef] [Green Version]
- Wigren, M.; Rattik, S.; Yao Mattisson, I.; Tomas, L.; Gronberg, C.; Soderberg, I.; Alm, R.; Sundius, L.; Ljungcrantz, I.; Bjorkbacka, H.; et al. Lack of ability to present antigens on major histocompatibility complex class II molecules aggravates atherosclerosis in ApoE−/− mice. Circulation 2019, 139, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kobiyama, K.; Winkels, H.; Tse, K.; Miller, J.; Vassallo, M.; Wolf, D.; Ryden, C.; Orecchioni, M.; Dileepan, T.; et al. Regulatory CD4(+) T cells recognize MHC-II-restricted peptide epitopes of apolipoprotein B. Circulation 2018, 138, 1130–1143. [Google Scholar] [CrossRef]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Michel, N.A.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B100-reactive CD4(+) T-regulatory cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef]
- Subramanian, M.; Thorp, E.; Hansson, G.K.; Tabas, I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J. Clin. Investig. 2013, 123, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Cheong, C.; Dandamudi, D.B.; Park, C.G.; Rodriguez, A.; Mehandru, S.; Velinzon, K.; Jung, I.H.; Yoo, J.Y.; Oh, G.T.; et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 2011, 35, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, M.; Yamashita, T.; Sasaki, N.; Hirata, K. Dendritic cells in atherogenesis: Possible novel targets for prevention of atherosclerosis. J. Atheroscler. Thromb. 2012, 19, 953–961. [Google Scholar] [CrossRef]
- Hermansson, A.; Johansson, D.K.; Ketelhuth, D.F.; Andersson, J.; Zhou, X.; Hansson, G.K. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation 2011, 123, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Yamashita, T.; Sasaki, N.; Nakajima, K.; Kita, T.; Shinohara, M.; Ishida, T.; Hirata, K. Oral administration of an active form of vitamin D3 (calcitriol) decreases atherosclerosis in mice by inducing regulatory T cells and immature dendritic cells with tolerogenic functions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2495–2503. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008, 117, 503–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, H.L.; da Cunha, A.P.; Quintana, F.; Wu, H. Oral tolerance. Immunol. Rev. 2011, 241, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Gojova, A.; Brun, V.; Esposito, B.; Fournier, N.; Cottrez, F.; Tedgui, A.; Groux, H. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation 2003, 108, 1232–1237. [Google Scholar]
- van Puijvelde, G.H.; Hauer, A.D.; de Vos, P.; van den Heuvel, R.; van Herwijnen, M.J.; van der Zee, R.; van Eden, W.; van Berkel, T.J.; Kuiper, J. Induction of oral tolerance to oxidized low-density lipoprotein ameliorates atherosclerosis. Circulation 2006, 114, 1968–1976. [Google Scholar] [CrossRef] [Green Version]
- van Puijvelde, G.H.; van Es, T.; van Wanrooij, E.J.; Habets, K.L.; de Vos, P.; van der Zee, R.; van Eden, W.; van Berkel, T.J.; Kuiper, J. Induction of oral tolerance to HSP60 or an HSP60-peptide activates T cell regulation and reduces atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2677–2683. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Kasahara, K.; Emoto, T.; Matsumoto, T.; Mizoguchi, T.; Kitano, N.; Sasaki, N.; Hirata, K. Intestinal immunity and gut microbiota as therapeutic targets for preventing atherosclerotic cardiovascular diseases. Circ. J. 2015, 79, 1882–1890. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N.; Emoto, T.; Yamashita, T.; Watanabe, H.; Hayashi, T.; Tabata, T.; Hoshi, N.; Hatano, N.; Ozawa, G.; Sasaki, N.; et al. Bacteroides vulgatus and Bacteroides dorei reduce gut microbial lipopolysaccharide production and inhibit atherosclerosis. Circulation 2018, 138, 2486–2498. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Marko, L.; Hoges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G.; et al. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation 2019, 139, 1407–1421. [Google Scholar] [CrossRef]
- Belghith, M.; Bluestone, J.A.; Barriot, S.; Megret, J.; Bach, J.F.; Chatenoud, L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat. Med. 2003, 9, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A.; et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Steffens, S.; Burger, F.; Pelli, G.; Dean, Y.; Elson, G.; Kosco-Vilbois, M.; Chatenoud, L.; Mach, F. Short-term treatment with anti-CD3 antibody reduces the development and progression of atherosclerosis in mice. Circulation 2006, 114, 1977–1984. [Google Scholar] [CrossRef] [Green Version]
- Ochi, H.; Abraham, M.; Ishikawa, H.; Frenkel, D.; Yang, K.; Basso, A.S.; Wu, H.; Chen, M.L.; Gandhi, R.; Miller, A.; et al. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+ CD25− LAP+ T cells. Nat. Med. 2006, 12, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ochi, H.; Chen, M.L.; Frenkel, D.; Maron, R.; Weiner, H.L. Inhibition of autoimmune diabetes by oral administration of anti-CD3 monoclonal antibody. Diabetes 2007, 56, 2103–2109. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Yamashita, T.; Takeda, M.; Shinohara, M.; Nakajima, K.; Tawa, H.; Usui, T.; Hirata, K. Oral anti-CD3 antibody treatment induces regulatory T cells and inhibits the development of atherosclerosis in mice. Circulation 2009, 120, 1996–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foks, A.C.; Frodermann, V.; ter Borg, M.; Habets, K.L.; Bot, I.; Zhao, Y.; van Eck, M.; van Berkel, T.J.; Kuiper, J.; van Puijvelde, G.H. Differential effects of regulatory T cells on the initiation and regression of atherosclerosis. Atherosclerosis 2011, 218, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.N.; Kyaw, T.S.; Kanellakis, P.; To, K.; Tipping, P.; Toh, B.H.; Bobik, A.; Agrotis, A. Cytokine therapy with interleukin-2/anti-interleukin-2 monoclonal antibody complexes expands CD4+CD25+Foxp3+ regulatory T cells and attenuates development and progression of atherosclerosis. Circulation 2012, 126, 1256–1266. [Google Scholar] [CrossRef] [Green Version]
- Korn, T.; Reddy, J.; Gao, W.; Bettelli, E.; Awasthi, A.; Petersen, T.R.; Backstrom, B.T.; Sobel, R.A.; Wucherpfennig, K.W.; Strom, T.B.; et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 2007, 13, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Sasaki, N.; Yamashita, T.; Kita, T.; Yodoi, K.; Sasaki, Y.; Takeda, M.; Hirata, K. CD3 antibody and IL-2 complex combination therapy inhibits atherosclerosis by augmenting a regulatory immune response. J. Am. Heart Assoc. 2014, 3, e000719. [Google Scholar] [CrossRef] [Green Version]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P., 3rd; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef] [Green Version]
- Saadoun, D.; Rosenzwajg, M.; Joly, F.; Six, A.; Carrat, F.; Thibault, V.; Sene, D.; Cacoub, P.; Klatzmann, D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N. Engl. J. Med. 2011, 365, 2067–2077. [Google Scholar] [CrossRef]
- Zhao, T.X.; Kostapanos, M.; Griffiths, C.; Arbon, E.L.; Hubsch, A.; Kaloyirou, F.; Helmy, J.; Hoole, S.P.; Rudd, J.H.F.; Wood, G.; et al. Low-dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndromes (LILACS): Protocol and study rationale for a randomised, double-blind, placebo-controlled, phase I/II clinical trial. BMJ. Open 2018, 8, e022452. [Google Scholar] [CrossRef]
- Zhao, T.X.; Newland, S.A.; Mallat, Z. 2019 ATVB Plenary Lecture: Interleukin-2 Therapy in Cardiovascular Disease: The Potential to Regulate Innate and Adaptive Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 853–864. [Google Scholar] [CrossRef]
- Schwarz, T. Mechanisms of UV-induced immunosuppression. Keio J. Med. 2005, 54, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Morita, A. Current developments in phototherapy for psoriasis. J. Dermatol. 2018, 45, 287–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, N.; Yamashita, T.; Kasahara, K.; Fukunaga, A.; Yamaguchi, T.; Emoto, T.; Yodoi, K.; Matsumoto, T.; Nakajima, K.; Kita, T.; et al. UVB exposure prevents atherosclerosis by regulating immunoinflammatory responses. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, A.; Khaskhely, N.M.; Ma, Y.; Sreevidya, C.S.; Taguchi, K.; Nishigori, C.; Ullrich, S.E. Langerhans cells serve as immunoregulatory cells by activating NKT cells. J. Immunol. 2010, 185, 4633–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, A.; Noordegraaf, M.; Maeda, A.; Torii, K.; Clausen, B.E.; Schwarz, T. Langerhans cells are required for UVR-induced immunosuppression. J. Investig. Dermatol. 2010, 130, 1419–1427. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Sasaki, N.; Yamashita, T.; Mizoguchi, T.; Emoto, T.; Amin, H.Z.; Yodoi, K.; Matsumoto, T.; Kasahara, K.; Yoshida, N.; et al. Ultraviolet B exposure inhibits angiotensin II-induced abdominal aortic aneurysm formation in mice by expanding CD4+Foxp3+ regulatory T cells. J. Am. Heart Assoc. 2017, 6, 007024. [Google Scholar] [CrossRef]
- Furuhashi, T.; Saito, C.; Torii, K.; Nishida, E.; Yamazaki, S.; Morita, A. Photo(chemo)therapy reduces circulating Th17 cells and restores circulating regulatory T cells in psoriasis. PLoS ONE 2013, 8, e54895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyama, S.; Murase, K.; Sato, T.; Hashimoto, A.; Tatekoshi, A.; Horiguchi, H.; Kamihara, Y.; Ono, K.; Kikuchi, S.; Takada, K.; et al. Narrowband ultraviolet B phototherapy ameliorates acute graft-versus-host disease by a mechanism involving in vivo expansion of CD4+CD25+Foxp3+ regulatory T cells. Int. J. Hematol. 2014, 99, 471–476. [Google Scholar] [CrossRef]
- Nissen, S.E.; Nicholls, S.J.; Sipahi, I.; Libby, P.; Raichlen, J.S.; Ballantyne, C.M.; Davignon, J.; Erbel, R.; Fruchart, J.C.; Tardif, J.C.; et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: The ASTEROID trial. JAMA 2006, 295, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Trogan, E.; Feig, J.E.; Dogan, S.; Rothblat, G.H.; Angeli, V.; Tacke, F.; Randolph, G.J.; Fisher, E.A. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef] [Green Version]
- Feig, J.E.; Parathath, S.; Rong, J.X.; Mick, S.L.; Vengrenyuk, Y.; Grauer, L.; Young, S.G.; Fisher, E.A. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 2011, 123, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.; Yamashita, T.; Kita, T.; Takeda, M.; Sasaki, N.; Kasahara, K.; Shinohara, M.; Rikitake, Y.; Ishida, T.; Yokoyama, M.; et al. Orally administered eicosapentaenoic acid induces rapid regression of atherosclerosis via modulating the phenotype of dendritic cells in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1963–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoInt. analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Kita, T.; Yamashita, T.; Sasaki, N.; Kasahara, K.; Sasaki, Y.; Yodoi, K.; Takeda, M.; Nakajima, K.; Hirata, K. Regression of atherosclerosis with anti-CD3 antibody via augmenting a regulatory T-cell response in mice. Cardiovasc. Res. 2014, 102, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Schlegel, M.P.; Afonso, M.S.; Brown, E.J.; Rahman, K.; Weinstock, A.; Sansbury, B.E.; Corr, E.M.; van Solingen, C.; Koelwyn, G.J.; et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ. Res. 2020, 127, 335–353. [Google Scholar] [CrossRef]
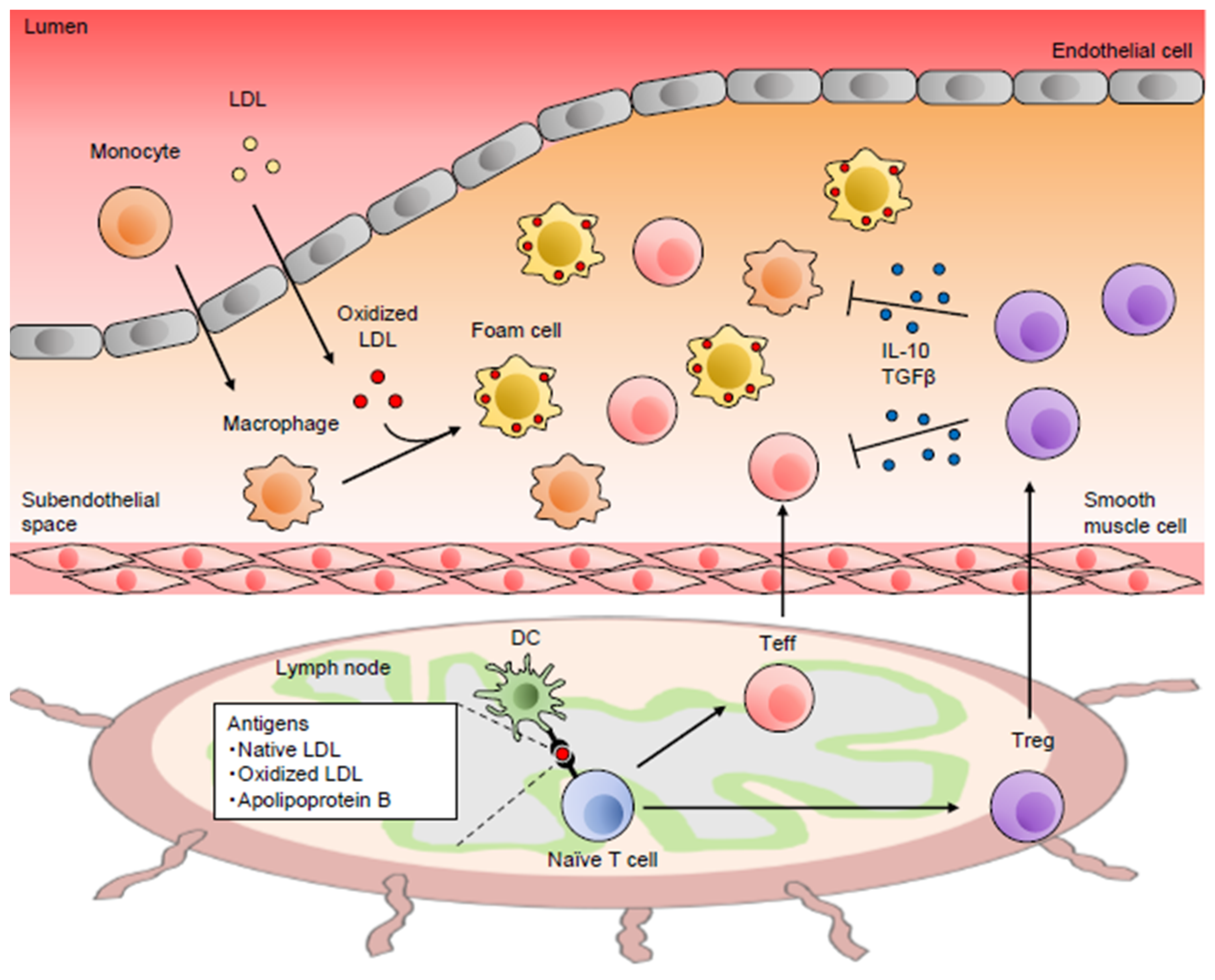
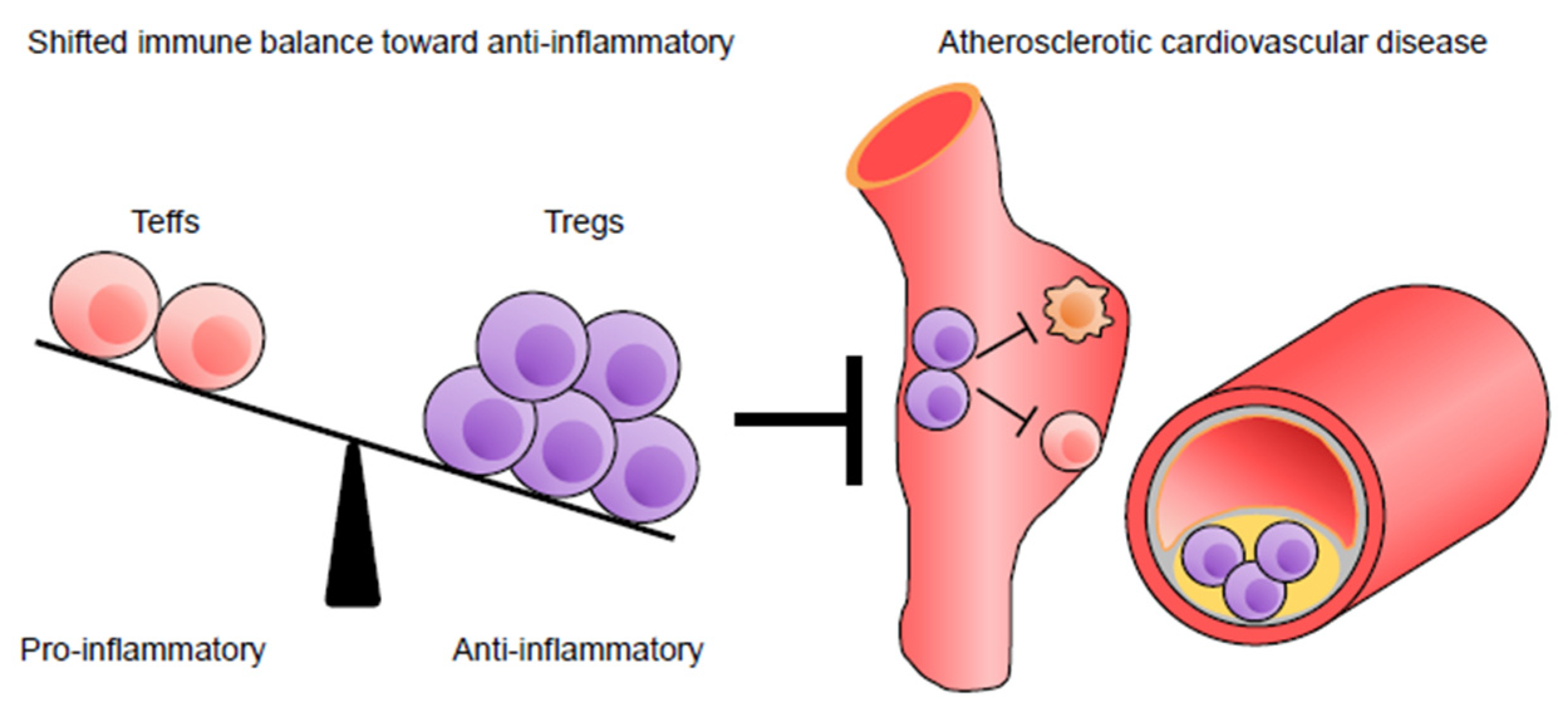

{kind=link}
{kind=link}
| Possible Protective Role of Tregs in Atherosclerosis | ||
| Description | References | |
| Mouse | Genetic deficiency of the CD80/CD86–CD28 signaling decreases CD4+CD25+ Tregs in lymphoid tissues and exacerbates atherosclerosis in Apoe−/− or Ldlr−/− mice. | [51] |
| Adoptive transfer of CD4+CD25+ Tregs attenuates the development of atherosclerosis in Treg-competent Apoe−/− mice. | [52] | |
| Genetic deletion of Foxp3+ Tregs accelerates atherosclerosis development in Ldlr−/− mice. | [55] | |
| Human | Low numbers of FOXP3+ Tregs are detected in all the progression stages of atherosclerotic plaques. | [56,57] |
| Peripheral Treg numbers are reduced in CAD patients | [58,61,66] | |
| Possible mechanisms of Treg-mediated atheroprotection | ||
| Cytokine secretion | Overexpression of IL-10 in T cells inhibits the development of atherosclerosis. | [74] |
| Genetic deletion of TGF-β signaling in T cells dramatically accelerates atherosclerotic lesion development with unstable plaque phenotype. | [75] | |
| Cell–cell contact | Overexpression of CTLA-4 in T cells inhibits atherosclerosis development by downregulating the CD80 and CD86 expression on DCs. | [34] |
| Efferocytosis | Tregs enhance apoptotic cell clearance by macrophages. | [76] |
| Treg immune responses under hypercholesterolemia | ||
| Cell number | The proportion of splenic CD4+Foxp3+ Tregs is markedly increased. | [77] |
| Treg differentiation is promoted in the liver under mild hypercholesterolemia. | [79] | |
| Function | The expression of Treg surface molecules related to migratory function is decreased. | [77] |
| Hypercholesterolemia increases in vitro Treg suppressive activity. | [78] | |
| Hypercholesterolemia modulates the intracellular metabolism of Tregs and promotes their migration towards atherosclerotic aortas. | [80] | |
| CD4+Foxp3+ Tregs differentiate into Th1-like cells in the aorta and secondary lymphoid tissues and become dysfunctional. | [85] | |
| Strategies | Treatment | Immune Effects | References |
|---|---|---|---|
| Vaccination | Treatment with native LDL, oxidized LDL, or ApoB-derived peptides | Induction of antigen-specific Tregs | [16] |
| Modulation of DC functions | Transfer of ApoB100-plused tolerogenic DCs | Promoted antigen-specific CD4+Foxp3+ Treg responses and suppressed pathogenic T cell responses to ApoB100 | [93] |
| Oral administration of active form of vitaminD3 (calcitriol) | Increased tolerogenic DCs and CD4+Foxp3+ Tregs | [94] | |
| Modulation of intestinal immunity | Oral tolerance induction to oxidized LDL or heat shock protein 60 | Increased CD4+CD25+Foxp3+ Tregs and promoted production of TGF-β or IL-10 in mesenteric lymph nodes | [98,99] |
| Oral administration of short-chain fatty acid propionate | Suppressed inflammatory responses mainly dependent on Tregs | [105] | |
| Treatment with antibodies and cytokines | Intravenous administration of anti-CD3 monoclonal antibodies | Increased CD4+CD25+Foxp3+ Tregs and suppressed Teff immune responses | [108,134] |
| Oral administration of anti-CD3 monoclonal antibodies | Increased CD4+LAP+ or CD4+Foxp3+ Tregs in mesenteric lymph nodes and suppressed Teff immune responses | [111] | |
| Treatment with recombinant mouse IL-2/anti-IL-2 monoclonal antibody complexes | Increased CD4+CD25+Foxp3+ Tregs and suppressed Teff immune responses | [112,113] | |
| Combination treatment with anti-CD3 monoclonal antibodies and IL-2 complexes | Increased CD4+Foxp3+ Tregs with activated phenotype | [115] | |
| UVB-based phototherapy | UVB irradiation to the skin | Increased CD4+Foxp3+ Tregs and decreased IFN-γ production from T cells | [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, T.; Sasaki, N.; Rikitake, Y. Recent Advances on the Role and Therapeutic Potential of Regulatory T Cells in Atherosclerosis. J. Clin. Med. 2021, 10, 5907. https://doi.org/10.3390/jcm10245907
Tanaka T, Sasaki N, Rikitake Y. Recent Advances on the Role and Therapeutic Potential of Regulatory T Cells in Atherosclerosis. Journal of Clinical Medicine. 2021; 10(24):5907. https://doi.org/10.3390/jcm10245907
Chicago/Turabian StyleTanaka, Toru, Naoto Sasaki, and Yoshiyuki Rikitake. 2021. "Recent Advances on the Role and Therapeutic Potential of Regulatory T Cells in Atherosclerosis" Journal of Clinical Medicine 10, no. 24: 5907. https://doi.org/10.3390/jcm10245907
APA StyleTanaka, T., Sasaki, N., & Rikitake, Y. (2021). Recent Advances on the Role and Therapeutic Potential of Regulatory T Cells in Atherosclerosis. Journal of Clinical Medicine, 10(24), 5907. https://doi.org/10.3390/jcm10245907