
Autoimmune Diseases of Digestive Organs—A Multidisciplinary Challenge: A Focus on Hepatopancreatobiliary Manifestation

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Autoimmune Digestive System Diseases Associated with HLA
2.1. Celiac Disease
2.2. Inflammatory Bowel Disease (IBD)
2.3. Autoimmune Enteropathy (AIE)
2.4. Autoimmune Hepatitis (AIH)
2.5. Primary Biliary Cholangitis (PBC)
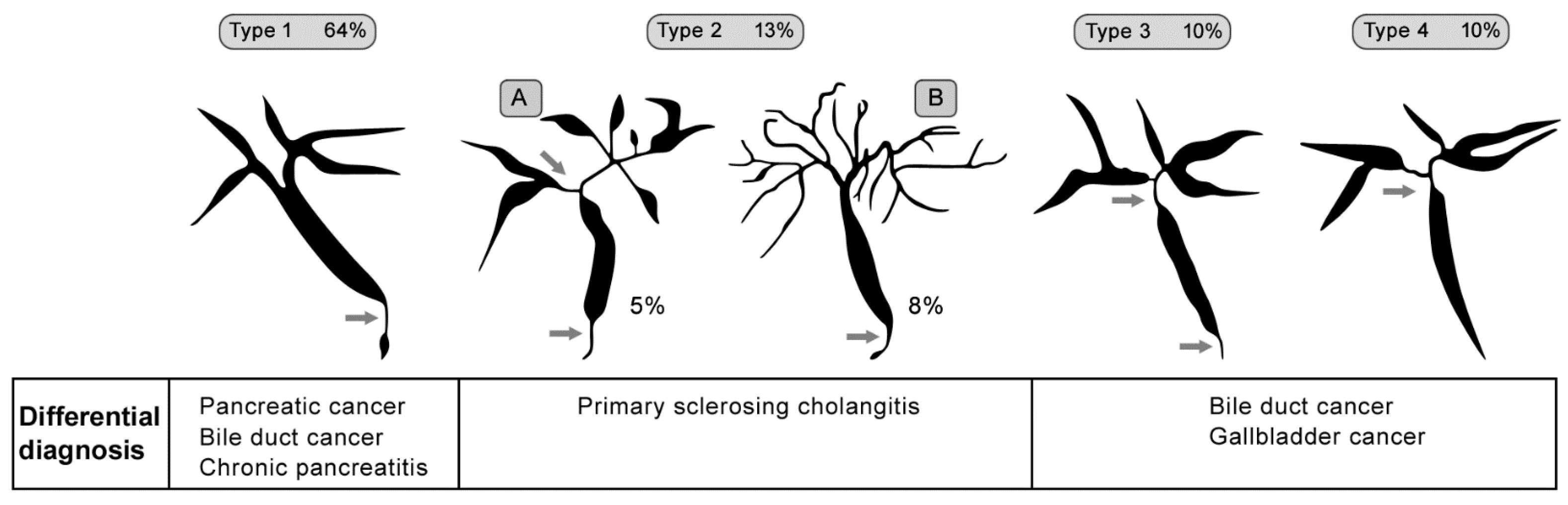
2.6. Primary Sclerosing Cholangitis (PSC)
3. Immunoglobulin G4-Related Diseases (IgG4-RD)
3.1. Autoimmune Pancreatitis (AIP)
3.2. IgG4-Related Hepatobiliary Disease
3.3. Other IgG4-Related Gastrointestinal Diseases
4. Other Autoimmune Gastrointestinal Diseases
Autoimmune Gastritis (AIG)
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Marsh, S.G.; Albert, E.D.; Bodmer, W.F.; Bontrop, R.E.; Dupont, B.; Erlich, H.A.; Geraghty, D.E.; Hansen, J.A.; Mach, B.; Mayr, W.R.; et al. Nomenclature for factors of the HLA system, 2002. Tissue Antigens 2002, 60, 407–464. [Google Scholar] [CrossRef]
- Cassinotti, A.; Birindelli, S.; Clerici, M.; Trabattoni, D.; Lazzaroni, M.; Ardizzone, S.; Colombo, R.; Rossi, E.; Porro, G.B. HLA and autoimmune digestive disease: A clinically oriented review for gastroenterologists. Am. J. Gastroenterol. 2009, 104, 195–217, 194, 218. [Google Scholar] [CrossRef] [PubMed]
- Grønbaek, L.; Otete, H.; Ban, L.; Crooks, C.; Card, T.; Jepsen, P.; West, J. Incidence, prevalence and mortality of autoimmune hepatitis in England 1997–2015. A population-based cohort study. Liver Int. 2020, 40, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Grønbæk, L.; Vilstrup, H.; Jepsen, P. Autoimmune hepatitis in Denmark: Incidence, prevalence, prognosis, and causes of death. A nationwide registry-based cohort study. J. Hepatol. 2014, 60, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Wijmenga, C. Coeliac disease and autoimmune disease-genetic overlap and screening. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Husby, S.; Koletzko, S.; Korponay-Szabó, I.; Kurppa, K.; Mearin, M.L.; Ribes-Koninckx, C.; Shamir, R.; Troncone, R.; Auricchio, R.; Castillejo, G.; et al. European Society Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for Diagnosing Coeliac Disease 2020. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 141–156. [Google Scholar] [CrossRef]
- Ludvigsson, J.F.; Bai, J.C.; Biagi, F.; Card, T.R.; Ciacci, C.; Ciclitira, P.J.; Green, P.H.; Hadjivassiliou, M.; Holdoway, A.; van Heel, D.A.; et al. Diagnosis and management of adult coeliac disease: Guidelines from the British Society of Gastroenterology. Gut 2014, 63, 1210–1228. [Google Scholar] [CrossRef]
- Zubillaga, P.; Vidales, M.C.; Zubillaga, I.; Ormaechea, V.; García-Urkía, N.; Vitoria, J.C. HLA-DQA1 and HLA-DQB1 genetic markers and clinical presentation in celiac disease. J. Pediatr. Gastroenterol. Nutr. 2002, 34, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Sciurti, M.; Fornaroli, F.; Gaiani, F.; Bonaguri, C.; Leandro, G.; Di Mario, F.; De Angelis, G.L. Genetic susceptibilty and celiac disease: What role do HLA haplotypes play? Acta Biomed. 2018, 89, 17–21. [Google Scholar] [CrossRef]
- Liu, E.; Dong, F.; Barón, A.E.; Taki, I.; Norris, J.M.; Frohnert, B.I.; Hoffenberg, E.J.; Rewers, M. High Incidence of Celiac Disease in a Long-term Study of Adolescents With Susceptibility Genotypes. Gastroenterology 2017, 152, 1329–1336.e1321. [Google Scholar] [CrossRef] [PubMed]
- Granito, A.; Zauli, D.; Muratori, P.; Muratori, L.; Grassi, A.; Bortolotti, R.; Petrolini, N.; Veronesi, L.; Gionchetti, P.; Bianchi, F.B.; et al. Anti-Saccharomyces cerevisiae and perinuclear anti-neutrophil cytoplasmic antibodies in coeliac disease before and after gluten-free diet. Aliment. Pharmacol. Ther. 2005, 21, 881–887. [Google Scholar] [CrossRef]
- Granito, A.; Muratori, L.; Muratori, P.; Guidi, M.; Lenzi, M.; Bianchi, F.B.; Volta, U. Anti-saccharomyces cerevisiae antibodies (ASCA) in coeliac disease. Gut 2006, 55, 296. [Google Scholar]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Oryoji, D.; Hisamatsu, T.; Tsuchiya, K.; Umeno, J.; Ueda, S.; Yamamoto, K.; Matsumoto, T.; Watanabe, M.; Hibi, T.; Sasazuki, T. Associations of HLA class I alleles in Japanese patients with Crohn’s disease. Genes Immun. 2015, 16, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, B.M. Role of HLA typing on Crohn’s disease pathogenesis. Ann. Med. Surg. 2015, 4, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Kuna, A.T. Serological markers of inflammatory bowel disease. Biochem. Med. 2013, 23, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Bouzid, D.; Kammoun, A.; Amouri, A.; Mahfoudh, N.; Haddouk, S.; Tahri, N.; Makni, H.; Masmoudi, H. Inflammatory bowel disease: Susceptibility and disease heterogeneity revealed by human leukocyte antigen genotyping. Genet. Test. Mol. Biomark. 2012, 16, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Halling, M.L.; Kjeldsen, J.; Knudsen, T.; Nielsen, J.; Hansen, L.K. Patients with inflammatory bowel disease have increased risk of autoimmune and inflammatory diseases. World J. Gastroenterol. 2017, 23, 6137–6146. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Bonovas, S.; Doherty, G.; Kucharzik, T.; Gisbert, J.P.; Raine, T.; Adamina, M.; Armuzzi, A.; Bachmann, O.; Bager, P.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohns Colitis 2020, 14, 4–22. [Google Scholar] [CrossRef]
- Van Rheenen, P.F.; Aloi, M.; Assa, A.; Bronsky, J.; Escher, J.C.; Fagerberg, U.L.; Gasparetto, M.; Gerasimidis, K.; Griffiths, A.; Henderson, P.; et al. The Medical Management of Paediatric Crohn’s Disease: An ECCO-ESPGHAN Guideline Update. J. Crohns Colitis 2021, 15, 171–194. [Google Scholar] [CrossRef]
- Moroncini, G.; Calogera, G.; Benfaremo, D.; Gabrielli, A. Biologics in Inflammatory Immune-mediated Systemic Diseases. Curr. Pharm. Biotechnol. 2017, 18, 1008–1016. [Google Scholar] [CrossRef]
- Baker, K.F.; Isaacs, J.D. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann. Rheum. Dis. 2018, 77, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Weiler-Normann, C.; Schramm, C.; Quaas, A.; Wiegard, C.; Glaubke, C.; Pannicke, N.; Möller, S.; Lohse, A.W. Infliximab as a rescue treatment in difficult-to-treat autoimmune hepatitis. J. Hepatol. 2013, 58, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E.S.; Kalaitzakis, E. Recent advances in the treatment of primary sclerosing cholangitis. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 413–425. [Google Scholar] [CrossRef]
- Chang, C.; Tanaka, A.; Bowlus, C.; Gershwin, M.E. The use of biologics in the treatment of autoimmune liver disease. Expert Opin. Investig. Drugs 2020, 29, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Crooks, B.; Barnes, T.; Limdi, J.K. Vedolizumab in the treatment of inflammatory bowel disease: Evolving paradigms. Drugs Context 2020, 9, 2019-10-2. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Akram, S.; Murray, J.A.; Pardi, D.S.; Alexander, G.L.; Schaffner, J.A.; Russo, P.A.; Abraham, S.C. Adult autoimmune enteropathy: Mayo Clinic Rochester experience. Clin. Gastroenterol. Hepatol. 2007, 5, 1282–1290, 1245. [Google Scholar] [CrossRef]
- Russo, P.; Alvarez, F. Autoimmune enteropathy: A review. Clin. Appl. Immunol. Rev. 2002, 2, 203–216. [Google Scholar] [CrossRef]
- Bishu, S.; Arsenescu, V.; Lee, E.Y.; Vargas, H.D.; de Villiers, W.J.; Arsenescu, R. Autoimmune enteropathy with a CD8+ CD7- T-cell small bowel intraepithelial lymphocytosis: Case report and literature review. BMC Gastroenterol. 2011, 11, 131. [Google Scholar] [CrossRef]
- Montalto, M.; D’Onofrio, F.; Santoro, L.; Gallo, A.; Gasbarrini, A.; Gasbarrini, G. Autoimmune enteropathy in children and adults. Scand. J. Gastroenterol. 2009, 44, 1029–1036. [Google Scholar] [CrossRef]
- Patey-Mariaud de Serre, N.; Canioni, D.; Ganousse, S.; Rieux-Laucat, F.; Goulet, O.; Ruemmele, F.; Brousse, N. Digestive histopathological presentation of IPEX syndrome. Mod. Pathol. 2009, 22, 95–102. [Google Scholar] [CrossRef]
- Kobayashi, I.; Kubota, M.; Yamada, M.; Tanaka, H.; Itoh, S.; Sasahara, Y.; Whitesell, L.; Ariga, T. Autoantibodies to villin occur frequently in IPEX, a severe immune dysregulation, syndrome caused by mutation of FOXP3. Clin. Immunol. 2011, 141, 83–89. [Google Scholar] [CrossRef]
- Biagi, F.; Bianchi, P.I.; Trotta, L.; Corazza, G.R. Anti-goblet cell antibodies for the diagnosis of autoimmune enteropathy? Am. J. Gastroenterol. 2009, 104, 3112. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Autoimmune hepatitis--approach to diagnosis. MedGenMed 2006, 8, 55. [Google Scholar]
- Hennes, E.M.; Zeniya, M.; Czaja, A.J.; Parés, A.; Dalekos, G.N.; Krawitt, E.L.; Bittencourt, P.L.; Porta, G.; Boberg, K.M.; Hofer, H.; et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008, 48, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.; Berg, P.A.; Bianchi, F.B.; Bianchi, L.; Burroughs, A.K.; Cancado, E.L.; Chapman, R.W.; Cooksley, W.G.; Czaja, A.J.; Desmet, V.J.; et al. International Autoimmune Hepatitis Group Report: Review of criteria for diagnosis of autoimmune hepatitis. J. Hepatol. 1999, 31, 929–938. [Google Scholar] [CrossRef]
- Sucher, E.; Sucher, R.; Gradistanac, T.; Brandacher, G.; Schneeberger, S.; Berg, T. Autoimmune Hepatitis-Immunologically Triggered Liver Pathogenesis-Diagnostic and Therapeutic Strategies. J. Immunol. Res. 2019, 2019, 9437043. [Google Scholar] [CrossRef]
- Muratori, P.; Czaja, A.J.; Muratori, L.; Pappas, G.; Maccariello, S.; Cassani, F.; Granito, A.; Ferrari, R.; Mantovani, V.; Lenzi, M.; et al. Genetic distinctions between autoimmune hepatitis in Italy and North America. World J. Gastroenterol. 2005, 11, 1862–1866. [Google Scholar] [CrossRef]
- Pape, S.; Schramm, C.; Gevers, T.J. Clinical management of autoimmune hepatitis. United Eur. Gastroenterol. J. 2019, 7, 1156–1163. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.K.K.; Viken, M.K.; Wittig, M.; Holm, K.; Folseraas, T.; Mucha, S.; Melum, E.; Hov, J.R.; Lazaridis, K.N.; Juran, B.D.; et al. HLA haplotypes in primary sclerosing cholangitis patients of admixed and non-European ancestry. HLA 2017, 90, 228–233. [Google Scholar] [CrossRef]
- Selmi, C.; Mayo, M.J.; Bach, N.; Ishibashi, H.; Invernizzi, P.; Gish, R.G.; Gordon, S.C.; Wright, H.I.; Zweiban, B.; Podda, M.; et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: Genetics, epigenetics, and environment. Gastroenterology 2004, 127, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Karlsen, T.H.; Lindor, K.D.; Adams, D.H. Primary sclerosing cholangitis. Lancet 2013, 382, 1587–1599. [Google Scholar] [CrossRef]
- Gochanour, E.; Jayasekera, C.; Kowdley, K. Primary Sclerosing Cholangitis: Epidemiology, Genetics, Diagnosis, and Current Management. Clin. Liver Dis. (Hoboken) 2020, 15, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Kowdley, K.V.; Harrison, M.E. ACG Clinical Guideline: Primary Sclerosing Cholangitis. Am. J. Gastroenterol. 2015, 110, 646–659; quiz 660. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef]
- Aabakken, L.; Karlsen, T.H.; Albert, J.; Arvanitakis, M.; Chazouilleres, O.; Dumonceau, J.M.; Färkkilä, M.; Fickert, P.; Hirschfield, G.M.; Laghi, A.; et al. Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guideline. Endoscopy 2017, 49, 588–608. [Google Scholar] [CrossRef]
- Culver, E.L.; Chapman, R.W. Systematic review: Management options for primary sclerosing cholangitis and its variant forms—IgG4-associated cholangitis and overlap with autoimmune hepatitis. Aliment. Pharmacol. Ther. 2011, 33, 1273–1291. [Google Scholar] [CrossRef]
- Goode, E.C.; Clark, A.B.; Mells, G.F.; Srivastava, B.; Spiess, K.; Gelson, W.T.H.; Trivedi, P.J.; Lynch, K.D.; Castren, E.; Vesterhus, M.N.; et al. Factors Associated With Outcomes of Patients with Primary Sclerosing Cholangitis and Development and Validation of a Risk Scoring System. Hepatology 2019, 69, 2120–2135. [Google Scholar] [CrossRef]
- Backhus, J.; Seufferlein, T.; Perkhofer, L.; Hermann, P.C.; Kleger, A. IgG4-Related Diseases in the Gastrointestinal Tract: Clinical Presentation, Diagnosis and Treatment Challenges. Digestion 2019, 100, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Takahashi, H. IgG4-Related Disease in Organs Other than the Hepatobiliary-Pancreatic System. Semin. Liver Dis. 2016, 36, 274–282. [Google Scholar] [CrossRef]
- Mahajan, V.S.; Mattoo, H.; Deshpande, V.; Pillai, S.S.; Stone, J.H. IgG4-related disease. Annu. Rev. Pathol. 2014, 9, 315–347. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, H.; Mahajan, V.S.; Della-Torre, E.; Sekigami, Y.; Carruthers, M.; Wallace, Z.S.; Deshpande, V.; Stone, J.H.; Pillai, S. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J. Allergy Clin. Immunol. 2014, 134, 679–687. [Google Scholar] [CrossRef]
- Wallace, Z.S.; Mattoo, H.; Carruthers, M.; Mahajan, V.S.; Della Torre, E.; Lee, H.; Kulikova, M.; Deshpande, V.; Pillai, S.; Stone, J.H. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann. Rheum. Dis. 2015, 74, 190–195. [Google Scholar] [CrossRef]
- Löhr, J.M.; Beuers, U.; Vujasinovic, M.; Alvaro, D.; Frøkjær, J.B.; Buttgereit, F.; Capurso, G.; Culver, E.L.; de-Madaria, E.; Della-Torre, E.; et al. European Guideline on IgG4-related digestive disease—UEG and SGF evidence-based recommendations. United Eur. Gastroenterol. J. 2020, 8, 637–666. [Google Scholar] [CrossRef] [PubMed]
- Shimosegawa, T.; Chari, S.T.; Frulloni, L.; Kamisawa, T.; Kawa, S.; Mino-Kenudson, M.; Kim, M.H.; Klöppel, G.; Lerch, M.M.; Löhr, M.; et al. International consensus diagnostic criteria for autoimmune pancreatitis: Guidelines of the International Association of Pancreatology. Pancreas 2011, 40, 352–358. [Google Scholar] [CrossRef]
- Schneider, A.; Michaely, H.; Weiss, C.; Hirth, M.; Rückert, F.; Wilhelm, T.J.; Schönberg, S.; Marx, A.; Singer, M.V.; Löhr, J.M.; et al. Prevalence and Incidence of Autoimmune Pancreatitis in the Population Living in the Southwest of Germany. Digestion 2017, 96, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chari, S.; Smyrk, T.C.; Deshpande, V.; Klöppel, G.; Kojima, M.; Liu, X.; Longnecker, D.S.; Mino-Kenudson, M.; Notohara, K.; et al. Autoimmune pancreatitis (AIP) type 1 and type 2: An international consensus study on histopathologic diagnostic criteria. Pancreas 2011, 40, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.J. Autoimmune Pancreatitis—A Riddle Wrapped in an Enigma. Dig. Dis. 2016, 34, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, L.D.; Farooq, A.; Bano, F.; Kleeff, J.; Baron, R.; Raraty, M.; Ghaneh, P.; Sutton, R.; Whelan, P.; Campbell, F.; et al. Differentiation of Autoimmune Pancreatitis from Pancreatic Cancer Remains Challenging. World J. Surg. 2019, 43, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.C.; Chang, M.C.; Chen, C.H.; Tsai, I.L.; Wang, S.Y.; Kuo, Y.P.; Chang, Y.T. High accuracy differentiating autoimmune pancreatitis from pancreatic ductal adenocarcinoma by immunoglobulin G glycosylation. Clin. Proteom. 2019, 16, 1–10. [Google Scholar] [CrossRef]
- Björnsson, E.; Chari, S.T.; Smyrk, T.C.; Lindor, K. Immunoglobulin G4 associated cholangitis: Description of an emerging clinical entity based on review of the literature. Hepatology 2007, 45, 1547–1554. [Google Scholar] [CrossRef]
- Kamisawa, T.; Nakazawa, T.; Tazuma, S.; Zen, Y.; Tanaka, A.; Ohara, H.; Muraki, T.; Inui, K.; Inoue, D.; Nishino, T.; et al. Clinical practice guidelines for IgG4-related sclerosing cholangitis. J. Hepatobiliary Pancreat. Sci. 2019, 26, 9–42. [Google Scholar] [CrossRef] [PubMed]
- Hubers, L.M.; Beuers, U. IgG4-related disease of the biliary tract and pancreas: Clinical and experimental advances. Curr. Opin. Gastroenterol. 2017, 33, 310–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deshpande, V.; Zen, Y.; Chan, J.K.; Yi, E.E.; Sato, Y.; Yoshino, T.; Klöppel, G.; Heathcote, J.G.; Khosroshahi, A.; Ferry, J.A.; et al. Consensus statement on the pathology of IgG4-related disease. Mod. Pathol. 2012, 25, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
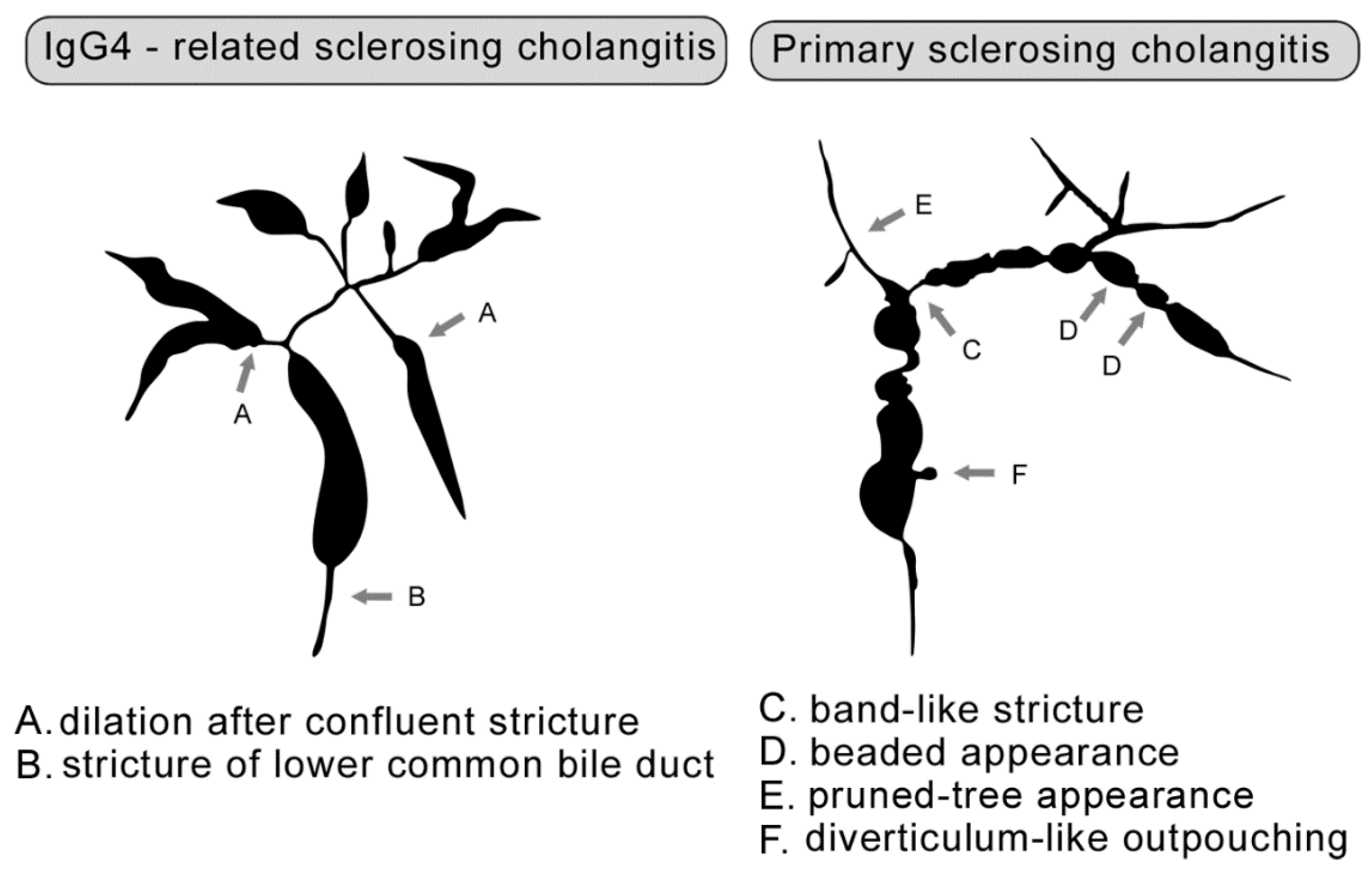
- Nakazawa, T.; Naitoh, I.; Hayashi, K.; Okumura, F.; Miyabe, K.; Yoshida, M.; Yamashita, H.; Ohara, H.; Joh, T. Diagnostic criteria for IgG4-related sclerosing cholangitis based on cholangiographic classification. J. Gastroenterol. 2012, 47, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Ohara, H.; Okazaki, K.; Tsubouchi, H.; Inui, K.; Kawa, S.; Kamisawa, T.; Tazuma, S.; Uchida, K.; Hirano, K.; Yoshida, H.; et al. Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012. J. Hepatobiliary Pancreat. Sci. 2012, 19, 536–542. [Google Scholar] [CrossRef]
- Chung, H.; Watanabe, T.; Kudo, M.; Maenishi, O.; Wakatsuki, Y.; Chiba, T. Identification and characterization of IgG4-associated autoimmune hepatitis. Liver Int. 2010, 30, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Umemura, T.; Zen, Y.; Hamano, H.; Joshita, S.; Ichijo, T.; Yoshizawa, K.; Kiyosawa, K.; Ota, M.; Kawa, S.; Nakanuma, Y.; et al. Clinical significance of immunoglobulin G4-associated autoimmune hepatitis. J. Gastroenterol. 2011, 46 (Suppl. S1), 48–55. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, F.; Ciccone, A.; Di Ruscio, M.; Vernia, F.; Cipolloni, G.; Coletti, G.; Calvisi, G.; Frieri, G.; Latella, G. IgG4-Related Disease Mimicking Crohn’s Disease: A Case Report and Review of Literature. Dig. Dis. Sci. 2018, 63, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Kamisawa, T.; Kuruma, S.; Tabata, T.; Chiba, K.; Iwasaki, S.; Endo, Y.; Kuwata, G.; Koizumi, K.; Shimosegawa, T.; et al. Immunoglobulin G4-related gastrointestinal diseases, are they immunoglobulin G4-related diseases? World J. Gastroenterol. 2013, 19, 5769–5774. [Google Scholar] [CrossRef]
- Sánchez-Oro, R.; Alonso-Muñoz, E.M.; Martí Romero, L. Review of IgG4-related disease. Gastroenterol. Hepatol. 2019, 42, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Topal, F.; Sarıtaş Yüksel, E.; Ekinci, N.; Pekdiker, M.; Cakalağaoğlu, F.; Alper, E.; Unsal, B. The prevalence of IgG4-positive plasma cell infiltrates in inflammatory bowel disease patients without autoimmune pancreatitis. Turk. J. Gastroenterol. 2014, 25, 558–562. [Google Scholar] [CrossRef]
- Obiorah, I.; Hussain, A.; Palese, C.; Azumi, N.; Benjamin, S.; Ozdemirli, M. IgG4-related disease involving the esophagus: A clinicopathological study. Dis. Esophagus 2017, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.; Woo, J.Y.; Kim, J.W.; Hong, H.S.; Yang, I.; Lee, Y.; Hwang, D.; Min, S.J. An immunoglobulin G4-related sclerosing disease of the small bowel: CT and small bowel series findings. Korean J. Radiol. 2013, 14, 776–780. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, S.B.; Lim, C.H.; Cha, M.G.; Kang, W.K. IgG4-related disease of the rectum. Ann. Surg. Treat. Res. 2016, 90, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.N.; Appelman, H.D. Autoimmune Gastritis. Arch. Pathol. Lab. Med. 2019, 143, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Neumann, W.L.; Coss, E.; Rugge, M.; Genta, R.M. Autoimmune atrophic gastritis--pathogenesis, pathology and management. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Lenti, M.V.; Rugge, M.; Lahner, E.; Miceli, E.; Toh, B.H.; Genta, R.M.; De Block, C.; Hershko, C.; Di Sabatino, A. Autoimmune gastritis. Nat. Rev. Dis. Primers 2020, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Lahner, E.; Zagari, R.M.; Zullo, A.; Di Sabatino, A.; Meggio, A.; Cesaro, P.; Lenti, M.V.; Annibale, B.; Corazza, G.R. Chronic atrophic gastritis: Natural history, diagnosis and therapeutic management. A position paper by the Italian Society of Hospital Gastroenterologists and Digestive Endoscopists [AIGO], the Italian Society of Digestive Endoscopy [SIED], the Italian Society of Gastroenterology [SIGE], and the Italian Society of Internal Medicine [SIMI]. Dig. Liver Dis. 2019, 51, 1621–1632. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| 1. Adult-onset chronic diarrhea (>6 weeks duration) |
| 2. Malabsorption |
|
| 4. Exclusion of other causes of villous atrophy, including celiac disease, refractorysprue and intestinal lymphoma |
| 5. Anti-enterocyte antibodies |
| Criteria | Cut-off | Points |
|---|---|---|
| ANA or SMA | ≥1:40 | 1 |
| ANA or SMA | ≥1:80 | |
| or LKM | ≥1:40 | 2 (max. 2 points for all antibodies) |
| or SLA | Positive | |
| IgG | >Upper normal limit | 1 |
| >1.10 times upper normal limit | 2 | |
| Liver histology (evidence of hepatitis is a necessary condition) | Compatible with AIH Typical AIH | 1 2 |
| Absence of viral hepatitis | Yes | 2 ≥6: probable AIH ≥7: definite AIH |
| 1. Clinical examination |
|
|
|
|
2. Imaging
|
| 3. Assessing serum IgG4 concentration (upper level of normal = 135 mg/dL, but only levels higher than 4× the upper level seems to have clear diagnostic value) 4. Presence of 3 major histopathological characteristics
|
| Criteria | Description |
|---|---|
| Pancreas histology (H) | Lymphoplasmacytic sclerosing pancreatitis (LPSP, core biopsy/resection). At least 3 of the following: periductal lymphoplasmacytic infiltrate without granulocytic infiltration, obliterative phlebitis, storiform fibrosis, abundant (>10 cells/high-power field) IgG4-positive cells |
| Parenchymal imaging (P) | Typical: diffuse enlargement with delayed enhancement (sometimes associated with ring-like enhancement) |
| Ductal imaging (D) | Long (>1/3 length of the main pancreatic duct) or multiple strictures without marked upstream dilation |
| Serology (S) | IgG4 > 2× upper normal limit |
| Other organ involvement (OOI) | 1 or 2
|
| Response to steroid therapy (Rt) | Rapid (≤2 weeks) radiologically demonstrable resolution or marked improvement in pancreatic/extrapancreatic manifestations |
| Criteria | Description |
|---|---|
| Histology (H) | Idiopathic duct centric pancreatitis (IDCP): Both of the following:
|
| Parenchymal imaging (P) | Typical: diffuse enlargement with delayed enhancement (sometimes associated with rim-like enhancement) |
| Ductal imaging (D) | Long (>1/3 length of the main pancreatic duct) or multiple strictures without marked upstream dilatation |
| Other organ involvement (OOI) | Clinically diagnosed inflammatory bowel disease |
| Response to steroid therapy (Rt) | Rapid (≤2 weeks) radiologically demonstrable resolution or marked improvement in pancreatic manifestations |
| Type 1 (LPSP) | Type 2 (IDCP) | |
|---|---|---|
| IgG4-RD | Yes | No |
| Prevalence | Asia > USA/Europe | USA/Europe > Asia |
| Sex | M > F | M = F |
| Worldwide percentage (%) | >90 | <10 |
| Age predominance (years) | >50 | 30–50 |
| Initial icterus (%) | >60 | <30 |
| Acute abdominal pain (%) | <30 | >60 |
| Elevated serum IgG4 (%) | >70 | <10 |
| Histopathology | Storiform fibrosis, LPSP, obliterative phlebitis | IDCP, GEL |
| Affection of other organs | Yes | No |
| Association with IBD (%) | <10 | >40 |
| Steroid response (%) | >90 | >90 |
| Relapse after steroid therapy (%) | >40 | <10 |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunovsky, L.; Dite, P.; Jabandziev, P.; Kala, Z.; Vaculova, J.; Andrasina, T.; Hrunka, M.; Bojkova, M.; Trna, J. Autoimmune Diseases of Digestive Organs—A Multidisciplinary Challenge: A Focus on Hepatopancreatobiliary Manifestation. J. Clin. Med. 2021, 10, 5796. https://doi.org/10.3390/jcm10245796
Kunovsky L, Dite P, Jabandziev P, Kala Z, Vaculova J, Andrasina T, Hrunka M, Bojkova M, Trna J. Autoimmune Diseases of Digestive Organs—A Multidisciplinary Challenge: A Focus on Hepatopancreatobiliary Manifestation. Journal of Clinical Medicine. 2021; 10(24):5796. https://doi.org/10.3390/jcm10245796
Chicago/Turabian StyleKunovsky, Lumir, Petr Dite, Petr Jabandziev, Zdenek Kala, Jitka Vaculova, Tomas Andrasina, Matej Hrunka, Martina Bojkova, and Jan Trna. 2021. "Autoimmune Diseases of Digestive Organs—A Multidisciplinary Challenge: A Focus on Hepatopancreatobiliary Manifestation" Journal of Clinical Medicine 10, no. 24: 5796. https://doi.org/10.3390/jcm10245796
APA StyleKunovsky, L., Dite, P., Jabandziev, P., Kala, Z., Vaculova, J., Andrasina, T., Hrunka, M., Bojkova, M., & Trna, J. (2021). Autoimmune Diseases of Digestive Organs—A Multidisciplinary Challenge: A Focus on Hepatopancreatobiliary Manifestation. Journal of Clinical Medicine, 10(24), 5796. https://doi.org/10.3390/jcm10245796