1. Introduction
Myotonic Dystrophy type 1 (DM1) is an autosomal dominant inherited muscular dystrophy with a multi-systemic nature. Patients display a wide variety of symptoms, including muscle weakness, myotonia, respiratory failure, cardiac conduction defects, cataracts, and endocrine disturbances. In addition, the age of onset varies greatly, from birth up to >70 years. The cause of the variability in clinical manifestation is poorly understood. DM1 is viewed as an RNA gain of function disorder, wherein a CTG expansion in the 3′ untranslated region of the myotonic dystrophy protein kinase (
DMPK) gene causes the accumulation of expanded transcripts as intranuclear RNA foci, which sequester a number of splicing factors, resulting in loss of function and downstream deregulation of the alternative splicing of several genes [
1]. In recent years, the view of DM1 as solely an RNA gain-of-function disorder has changed. Several new discoveries, such as antisense transcription and repeat associated non-ATG (RAN) translation, have added to the complexity of this disease. DM1 antisense (DM1-AS) transcription was first described by Cho and collaborators in 2005 [
2]. They reported an antisense transcript, emanating from the adjacent SIX5 regulatory region, which was converted into 21 nt siRNAs. These siRNAs were proposed to have a regulatory role in heterochromatin formation. Huguet and collaborators, however, showed that DM1-AS transcription extends across the CAG repeat, as they could detect DM1-AS RNA after the repeat in the 3′ region of DM1 tissue and they also showed the presence of antisense RNA foci, which did not co-localize with sense RNA foci, in adult mouse models [
3]. Similar results were found by Michel and collaborators in human fetal samples [
4]. The inclusion of the expanded repeat was confirmed by Gudde and collaborators in 2017, although they found DM1-AS transcripts to be low in abundance and with varying lengths, both including and excluding the CAG repeat [
5]. In addition, the DM1-AS strand was found by Zu and collaborators to give rise to RAN-translated polyglutamine stretches [
6]. RAN translation is a typical phenomenon seen in repeat expansion disorders, in which peptides are produced from all frames without ATG start codon recognition. Zu and collaborators discovered a novel polyglutamine (polyGln) RAN protein expressed from the antisense CAG expansion transcript of the
DMPK gene [
6]. Nuclear polyGln RAN protein aggregates were found at a low frequency in a DM1 patient’s myoblasts and skeletal muscle (
n = 1) and at a higher frequency in leukocytes from peripheral blood (
n = 1) [
6]. The nuclear aggregates co-localized with caspase-8, an early indicator of polyGln-induced apoptosis. This suggests that RAN proteins may be an additional mechanism of cytotoxicity in DM1 cells.
Since its first discovery in 2011 by Zu and collaborators, RAN translation has been extensively studied in multiple expansion disorders and great advances have been made [
7]. However, the contribution of RAN translation to DM1 pathology has not been further studied since its first report in 2011. Much remains unknown regarding the presence of RAN translation and its mechanism in DM1. Is it equally present across patients, and is its distribution across tissues similar? To what extent does it contribute to the pathology of the disease? In order to further enhance our knowledge of RAN translation in DM1, we decided to study the presence of RAN translation in DM1 primary cell cultures—myoblasts, skin fibroblasts and lymphoblastoids—derived from ten DM1 patients, with a heterogeneous display of subtypes. The RAN-translated polyGln has been described to originate from the antisense strand of the
DMPK gene. We therefore validated the presence of DM1-AS transcription in our three patient-derived cellular models and lower expression levels were found in patients compared to controls. Additionally, we found that the DM1-AS transcripts were found in both the nucleus and the cytoplasm, of which at least a portion contained the expanded repeat, as shown by the presence of antisense RNA foci in patients. However, the polyGln RAN protein was not present in patient-derived DM1 cells, or was present in such low quantities that it was below the detection limit of the currently available techniques.
2. Materials and Methods
2.1. Samples
This study was approved by the Ethics Committee of the University Hospital Germans Trias i Pujol and was performed in accordance with the Declaration of Helsinki for Human Research. Written informed consent was obtained from all participants. The study included ten patients with DM1 and thirteen controls with no previous family history of neuromuscular disorders (recruited from the traumatology department, in whom surgery was needed). DM1 diagnosis was confirmed or discarded via triplet-primed PCR in all the study participants. Clinical information of DM1 patients was obtained from medical records and updated at the last visit. We obtained three different samples from eight patients and eleven controls: blood, muscle biopsy, and skin biopsy. The other two patients and two controls only provided a blood sample. All samples were simultaneously obtained from patients with confirmed juvenile, adult or late-onset DM1. The muscle biopsy was obtained from biceps brachii (n = 7) or vastus lateralis (n = 1) of DM1 patients and from intrinsic forearm or hand muscles of eleven non-DM1 patients. Skin biopsy was obtained with a 0.5 cm skin punch.
2.2. Small Pool PCR for Sizing the CTG Repeat
Total genomic DNA was extracted from peripheral blood samples, as previously described [
8]. To estimate the length of the expanded mode allele, small-pool PCR (SP-PCR) was carried out with small amounts of input DNA (300 pg), using flanking primers DM-C and DM-DR, as previously described [
9,
10]. PCR was performed using a Custom PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 69 mM 2-mercaptoethanol, and Taq polymerase (Sigma-Aldrich, Gillingham, UK) at 1 unit per 10 µL. All reactions were supplemented with 5% DMSO and the annealing temperature was 63.5 °C. DNA fragments were resolved by electrophoresis on a 1% agarose gel, and Southern blot hybridized as described in references [
9,
10]. Autoradiographic images were scanned and the CTG size of the mode was estimated through comparison against the molecular weight ladder using GelAnalyzer 19.1 software (
www.gelanalyzer.com, by Istvan Lazar Jr. and Istvan Lazar Sr.).
2.3. Cell Culture
Myoblasts were isolated from the biopsied tissue by CD56 magnetic separation. Myoblasts were grown until 60–70% confluency on 0.1% gelatin-coated coverslips in six-well tissue-culture plates in a proliferation medium containing DMEM supplemented with 10% FBS, 22% M-199, 1× PSF, insulin 1.74 μM, l-glutamine 2 mM, FGF 1.39 nM and EGF 0.135 mM. Skin fibroblasts were isolated from biopsied tissue using the explant method. Skin fibroblasts were grown until 70% confluency on 0.5% poly-d-lysin coated coverslips in a six-well tissue culture plate in a proliferation medium containing DMEM, supplemented with 10% FBS and 1× PSF. Peripheral blood mononuclear cells were isolated from blood collected in heparin tubes according to the Ficoll gradient and immortalized using Epstein–Barr virus. Lymphoblastoids were grown until 70% confluency on 0.1% poly-d-lysin coated coverslips in a six-well tissue culture plate in a proliferation medium containing RPMI, supplemented with 10% FBS, 1× PSF and 2 mM l-glutamine. HEK293 cells were cultured until 80% confluency on 0.5% poly-d-lysin coated coverslips in a six-well tissue culture plate in MEM, supplemented with 5% horse serum, 5% FBS, 1% Glutamine and 1% PSF. In addition to the coverslips, cell pellets from each of the cell types were extracted and stored for later use in RNA and protein studies.
2.4. cDNA Construct Huntingtin and HEK293 Transfection
The Q17 vector was generously provided to us by R.P.V.-M.. In short, the construct is part of the cDNA of human huntingtin (585 amino acids), containing 17 CAGs, and was cloned into a pcDNA3.1-Gateway. The human huntingtin with the 17 CAGs was in frame with mCherry, a fluorescent. DNA transfections were performed when HEK293 cells reached 80% confluency using Lipofectamine 2000 reagent (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. DNA:lipofectamine ratio was 1:2 and incubation lasted 48 h.
2.5. RNA Isolation and Subcellular Fractionation
Total RNA from cultured myoblast, skin fibroblast and lymphoblastoid cell lines was isolated using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) or the RNeasy plus mini kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Total RNA from a healthy human heart was used as a positive control in all RT-PCR analyses (AM7966, Thermo Fisher Scientific, Waltham, MA, USA).
For RNA isolation from the subcellular fractions, skin fibroblasts were grown to 80% confluency, collected through trypsinization and pelleted through centrifugation at 2000 rpm for 5 min at 4 °C. Cell pellets were washed twice with ice-cold PBS. Cells were resuspended in ice-cold cell disruption buffer (10 mM KCl, 1.5 mM MgCl
2, 20 mM Tris-Cl (pH 7.5), 1 mM DTT) and incubated on ice for 10 min [
11]. Samples were homogenized using a tissue disruptor for 30 s and then Triton X-100 was added to a final concentration of 0.1%. The lysate was centrifuged at 2000 rpm for 5 min at 4 °C, after which the supernatant (cytoplasmic fraction) was removed. Both cytoplasmic and nuclear fraction underwent RNA isolation by the RNeasy plus mini kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions.
2.6. RT-PCR Analysis for DM1-AS Transcript Detection
For detection of the DM1-AS transcripts, a similar strategy to Zu et al. was followed [
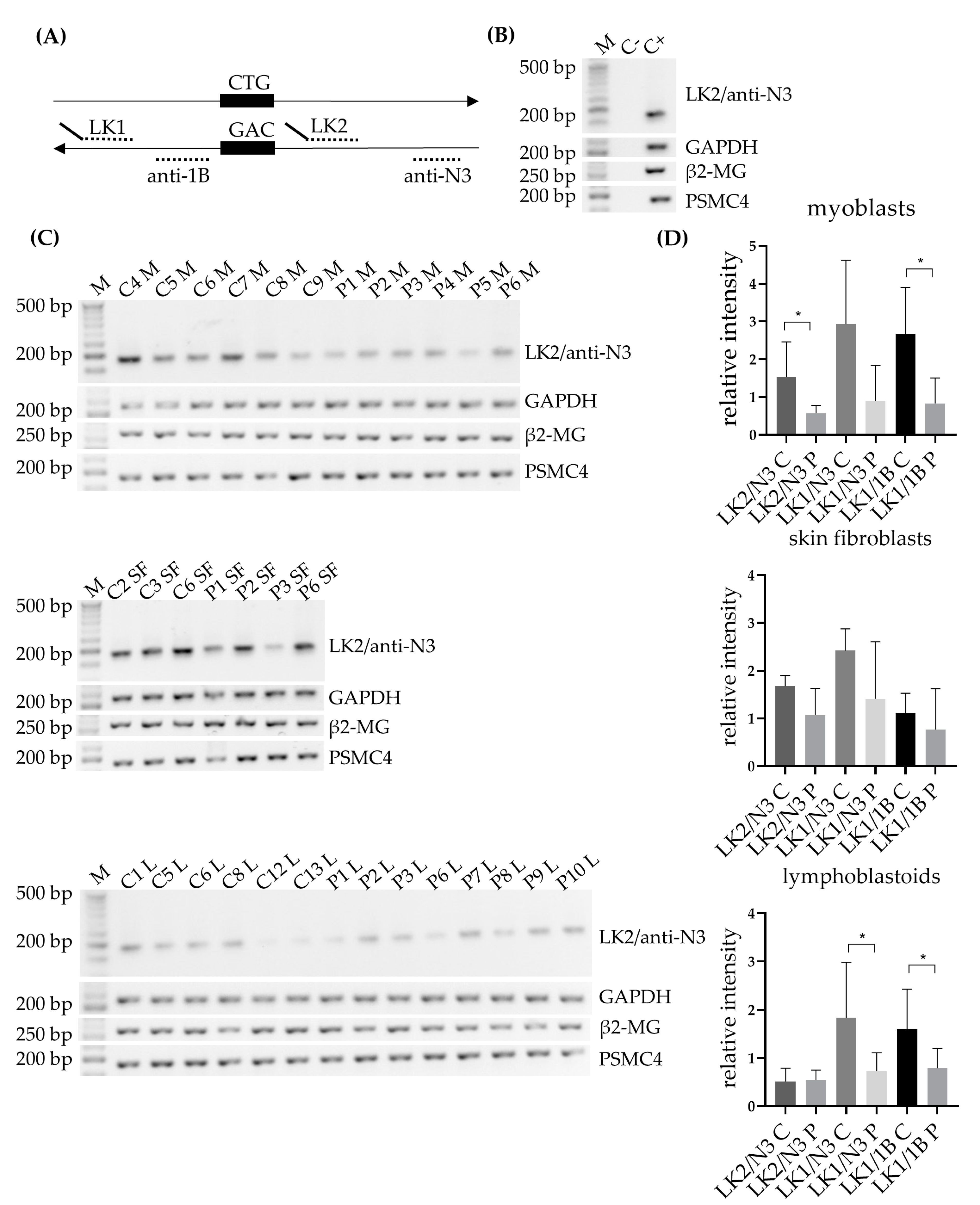
6], with some minor changes. In brief, an equivalent of 1 µg total RNA was subjected to cDNA synthesis using SuperScript IV reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA) at 55 °C, using random hexamers (50 µM, N8080127, Thermo Fisher Scientific, Waltham, MA, USA). The subsequent PCR was carried out using DM1-AS specific primers LK1, together with anti-N3 or anti-1B, and LK2, together with anti-N3 (
Figure 1A; primer details and PCR conditions can be found in
Table S1). Both LK1 and LK2 contained a linker sequence, for which a specific primer was created and used in some of the PCR reactions. For endogenous controls GAPDH, β2-microglobulin (β2-MG) and PSMC4 a similar approach was followed (primer details and PCR condition found in
Table S1).
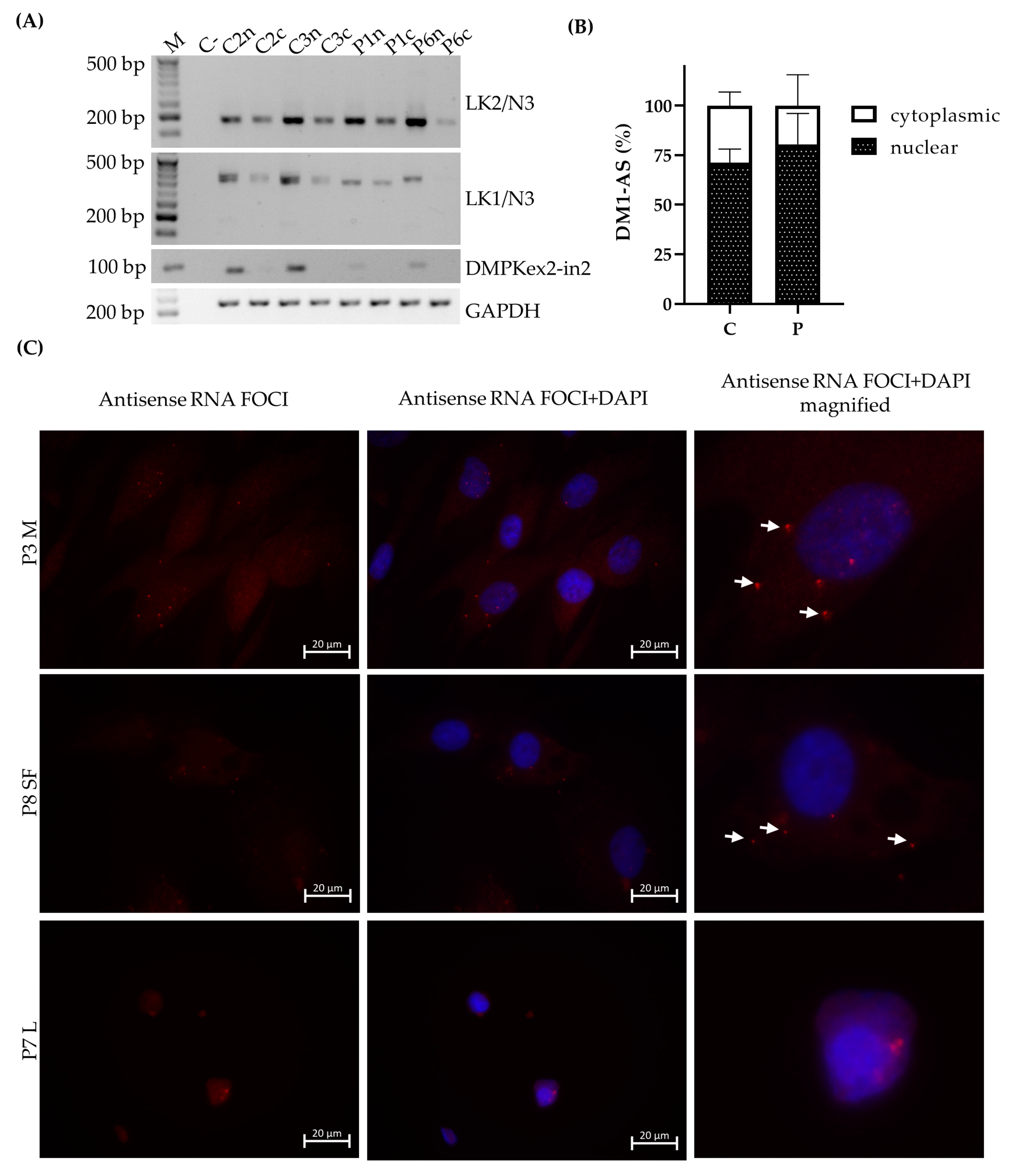
To analyze nuclear and cytoplasmic RNA fractions, 150 ng RNA was used for the RT reaction for both fractions. RNA was reverse transcribed using SuperScript IV reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA) at 55 °C, using random hexamers (50 µM, N8080127, Thermo Fisher Scientific, Waltham, MA, USA). For RT-PCR analyses, the same approach was used as described above with primer combinations LK1/anti-N3 and LK2/anti-N3. As a nuclear marker, pre-mRNA DMPK was used and GAPDH as an endogenous control (primers and PCR conditions in
Table S1).
2.7. Immunoblotting
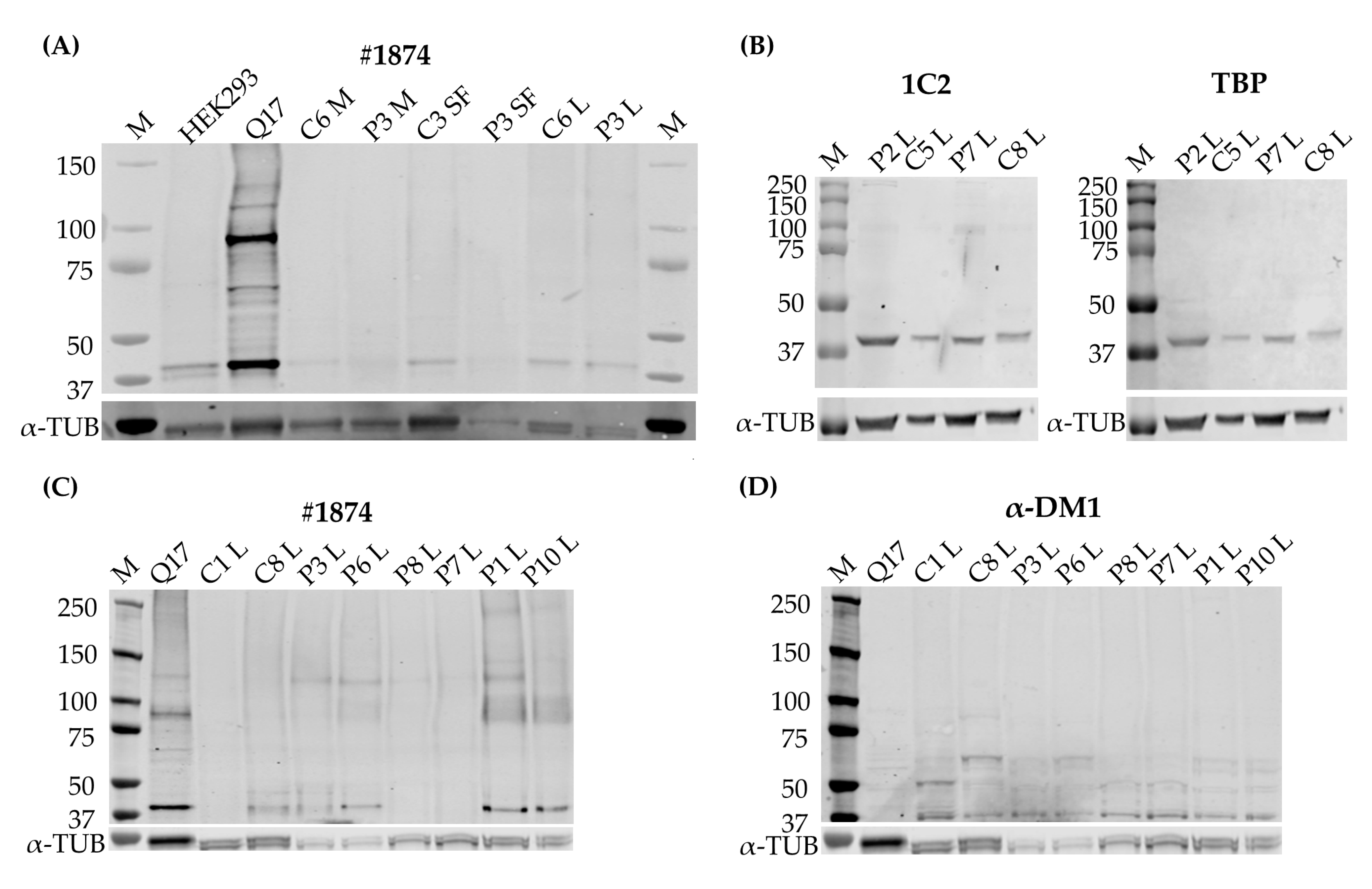
The cell pellets collected from the cell cultures were lysed in a RIPA buffer, supplemented with a cOmplete™ Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and homogenized with a tissue disruptor. After centrifugation, the supernatant was collected and the protein concentration determined with a DC™ Protein Assay Kit II (Bio-Rad Laboratories, Hercules, CA, USA). 20–70 µg of protein was separated on a 3–8% gradient Tris-Acetate gel or an 8% acrylamide gel. Gel proteins were transferred by the iBlot2 system (Thermo Fisher Scientific, Waltham, MA, USA) for Tris-Acetate gels to a nitrocellulose membrane and by wet transfer to a PVDF membrane for the acrylamide gels (Merck, Darmstadt, Germany). Membranes were blocked for one hour in Intercept (TBS) blocking buffer (LI-COR, Lincoln, NE, USA). Immunoblotting was performed with α-DM1 antibody (1:1000, kindly provided by Laura Ranum), 1C2 (clone 5TF1-1C2, 1:250-1:1000, Merck, Darmstadt, Germany), #1874 (clone 3B5H10, Merck, Darmstadt, Germany, 1:1000), TATA-box-binding protein (TBP) (1:250-1:1000, Abcam, Cambridge, UK) or α-tubulin (1:5000, Merck, Darmstadt, Germany) overnight at 4 °C. Appropriate secondary antibodies, anti-rabbit 1:8000 conjugated with IRDye 680RD and anti-mouse 1:8000 conjugated with IRDye 800CW or IRDye 680RD (Thermo Fisher Scientific, Waltham, MA, USA), were used. Band pattern was revealed with an Odyssey Imager (LI-QOR, Lincoln, NE, USA). For favoring the detection of bigger proteins, one of the membranes was stripped with a mild stripping buffer (20 mM glycine, 3 mM SDS, 0.1% Tween 20 in deionized water, with a pH of 2.2), after which the membrane was cut below the 50kD marker and re-blocked and probed with #1874.
2.8. Fluorescence In Situ Hybridization (FISH) and Immunofluorescence
Myoblasts, skin fibroblasts and lymphoblastoids grown on coverslips were fixed in 4% paraformaldehyde for 30 min and permeabilized with 0.3% Triton X-100 at 4 °C for 5 min (0.1% Triton X-100 for lymphoblastoids).
For the FISH, coverslips were incubated with 30% formamide in 2× SSC buffer for 30 min at room temperature, followed by an overnight incubation at 37 °C in darkness with a hybridization buffer, containing 0.01 µM Cy3-labelled (CAG)10 or (CTG)10 probe, 30% formamide, 1% dextran sulfate, 0.02% BSA and 2 mM vanadyl in 2× SSC buffer. The following day, the coverslips were washed with 30% formamide in 2× SSC buffer at 45 °C, 1× SSC buffer at 37 °C and 1× PBS at room temperature, and mounted with ProLong Gold Anti-Fade Mountant with DAPI (Thermo Fisher Scientific, Waltham, MA, USA).
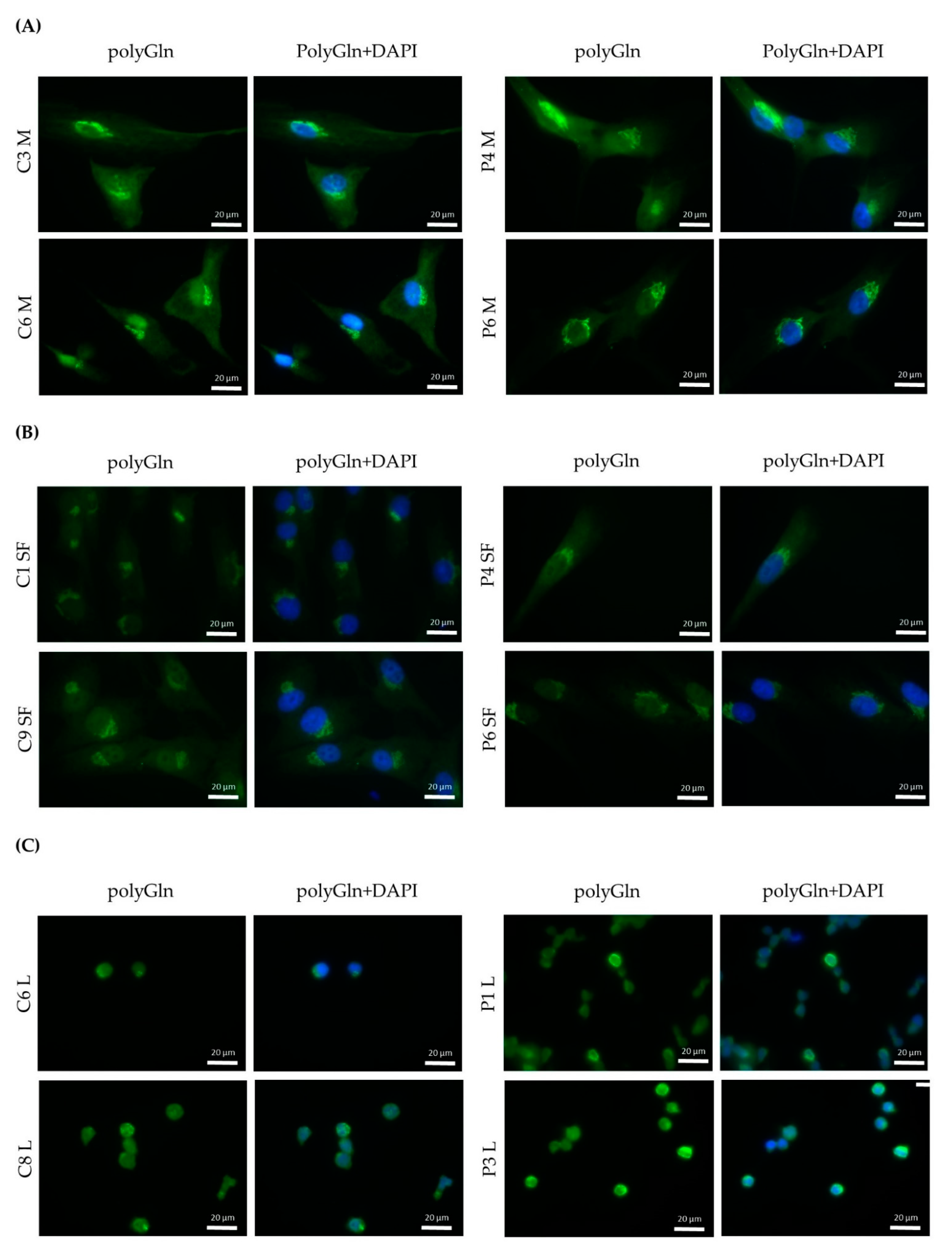
For immunofluorescence, three approaches were followed: a simple immunofluorescence for the detection of RAN-translated polyglutamine protein in all three primary cell cultures, a single-step double immunofluorescence to compare one of the commercial antibodies with the custom antibody used and a two-step double immunofluorescence to assess the co-localization of the found protein with the Golgi apparatus.
For the simple immunofluorescence, coverslips with myoblasts, skin fibroblasts or lymphoblastoids were blocked for 1 h in 5% filtered NGS and incubated overnight at 4 °C with either α-DM1 (1:200, kindly provided by Laura Ranum), 1C2 (Merck, Darmstadt, Germany, ref MAB1574, 1:1000) or #1874 (Merck, Darmstadt, Germany, ref P1874, 1:1000). Next, cells were washed and incubated with goat anti-rabbit conjugated with alexa fluor 488 (Thermo Fisher Scientific, Waltham, MA, USA) for α-DM1 and goat anti-mouse conjugated with alexa fluor 488 (Thermo Fisher Scientific, Waltham, MA, USA) for 1C2 and #1874 for 1 h at room temperature, in darkness. After another round of washes in 1× PBS supplemented with 0.025% Tween-20, coverslips were mounted with ProLong Gold or Diamond Anti-Fade Mountant with DAPI (Thermo Fisher Scientific, Waltham, MA, USA).
For the single-step double immunofluorescence, coverslips were blocked for 1 h in 5% filtered NGS and incubated overnight at 4 °C with two antibodies in the same mix, α-DM1 (1:500 for myoblasts and skin fibroblasts and 1:5000 for lymphoblastoids, kindly provided by Laura Ranum) and 1C2 (1:500, Merck, Darmstadt, Germany). Next, cells were washed and incubated with goat anti-rabbit conjugated with alexa fluor 488 (Thermo Fisher Scientific, Waltham, MA, USA) and goat anti-mouse conjugated with alexa fluor 594 (Thermo Fisher Scientific, Waltham, MA, USA). After another round of washes in 1× PBS, coverslips were mounted with ProLong Gold Anti-Fade Mountant with DAPI (Thermo Fisher Scientific, Waltham, MA, USA).
For the two-step immunofluorescence, coverslips, containing either skin fibroblasts or myoblasts, were blocked for 30 min with the horse blocking solution, containing 5% normal horse serum, 10% normal human serum and 0.02% bovine serum albumin in 1× TBS. Subsequently, the coverslips were incubated overnight at 4 °C with 1C2 (1:200, Merck, Darmstadt, Germany). The following day, the cells were washed three times for 5 min with 1× TBS and incubated at room temperature for 1 h with biotin-labeled horse anti-mouse (1:2000, Thermo Fisher Scientific, Waltham, MA, USA). After another round of washes, the cells were incubated at room temperature for 1 h with streptavidin conjugated with alexa fluor 594 (1:2000, Thermo Fisher Scientific, Waltham, MA, USA). Next, the cells were washed and blocked with the goat blocking solution, containing 5% normal goat serum, 10% normal human serum, 10% Goat anti-horse IgG (Thermo Fisher Scientific, Waltham, MA, USA) and 0.02% bovine serum albumin in 1× TBS. Subsequently, the coverslips were incubated overnight at 4 °C with TGN-38 (1:200, Merck, Darmstadt, Germany). The following day, the cells were washed three times for 5 min with 1× TBS and incubated at room temperature for 1 h with goat anti-mouse conjugated with alexa fluor 488 (1:500, Thermo Fisher Scientific, Waltham, MA, USA). After another round of washes, the cells were mounted with ProLong Gold Anti-Fade Mountant with DAPI (Thermo Fisher Scientific, Waltham, MA, USA).
2.9. Image and Statistical Analysis
To assess DM1-AS expression and protein quantity, intensity measurements were taken using ImageJ software, and normalized against the endogenous controls. Differences in transcript expression were calculated using the Mann–Whitney’s non-parametric U test with Graphpad Prism 9.1.2 software; significance level was set at 0.05.
4. Discussion
We studied the presence of antisense transcription and polyGln RAN protein in three primary cell cultures of patients with DM1, namely myoblast, skin fibroblast and lymphoblastoid cell lines, in order to further elucidate its contribution to DM1 pathology.
The presence of antisense transcription, the origin of RAN-translated polyGln, was validated in our three primary cell cultures with three different primer combinations, and lower levels of expression were observed in DM1 patients compared to controls, which reached significance for LK2/anti-N3 and LK1/anti-1B in myoblasts and LK1/anti-N3 and LK1/anti-1B in lymphoblastoids. Of note, the LK1/anti-N3 primer combination, which encompasses the repeat region, revealed that it primarily detected the smaller transcripts, most likely corresponding to the wild-type allele. Only P6 showed an additional band in myoblasts and skin fibroblasts, which, based on the CTG repeat size, could correspond to the expanded repeat. P6 had the smallest expanded repeat (130 repeats) in our cohort and it could be that the other patients, carrying much larger expanded repeats, could not be detected by this method. This could be a potential explanation for the lower levels of expression seen in patients compared to controls. However, the lower levels were also observed with the other two primer combinations that did not encompass the expanded repeat, making it highly unlikely that the lower levels seen were solely due to the binding of the wild-type allele. DM1-AS expression has only been studied by a handful of other groups. Zu and collaborators showed its presence in a heart sample of a DM1 patient and a healthy control; however, the expression patterns were hard to interpret, since an endogenous control was lacking [
6]. A study by Gudde and collaborators showed a slightly higher expression in muscle biopsies of DM1 patients when studying RNA-seq data from the myotonic dystrophy deep sequencing data repository [
5]. They did, however, mention that globally no obvious differences in read density were observed between DM1 patients and controls. However, when stratified based on inferred MBNL concentration, the most severely affected patients showed a three-fold increase in DM1-AS expression compared to controls, which was in vast contrast to the lower expression levels found in our cohort [
5]. Another study, performed by Brouwer and collaborators, showed that in mouse models with increasing CTG repeat length, the DM1-AS transcription levels remained unchanged [
15]. Unfortunately, disease severity in Gudde and collaborators’ report was based on the inferred MBNL concentration of DM1 patients, and this was not available for our patients, which meant we could not do a similar stratification. We did, however, have extensive knowledge on the clinical phenotype of our DM1 cohort and had patients from three different clinical subtypes included in our studies, namely juvenile, adult and late-onset. Upon revision, a correlation between expression levels and clinical phenotype could not be found, based on for example, age of onset, muscle involvement (muscle weakness, myotonia) or CTG repeat size, the latter in agreement with the report of Brouwer and collaborators [
15]. The sample size of our cohort was rather small for such comparisons or stratification, hindering the analysis. To determine whether DM1-AS transcript expression is linked to disease severity, a bigger cohort is needed.
The presence of DM1-AS transcripts in DM1 cells does not necessarily mean that these transcripts can reach the cytoplasm and be RAN-translated. To further elucidate the localization of these transcripts, cellular fractionation was performed and revealed the presence of DM1-AS transcripts in the cytoplasm of both patients and controls, with a higher percentage in the nuclear fraction. The presence of cytoplasmic DM1-AS transcripts was previously reported by Gudde and collaborators, as they showed the presence of DM1-AS transcripts in the cytoplasmic fraction of myoblasts [
5]. However, in both the fractionated and unfractionated DM1-AS pool, it was unclear whether the transcripts possessed the expanded repeat. The LK1/anti-N3 combination already hinted that not all of the DM1-AS transcripts had the expanded repeat. This was also shown by Gudde and collaborators, who found a heterogeneous pool of DM1-AS transcript sizes, with and without the expanded repeat [
5]. To determine whether the DM1-AS transcripts in DM1 cells included the expanded repeat, we performed a FISH to detect antisense RNA foci and found that antisense RNA foci were present in both the nucleus and cytoplasm of DM1 cells, indicating that these DM1-AS transcripts contained the expanded repeat and RAN translation was therefore, hypothetically, possible. However, the number of antisense RNA foci compared to sense RNA foci was much lower and they were not present in all cells. In addition, the cytoplasmic antisense RNA foci were even rarer, with only approximately 10% of myoblasts and skin fibroblasts containing cytoplasmic RNA foci, indicating that the presence of DM1-AS transcripts with the expanded repeat in the cytoplasm was quite low. Previous reports on antisense RNA foci in DM1 have also shown that the amount of antisense RNA foci in the nucleus was less compared to sense RNA foci [
3,
4]. The polyGln RAN protein was undetectable in all three of our primary cell cultures via two different approaches. Western blots revealed a 42 kD polyGln-containing protein with the two commercial anti-polyGln antibodies, which was most likely TBP. In fact, the original immunogen for the 1C2 antibody was the general transcription factor TBP, which contains a 38-Gln stretch and therefore matches our results. It was shown, however, that although TBP will always show up on Western blots in both patients and controls, the antibody favored the binding of longer stretches of polyGln, such as were present in Huntington’s disease and cerebellar ataxia type 1 and 3 [
16]. Accordingly, a certain subset of lymphoblastoids did show a band that might correspond to the polyGln RAN protein, but the custom α-DM1 antibody showed a range of non-specific bands in both patients and controls and we were therefore unable to determine the origin of this protein with certainty. In addition, it is difficult to know the exact size of the polyGln RAN protein produced by the DM1-AS, as the disease is prone to somatic mosaicism. This means that cells of the same tissue can carry different CTG expansion sizes and it is therefore also possible to have a range of potential sizes for the protein originating from these transcripts [
17,
18]. However, Zu and collaborators showed a band just below 60 kD in a patient carrying 85 CTG·CAGs [
6]. Our patients carried expansions much larger than that, and when estimating the molecular weight based on the CTG expansion size, it was possible to have polyGln RAN proteins in the range we found within the lymphoblastoid cell lines. This will remain, however, hypothetical, as it seems we do not have a proper functional custom DM1 polyGln RAN antibody and no positive control available to test its functionality.
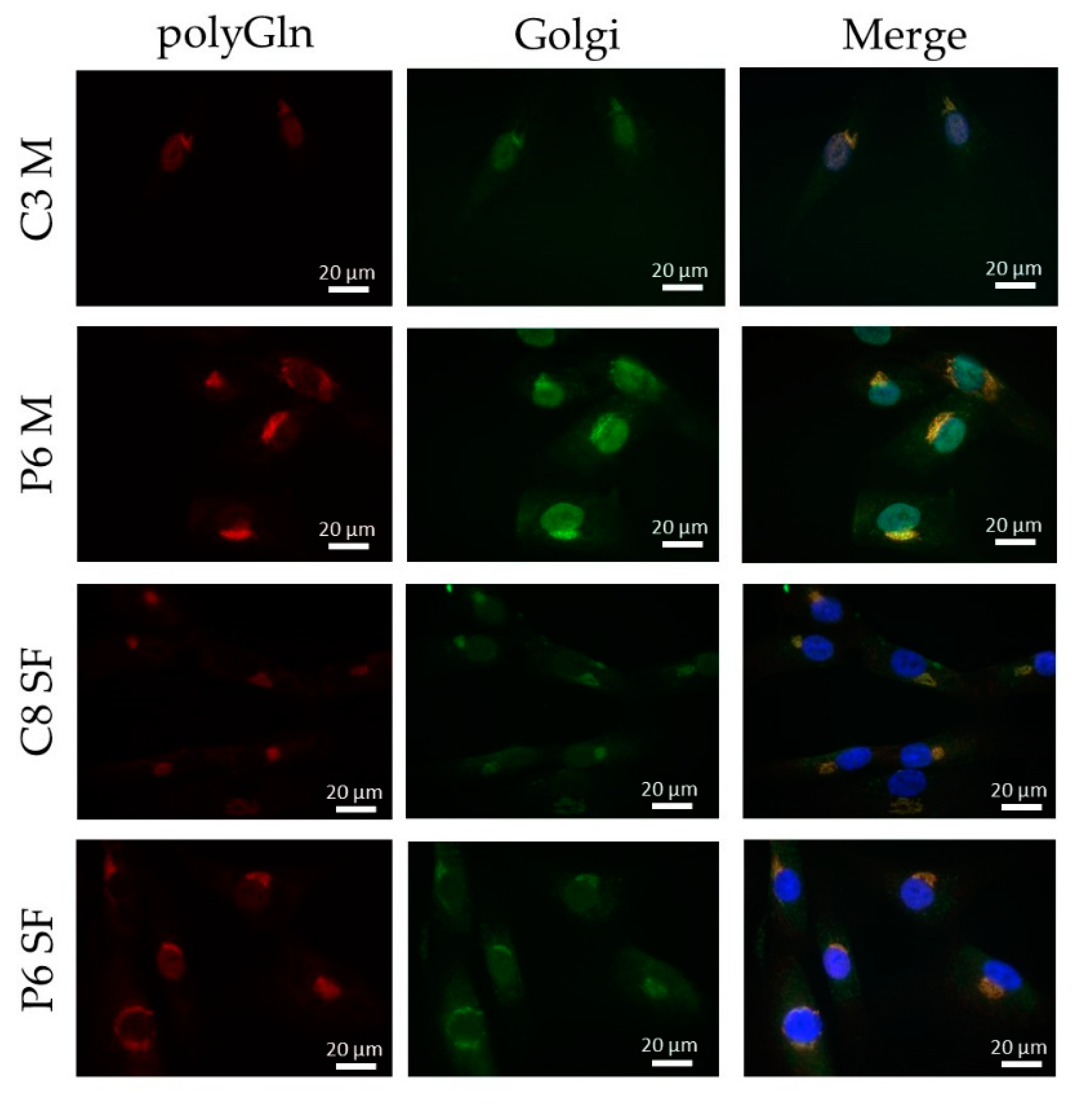
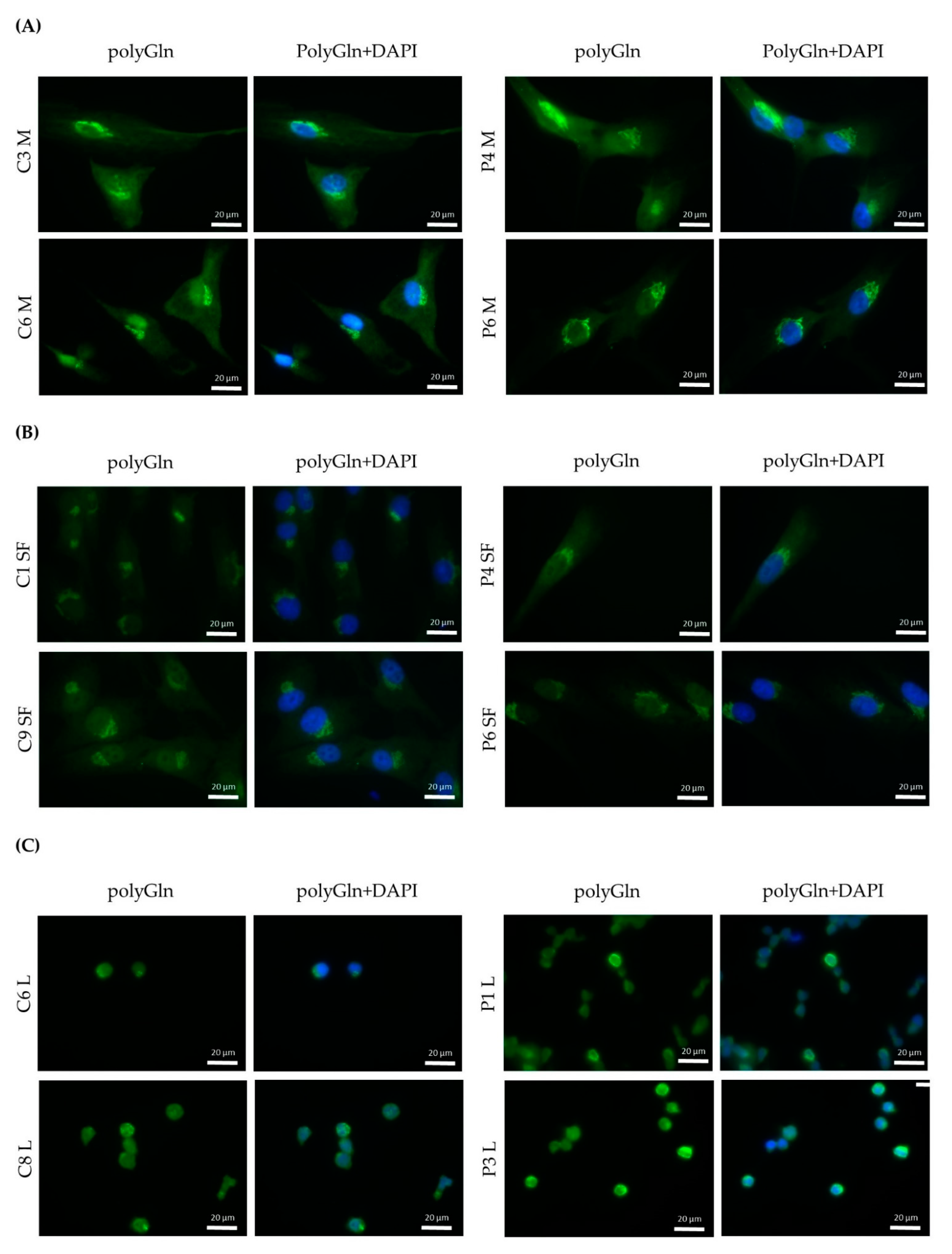
Immunofluorescence revealed a cytoplasmic aggregate surrounding the nucleus in myoblasts and skin fibroblasts with both commercial anti-polyGln antibodies, which was found to be co-localized with the Golgi apparatus. Since the aggregate was visible in both patients and controls and no apparent differences were seen, this might indicate the detection of another endogenous polyGln-containing protein. For example, ataxin-2, the product of the spinocerebellar ataxia type 2 gene, contains 22 glutamines and resides in the Golgi apparatus [
19]. In addition, our immunofluorescence did show staining of the nucleus at high antibody concentrations, which might be due to binding of the transcription factor TBP, also detected by the immunoblots (42 kD band). Taken together, this would mean that both commercial anti-polyGln antibodies bind to several endogenous polyGln-containing proteins, especially at higher antibody concentrations. However, no apparent differences were found between patients and controls across a wide range of concentrations, and the use of α-DM1 antibody did not reveal these similar aggregates. This is in vast contrast to the results previously reported by Zu and collaborators, as they found nuclear polyGln RAN protein aggregates at low frequencies in a DM1 patient’s myoblasts and skeletal muscle (
n = 1) and at higher frequencies in leukocytes from peripheral blood (
n = 1) [
6]. The 1C2 antibody was used in their experiments to validate the specificity of their custom α-DM1 antibody. Although one of the cell types we used was the same as theirs, i.e., myoblasts, neither antibody was able to find the polyGln RAN protein in our myoblast cell lines, nor in the other two primary cell cultures. In fact, although both types of antibodies showed a protein of approximately 42 kD, our simultaneous staining showed that it was not the same protein, indicating that the antibodies were not able to recognize the same proteins. This was surprising, as the commercial antibody was used to validate the custom antibody in the paper of Zu and collaborators [
6]. Our DM1-AS results suggested that the presence of DM1-AS transcripts containing the expanded repeat in the cytoplasm of DM1 cells is quite a rare occurrence. This highly affects the chance of producing polyGln RAN proteins. In addition, polyGln-containing proteins are very common in healthy subjects. Taking these two notions together, it might be plausible that with current techniques, sensitivity is too low to detect such low quantities of the polyGln RAN protein, which in addition is hindered by the presence of other polyGln containing proteins.
Although we were unable to detect polyGln RAN proteins in our DM1 cells, much progress has been made in other repeat expansion disorders displaying RAN translation, which could help in the field of DM1. Nine expansion disorders have been added since the first discovery of RAN translation in SCA8 and DM1: C9orf72 amyotrophic lateral sclerosis/frontotemporal dementia [
20,
21,
22], fragile X tremor/ataxia syndrome [
23], Huntington’s disease (HD) [
24,
25], spinocerebellar ataxia 3 and 31 [
26,
27], Fuchs’ endothelial corneal dystrophy [
28] and myotonic dystrophy type 2 (DM2) [
29]. Of these, SCA8, SCA3 and HD are the three repeat expansion disorders in which the RAN proteins originate from a CAG expansion, and can therefore result in polyGln RAN proteins. Interestingly, in vivo, none of these diseases show polyGln RAN proteins, but instead produce poly-alanine, and for HD additionally poly-serine RAN proteins. It might be interesting to include custom antibodies for the two additional homo-polymeric protein possibilities with regard to DM1. Although the name suggests a close relationship between DM1 and DM2, the underlying expansion in DM2 is a CCTG expansion and therefore results in complex poly-LPAC (sense) or poly-QAGR (antisense) RAN proteins, and is thus not hindered by the presence of endogenous polyGln proteins. The study was performed in autopsy brains, a tissue not yet studied for DM1, which might also be worth exploring.
In conclusion, DM1-AS transcript levels were lower in patients compared to controls and were present in both the nucleus and the cytoplasm of DM1 cells. Only a small portion of the DM1-AS transcripts contained the expanded repeat, substantially lowering the possibility of RAN translation in DM1. The polyGln RAN protein was not present in patient-derived DM1 cells, or was present in such low quantities that it is below the detection limit of the currently available techniques.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}