Abstract
Background: Acute pancreatitis (AP) is a serious, mechanistically not entirely resolved side effect of L-asparaginase-containing treatment for acute lymphoblastic leukemia (ALL). To find new candidate variations for AP, we conducted a genome-wide association study (GWAS). Methods: In all, 1,004,623 single-nucleotide variants (SNVs) were analyzed in 51 pediatric ALL patients with AP (cases) and 1388 patients without AP (controls). Replication used independent patients. Results: The top-ranked SNV (rs4148513) was located within the ABCC4 gene (odds ratio (OR) 84.1; p = 1.04 × 10−14). Independent replication of our 20 top SNVs was not supportive of initial results, partly because rare variants were neither present in cases nor present in controls. However, results of combined analysis (GWAS and replication cohorts) remained significant (e.g., rs4148513; OR = 47.2; p = 7.31 × 10−9). Subsequently, we sequenced the entire ABCC4 gene and its close relative, the cystic fibrosis associated CFTR gene, a strong AP candidate gene, in 48 cases and 47 controls. Six AP-associated variants in ABCC4 and one variant in CFTR were detected. Replication confirmed the six ABCC4 variants but not the CFTR variant. Conclusions: Genetic variation within the ABCC4 gene was associated with AP during the treatment of ALL. No association of AP with CFTR was observed. Larger international studies are necessary to more conclusively assess the risk of rare clinical phenotypes.
Keywords:
acute lymphoblastic leukemia; L-asparaginase; acute pancreatitis; polymorphism; SNV; ABCC4; CFTR 1. Introduction
Acute lymphoblastic leukemia (ALL) is the most common pediatric malignancy and represents approximately 25% of cancers and 80% of all leukemias diagnosed in children and adolescents [1,2]. Contemporary treatment extends over a period of 2 to 3 years and usually consists of combination chemotherapy, which is substituted in small proportions of patients by cranial irradiation or allogeneic hematopoietic stem cell transplantation [3,4]. Timely application of therapy is important to secure optimal treatment effect and outcome but is often compromised by undesired side effects leading to treatment interruptions. Early severe side effects related to the treatment of ALL encompass a variety of specific complications, such as bacterial, viral, and fungal infections; hemostaseological problems; and side effects that can be attributed to specific drugs [5]. Examples of drug-specific toxicities observed during the treatment of ALL are methotrexate-related encephalopathy, steroid-treatment-related avascular bone necrosis, topoisomerase-II-associated secondary acute myeloid leukemia, and acute pancreatitis (AP) developing in the context of L-asparaginase (L-asp) application [6,7,8,9,10].
The mechanism of action of L-asp is the depletion of the extracellular amino acid asparagine by the hydrolysis of asparagine to aspartic acid and ammonia. The depletion results in the inhibition of protein synthesis by malignant cells, such as lymphoblasts, leading to cell death due to the inability to synthesize endogenous asparagine. L-asp used for the treatment of ALL is derived from either Escherichia coli (E. coli) (native or PEGylated L-asp) or Erwinia chrysanthemi [7,8,11], both being associated with AP. The mechanism of AP in association with L-asp is poorly understood. Although L-asp is believed to be the main reason for developing AP, other cytotoxic chemotherapeutics, including 6-mercaptopurine, glucocorticoids, and cytarabine, have been associated with AP, as well [12,13,14,15]. Suggested published risk factors for developing AP associated with L-asp treatment include, for example, higher age at diagnosis, acute hypertriglyceridemia, and genetic polymorphisms [11,16,17,18]. Support for an underlying genetic predisposition comes from the observation that a few applications of L-asp are sufficient to initiate AP and that there is a high probability of recurrence after re-exposure to L-asp [11].
So far, genetic linkage and candidate gene studies have identified several genes (e.g., PRSS1, PRSS2, SPINK1, CTRC, CASR, and CFTR) that could be associated with chronic, hereditary, and hyperlipidemic pancreatitis. Until recently, no specific loci associated with AP had been identified [11,16,19]. However, meanwhile, genome-wide association studies (GWAS) have identified single-nucleotide variants in the genes CPA2, ULK2, and PRSS1 as being associated with L-asp-associated AP in pediatric ALL [20,21,22]. Here, we present our results from a GWAS on the etiology of AP in childhood ALL by comparing 51 patients with AP to 1388 control patients without symptoms of AP.
2. Materials and Methods
2.1. Study Individuals
Patients included in this study were 1 to 18 years of age and enrolled in the European AIEOP-BFM ALL 2000 multicenter clinical trial on the treatment of pediatric ALL conducted in Austria, Germany, Italy, and Switzerland [23,24]. Diagnostics and treatment in AIEOP-BFM ALL 2000 have been described previously [23,24,25,26,27]. Briefly, the AIEOP-BFM ALL 2000 patients were stratified into three branches (standard, intermediate, and high risk). Risk group stratification included minimal residual disease (MRD) analysis and required two MRD targets with sensitivities of ≤10−4. Standard-risk patients were MRD-negative on treatment days 33 (TP1) and 78 (TP2) and had no high-risk criteria. High-risk patients had residual disease (≥10−3) at TP2. MRD-intermediate-risk patients had positive MRD detection at either one or both time points but at a level of <10−3 at TP2. Although MRD analysis was the main stratification criterion in AIEOP-BFM ALL 2000, established high-risk parameters were also retained: patients with a poor response to prednisone or ≥5% leukemic blasts in the bone marrow on day 33 or positivity for a t(9;22) or t(4;11) or their molecular equivalents (BCR-ABL1 or MLL-AF4 gene fusions) were stratified into the high-risk group independent of their MRD results. Treatment details of AIEOP-BFM ALL 2000 are given in Table S1.
Diagnosis of AP was based on the presence of two of the following three clinical symptoms [28]: (1) abdominal pain consistent with acute pancreatitis (acute onset of a persistent, severe, epigastric pain often radiating to the back), (2) serum lipase activity (or amylase activity) at least three times greater than the upper limit of normal, and (3) characteristic findings of AP on abdominal computed tomography, magnetic resonance imaging, or transabdominal ultrasonography or surgical findings consistent with AP.
2.2. DNA Isolation
During the course of treatment, bone marrow and/or blood samples were collected for remission evaluation at defined time points. Morphologically leukemia-cell-free samples with MRD levels of ≤10−3 were selected from these time points and used for DNA isolation using previously described standard techniques [26,27,29]. DNA yielded by this procedure was regarded as a germline DNA surrogate.
2.3. Single-Nucleotide Variant (SNV) Genotyping for Genome-Wide Screening
The GWAS was conducted in 54 childhood ALL patients with AP (cases) and 1435 patients without AP (controls). DNA was genotyped using Human1M-Duo BeadChips (Illumina, San Diego, CA, USA) containing 1,048,711 SNV markers. To avoid false positive data, 44,088 SNVs were excluded due to poor call rate (CR) (<95%) and/or deviation from Hardy–Weinberg equilibrium in the controls (p > 0.001). Furthermore, 37 patients (cases/controls) were excluded due to poor genotyping (CR < 95%) and cryptical relationship (IBS-distance > 0.8). Additionally, a multidimensional scaling analysis (MDS) identified 13 patients (cases/controls) with a non-European background. These subjects were also excluded from the study (Figure S1). The quality control finally resulted in a cohort size of 51 cases and 1388 controls.
Two methodological approaches were used to identify candidate SNVs for AP in this GWAS. First, only SNVs with a p-value smaller than 1 × 10−7, a minimum of one genotyping call in each group of cases and controls, and no restriction of minor allele frequency (MAF) were included. The second approach differed from the first by only including those SNVs with a MAF of more than 0.5%. Minimal evidence of an overall inflation of the test statistics due to population stratification with a moderate genomic inflation factor (approach 1: λ = 1.09; approach 2: λ = 1.10) was found (Figure S2).
To confirm the top 20 SNVs from the GWAS, a replication analysis was conducted in an independent patient set of 54 AP cases (selected from both ALL BFM 2000 and AIEOP BFM ALL 2009 study cohorts) and 225 controls (patients with no history of AP from the ALL BFM 2000 cohort). Candidate SNVs were genotyped using the SNVlex multiplex and TaqMan technology (Applied Biosystems, Foster City, CA, USA).
2.4. Gene Sequencing
To fine-map ATP-binding cassette sub-family C member 4 (ABCC4); 281,605 base pairs) and to evaluate the ABCC4-related cystic fibrosis conductance regulator (CFTR); 188,702 base pairs) gene as a candidate for AP predisposition, the two genes were completely sequenced in a cohort of 48 cases and 47 controls selected from the above-described GWAS and replication cohorts depending on the availability of sufficient amounts of non-malignant DNA. Next-generation sequencing (NGS) was conducted on a HiSeq2000 platform (Illumina) using the HaloPlex Illumina 100 kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s recommendations. The reads were mapped against the human reference genome build hg19 using BWA [30], sorted, converted to bam format, and indexed with SAMtools [31]. Local realignment around InDels and base quality score recalibration were performed with the GATK [32] according to their best practice recommendations, followed by variant calling and variant quality score recalibration. Data were analyzed using the program Integrative Genomic Viewer version 2.3.25 (www.broadinstitute.org/igv/ (accessed on 20 October 2021)) [33,34]. For identification of potential candidate SNVs, regions with a poor sequencing rate (<90%) were excluded. Follow-up SNVs in independent patients from ALL BFM 2000 and AIEOP BFM ALL 2009 with available non-malignant DNA (most of which were part of the initial GWAS and replication cohorts) were analyzed by a Sanger sequencing using an automated fluorescent sequencer (Applied Biosystems 3730xl DNA Analyzer). All data referring to chromosomal positions were based on GRCh37/hg19 assembly.
2.5. Plotting
Regional association plots were created for the GWAS SNVs using a modified version of deBakker’s R script (Figure 1, Figures S3 and S4) by using GWAS SNVs as well as imputed SNVs (if possible). The imputation was done using gPLINK version 2.050 in combination with PLINK v1.07 (www.pngu.mgh.harvard.edu/purcell/plink/ (accessed on 20 October 2021)) [35]. For this purpose, genotypes of autosomal SNVs based on data of 1000 genomes were used. As an input for imputation, only SNVs from the GWAS that passed the above-mentioned quality controls were included.
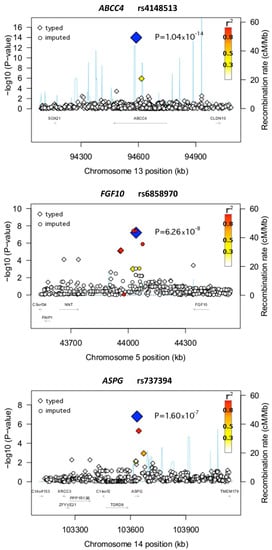
Figure 1.
Regional plots of the loci ABCC4, FGF10, and ASPG. Plots of the negative decadic logarithm of the combined p-values obtained in the GWAS are shown. The data were imputed with CEU haplotypes generated by the 1000 Genomes Project (August 2010 release) as a reference. A window of ±500 kb around the lead SNVs (blue solid diamonds) is indicated. The magnitude of the linkage disequilibrium with the central SNV measured by r2 is reflected by the color of each SNV symbol (color coding: see the upper-right corner of the plot). Recombination activity (in centimorgans (cM) per Mb) is depicted by a blue line. Positions are given as NCBI’s build coordinates.
2.6. Statistical Analyses
Associations between patient characteristics were evaluated using Fisher’s exact or χ2-tests. The GWAS was assessed using gPLINK. Associations of variations detected by NGS and the replication analyses in the respective cohorts used unconditional logistic regression analysis or Fisher’s exact test. Quality control and identity-by-state analysis of the GWAS data was evaluated by gPLINK and R statistics version 2.15.1 (www.r-project.org (accessed on 20 October 2021)). To estimate the European ancestry of the GWAS cohort, the multidimensional scaling analysis was evaluated using R statistics with HapMap CEU, YRI, and JRT/CHB cohorts as reference ancestral populations. Computations were performed using IBM SPSS statistics (IBM Corp., Version 21.0.0, Armonk, NY, USA) and R statistics.
3. Results
3.1. GWAS-Based Identification and Replication of Genomic SNVs Associated with AP
In our GWAS cohort, the incidence of AP was 3.6%, which was in the range of the reported incidence of childhood-ALL-therapy-associated pancreatitis (0.7–18%) [6,7,10]. One previously described clinical risk factor associated with AP development during the treatment of childhood ALL is higher patient age, which was also observed in our analysis (Table 1) [7,11,17,18]. No significant associations of AP with the treatment risk group were detected (Table 1).

Table 1.
Clinical characteristics of 1439 patients with ALL from trial AIEOP-BFM ALL 2000 (GWAS cohort) according to the acute pancreatitis (AP) status.
As mentioned above, our study used two methodological approaches to detect potential associations for developing AP. In the first approach, six SNVs fulfilled the predefined criteria for significance (Table 1; Figure 1 and Figure S3). An intronic SNV in the ABCC4 gene (rs4148513) demonstrated the strongest association with AP (p = 1.04 × 10−14; OR = 84.09) (Figure 1; Table 2). Of interest, besides rs4148513, another SNV in ABCC4 was independently and highly associated with AP in the GWAS (rs4148500; p = 7.23 × 10−6) (Table 2). Other genes with significant associations in the first GWAS approach included SEMA3D, C15orf41, COG5, ST7, and UPF1.
Table 2.
Top SNVs associated with AP identified by genome-wide association analysis and replicated by Sanger sequencing: approach 1.
In the second approach, 13 highly significant SNVs were identified (Table 3; Figure 1 and Figure S4). The SNV with the strongest association (rs6858970) was detected close to the fibroblast growth factor 10 (FGF10) gene (p = 6.26 × 10−8; OR = 8.61) (Figure 1; Table 3). Another highly associated SNV in this approach was rs737394 (p = 1.59 × 10−7; OR = 3.19), an SNV located on an intronic region of the asparaginase homolog (S. cerevisiae) (ASPG) gene (Figure 1; Table 3)). Other SNVs identified by the second approach were located on or in the vicinity of genes associated with mechanisms and pathways such as cell growth, cell differentiation, and cell death (Table 3).
Table 3.
Top SNVs associated with AP identified by genome-wide association analysis and replicated by Sanger sequencing: approach 2.
In total, 20 SNVs were detected by our two GWAS approaches. Six of them were found to be located in intergenic regions, whereas 14 SNVs were discovered directly on a gene (Table 2, Table 3, and Table S2). All of these 20 SNVs were genotyped in additional independent patient samples (54 cases with AP and 225 controls without AP). However, none of the 20 SNVs yielded significant results in replication experiments (Table 2 and Table 3). The most significant SNV of the GWAS from the first approach (rs4148513) was neither detected in an additional case nor detected in an additional control individual.
3.2. SNVs from Candidate Gene Studies and GWAS
We investigated all SNVs present on our array platform that were located on or in the vicinity of those genes previously associated with changes in susceptibility to pancreatitis, including CFTR, CTRC, PRSS2, SPINK1, CASR, and the recently reported variants in AP-associated carboxypeptidase A2-encoding gene CPA2, in unc-51 like autophagy activating kinase 2-encoding gene ULK2, and in serine protease 1-encoding gene PRSS1 [20,21,22] but could not replicate any of the previously described significant associations (Table S3).
3.3. Fine-Mapping of Potential AP-Associated Variants by Sequencing the ABCC4 and CFTR Genes
Out of the 20 SNVs, the 2 with the highest significance in the GWAS approach were located on the ABCC4 gene. ABCC4 is a member of the superfamily of ATP-binding cassette (ABC) transporters, which also includes CFTR. Since patients with cystic fibrosis are prone to developing pancreatic problems, including pancreatitis, CFTR is a relevant candidate gene for pancreatitis in non-CF patients. The relationship to ABCC4 as well as the candidate gene status of CFTR for AP led us to include both genes, ABCC4 and CFTR, in a targeted NGS-based sequencing approach applied to 48 cases with AP and 47 controls without AP. In total, seven SNVs were significantly associated with AP according to the significance criteria mentioned above (see Section 2; Table 4). All NGS-based SNVs with significant associations were confirmed by Sanger sequencing. Six of the seven variants were located on the ABCC4 gene and only one on the CFTR gene. One of the most significantly associated variants was the insertion rs34839857 (p = 1.0 × 10−2) in ABCC4, with 21 alleles present in the case group and 7 in controls. Results by genotype for the seven SNVs are given in Table S4 (Table S5 demonstrates the below-described replication and Table S6 the joint analysis of both cohorts used in fine-mapping analysis). Linkage disequilibrium (LD) analyses are demonstrated in Tables S7 and S8. The top candidate SNV from the GWAS showed no LD with any of the newly NGS identified ABCC4 SNVs.
Table 4.
Top ABCC4 and CFTR SNVs associated with AP identified through next-generation sequencing and replication analysis by Sanger sequencing.
4. Discussion
It is assumed that chemotherapeutic drugs (mainly L-asp) are the main trigger for AP in the therapeutic course of childhood ALL [6,7,8,9,10,11,13,36]. In our analyses, we were able to confirm higher age as a previously published risk factor for developing AP associated with L-asp treatment (Table 1) [7,11,17,18]. In contrast, we did not detect significant associations of AP with the treatment risk group. Several studies have analyzed the effect of risk stratification for ALL treatment as a risk factor for AP with controversial results [36,37,38]. The observed positive associations are most likely explained by higher doses of L-asp being applied in high-risk patients [36,37]. In comparison to standard- and intermediate-risk patients, our high-risk patients also received higher cumulative doses of L-asp (Table S1). Despite higher frequencies of AP in high-risk patients observed in our study, no significant differences could be detected. This is most likely due to a lack of power in our relatively small sample set.
In addition to demographic or clinical risk factors, there is evidence of genetic factors contributing to the pathophysiology of AP as a severe treatment complication. In our first GWAS approach with no restrictions on MAF, the strongest association was observed for an SNV located on the ABCC4 gene. ABCC4 belongs to the ABC transporter superfamily, which mediates the efflux of drugs and plays an important role in the development of drug resistance. ABCC4 itself is known to mediate the transport of different chemotherapeutic drugs out of the cell (e.g., 6-mercaptopurine and methotrexate) [39,40,41,42]. Therefore, variability in ABCC4 activity may affect pharmacokinetics of ABCC4 transport substrates and consequently modulate drug effects. Of importance in the context of our findings, ABCC4 is highly expressed in the pancreas [39,43]. In addition, in a recent study using a rat model to study AP, Ventimiglia and colleagues described a protective role of atrial natriuretic factor (ANF) mediated by cAMP extrusion through ABCC4 and suggested that the regulation of ABCC4 by ANF could be relevant to maintaining pancreatic acinar cell homeostasis [44].
The top-ranked SNV in our second GWAS approach, which included SNVs with a MAF of more than 0.5%, was located in the vicinity of FGF10, a gene belonging to the fibroblast growth factor family. Members of this group take part in the regulation of cell growth and cell differentiation. In addition, the FGF-family is suspected to be involved in pancreatic diseases such as pancreatic cancer, chronic pancreatitis, and acute pancreatitis [45,46,47]. The FGF10 gene itself is required for the normal development of the pancreas [47,48]. In a publication of Ishiwata et al., the authors proposed that FGF10 together with FGF7 may contribute to the regeneration and differentiation of acinar cells and the angiogenesis of AP [49]. However, despite FGF10 being a plausible candidate for a role in the pathophysiology of AP, our replication analysis did not support the initial findings.
As mentioned above, one of the most serious adverse events of L-asp treatment is AP. L-asp catalyzes the hydrolysis of asparagine into aspartate and ammonia. The human genome encodes at least three enzymes that can catalyze this reaction, asparaginase homolog (S. cerevisiae) (ASPG), aspartylglucosaminidase (AGA), and asparaginase like 1 (ASRGL1) [50]. Of interest, one SNV selected for further follow-up after our initial GWAS screen was located on the gene ASPG. This little studied gene has sequence similarity at the N-terminal domain with the E. coli types I and II asparaginase [51,52]. It has also been shown that HEK293 cells exhibit asparaginase activity when they are transfected with the cDNA of ASPG [53]. Although purely hypothetical, this initial finding, which did not hold in replication analysis, may justify some follow-up investigations of ASPG activity in the context of AP development.
We investigated all SNVs present on the GWAS SNV array that were located on or in the vicinity of the genes known to be associated with changes in susceptibility to pancreatitis, including CFTR, CTRC, PRSS2, SPINK1, and CASR, but did not find any significant association. Therefore, these previously described candidate genes for chronic pancreatitis may not play distinct roles in AP. However, we also failed to detect any association with CPA2, ULK2, and PSSR1, three recently reported AP-associated genes [20,21,22] (Table S3). Regarding this, our analyses may have been hampered by suboptimal SNV coverage of these candidates on our array (e.g., CFTR: 140 SNVs in or ±50 kb up and downstream of the gene) and the fact that hardly any of the few well-known SNVs previously associated with pancreatitis, including the top CPA2 SNV, were actually present on our platform. LD information on this CPA2 variant (rs199695765) could not be obtained, probably due to its rareness, so there can be no conclusions drawn from CPA2 variants present on O1MQR. However, one of the recently published PRSS1 variants was genotyped, showing no association to the AP phenotype (as shown in Table S3). The other published variant is not present on O1MQR but in perfect LD with the first one. The previously published ULK2 variant rs281366 was also not genotyped on O1MQR but Table S3 lists several SNVs, for example rs205111, rs9895806, and rs9914674, that are highly linked to the published variant. In summary, our GWAS setting could not replicate the associations of rare or common SNVs to the phenotype of AP that was identified in previously published GWA studies.
Replication of the 20 top candidate SNVs from our GWAS was, unfortunately, not successful. The reasons are manifold, including the fact that our GWAS included rare variants with a low MAF. GWAS analyses often begin by discarding all genotypes for SNVs with a MAF of less than 10%, which results in an enormous loss of data. Low-MAF SNVs are associated with technical and statistical problems, such as lower genotyping rates and inflated false-positive results [53]. The decision to include rare alleles in our analyses was based on the hypothesis that AP is a rare clinical phenotype and may be associated with rare SNVs. From a methodological perspective on GWAS analyses, our practical approach is supported by investigations demonstrating nominally significant results occurring significantly less often than expected for low-MAF SNVs, resulting in a conservative bias [54,55]. However, despite positive arguments to include SNVs of low MAF, our replication cohorts may have been virtually too small to reliably detect enough cases carrying rare variants. For example, the highest-ranked SNV in our GWAS (rs4148513) occurred in three cases and one control only and was not detected in a single individual of the entire validation cohort. Nevertheless, combined data from our GWAS and the validation cohorts still demonstrated strong associations of initially identified candidate variations with AP, supporting the assumption that the initially detected SNVs might truly play a role in the development of AP.
Lending additional support to our findings from initial experiments, we conducted fine-mapping of ABCC4 by sequencing the entire gene. ABCC4 was chosen because of our GWAS findings and its simultaneous candidate status based on biological function (see above). As a second candidate gene for pancreatitis, CFTR was chosen for sequencing [56,57,58]. CFTR also belongs to the ABC transporter superfamily and plays a role in water and salt transport at the plasma membrane of epithelial cells. Mutations in CFTR lead to cystic fibrosis (CF) commonly affecting the lungs, liver, intestine, and pancreas [59]. Moreover, variants within CFTR associated with pancreatitis were found in patients without additional symptoms of CF [19,60]. CFTR as a genetic risk factor for AP and chronic pancreatitis was linked with trypsin activation and survival in pancreatitis patients [60,61]. Of particular interest, in replication analysis of seven candidate SNVs in ABCC4 and CFTR detected through NGS, all six ABCC4 variants demonstrated similar effects regarding point estimates while the CFTR SNV did not. Its consistent behavior in our different analytical approaches, including genotype analysis, implies that ABCC4 might truly be associated with AP.
To conclude, for the first time, we were able to associate germline genetic variation in ABCC4 with the risk of AP during treatment for childhood ALL. Our results demonstrate that ABCC4 was consistently related to AP in GWAS as well as in fine-mapping analyses by NGS, supporting a true role of ABCC4 in the development of AP. However, our study on a rare phenotype in a rare disease also clearly demonstrates that international joint efforts are needed to more reliably assess genetic risk factors for AP and other rare toxicities observed in childhood ALL by using larger pooled patient cohorts.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jcm10214815/s1: Table S1. Treatment details of protocol AIEOP-BFM ALL 2000. Table S2. Additional information on top AP-associated SNV from GWAS and fine-mapping (NGS) analyses. Table S3. SNV within the CPA2, PRSS1 and ULK2 genes previously identified by GWAS analyses and their association with AP in our cohort. Table S4. Genotype frequencies and association with risk of AP for SNV derived from fine-mapping by NGS analyses in the initial cohort. Table S5. Genotype frequencies and association with risk of AP for SNV derived from fine-mapping by NGS analyses in the replication cohort. Table S6: Genotype frequencies and association with risk of AP for SNV derived from fine-mapping by NGS analyses in the combined cohort (initial and replication). Table S7. Linkage disequilibrium of top ABCC4 SNV from GWAS and fine-mapping analyses. Table S8. Linkage disequilibrium of CFTR SNV. Figure S1. Identification of individuals in the GWA scan of non-European ancestry. Figure S2. Quantile-quantile (Q-Q) plots showing observed vs. expected distribution of p-values for association of the GWAS-SNVs with Acute Pancreatitis (AP). Figure S3. Regional plots of the loci of the SNVs identified within the first approach of the GWAS of AP patients and controls (with the exception of the SNV located on the gene ABCC4, which is represented in Figure 1 in the article). Figure S4. Regional plots of the loci of the SNVs identified within the second approach of the GWAS of AP patients and controls (with the exception of the SNVs located on the genes FGF10 and ASPG, which are represented in Figure 1 in the article).
Author Contributions
C.R.B.: Data provision and the final approval of the manuscript. T.B.: Collection and assembly of data, data analysis and interpretation, laboratory analyses, manuscript writing, and the final approval of the manuscript. A.B.: Collection and assembly of data and the final approval of the manuscript. G.C.: Collection and assembly of data and the final approval of the manuscript. E.E.: Data analysis and interpretation and the final approval of the manuscript. M.F.: Data analysis and interpretation and the final approval of the manuscript. A.F.: Data interpretation and the final approval of the manuscript. L.H.: Data analysis and the final approval of the manuscript. R.S.H.: Data analysis and interpretation and the final approval of the manuscript. S.V.J.: Data analysis and the final approval of the manuscript. N.K.: Collection and assembly of data and the final approval of the manuscript. R.K.: Collection and assembly of data and the final approval of the manuscript. C.P.K.: Data interpretation and the final approval of the manuscript. A.M.: Collection and assembly of data and the final approval of the manuscript. B.-S.L.: Laboratory analysis, data interpretation, and the final approval of the manuscript. M.S.(Martin Schrappe): Collection and assembly of data and the final approval of the manuscript. P.S.: Data analysis, manuscript writing, and the final approval of the manuscript. M.S.(Martin Stanulla): Conception and design of study, collection, assembly and interpretation of data, supervision of research, manuscript writing, and the final approval of the manuscript. M.Z.: Data analysis, data interpretation, and the final approval of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The project was funded by Madeleine Schickedanz-Kinderkrebsstiftung, Deutsche José Carreras Leukämie-Stiftung (DJCLS R 15/04), TRANSCALL2, ERA-NET TRANSCAN/European Commission under the 7th Framework Programme (FP7), and Verein für krebskranke Kinder Hannover e.V.
Institutional Review Board Statement
The study was approved by the Institutional Review Boards of Hannover Medical School, Hannover (Nr. 2522), and the Medical Faculty of the Christian Albrechts University, Kiel, Germany (A 177/09), and conducted according to the guidelines of the Declaration of Helsinki.
Informed Consent Statement
Informed consent was obtained from the legal representatives of all subjects involved in the study.
Data Availability Statement
Datasets of the current study are not publicly available but are available from the corresponding author on reasonable request.
Acknowledgments
We thank all participating patients, their families, and all personnel involved in AIEOP-BFM ALL-BFM 2000.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef]
- Redaelli, A.; Laskin, B.L.; Stephens, J.M.; Botteman, M.F.; Pashos, C.L. A systematic literature review of the clinical and epidemiological burden of acute lymphoblastic leukaemia (ALL). Eur. J. Cancer Care 2005, 14, 53–62. [Google Scholar] [CrossRef]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Stanulla, M.; Schrappe, M. Treatment of childhood acute lymphoblastic leukemia. Semin. Hematol. 2009, 46, 52–63. [Google Scholar] [CrossRef]
- Vagace, J.M.; Gervasini, G. Chemotherapy toxicity in patients with acute leukemia. In Acute Leukemia—The Scientists Perspective and Challenge; Antica, M., Ed.; InTechOpen: Rijeka, Croatia, 2011; pp. 391–414. [Google Scholar]
- Alvarez, O.A.; Zimmerman, G. Pegaspargase-induced pancreatitis. Med. Pediatr. Oncol. 2000, 34, 200–205. [Google Scholar] [CrossRef]
- Knoderer, H.M.; Robarge, J.; Flockhart, D.A. Predicting asparaginase-associated pancreatitis. Pediatr. Blood Cancer 2007, 49, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Laugel, V.; Escande, B.; Entz-Werle, N.; Mazingue, F.; Ferster, A.; Bertrand, Y.; Missud, F.; Lutz, P. [Severe acute pancreatitis in children receiving asparaginase: Multicenter retrospective study]. Arch. Pediatr. 2005, 12, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sadoff, J.; Hwang, S.; Rosenfeld, D.; Ettinger, L.; Spigland, N. Surgical pancreatic complications induced by L-asparaginase. J. Pediatr. Surg. 1997, 32, 860–863. [Google Scholar] [CrossRef]
- Sahu, S.; Saika, S.; Pai, S.K.; Advani, S.H. L-asparaginase (Leunase) induced pancreatitis in childhood acute lymphoblastic leukemia. Pediatr. Hematol. Oncol. 1998, 15, 533–538. [Google Scholar] [CrossRef]
- Raja, R.A.; Schmiegelow, K.; Frandsen, T.L. Asparaginase-associated pancreatitis in children. Br. J. Haematol. 2012, 159, 18–27. [Google Scholar] [CrossRef]
- Altman, A.J.; Dinndorf, P.; Quinn, J.J. Acute pancreatitis in association with cytosine arabinoside therapy. Cancer 1982, 49, 1384–1386. [Google Scholar] [CrossRef] [Green Version]
- Halalsheh, H.; Bazzeh, F.; Alkayed, K.; Salami, K.; Madanat, F. 6-Mercaptopurine-induced recurrent acute pancreatitis in children with acute lymphoblastic leukemia/lymphoma. J. Pediatr. Hematol. Oncol. 2013, 35, 470–472. [Google Scholar] [CrossRef]
- Riemenschneider, T.A.; Wilson, J.F.; Vernier, R.L. Glucocorticoid-induced pancreatitis in children. Pediatrics 1968, 41, 428–437. [Google Scholar]
- Varma, M.R.; Mathew, S.; Krishnadas, D.; Vinayakumar, K.R. Imatinib-induced pancreatitis. Indian J. Pharmacol. 2010, 42, 50–52. [Google Scholar] [CrossRef] [Green Version]
- Whitcomb, D.C. Genetic aspects of pancreatitis. Annu. Rev. Med. 2010, 61, 413–424. [Google Scholar] [CrossRef]
- Kearney, S.L.; Dahlberg, S.E.; Levy, D.E.; Voss, S.D.; Sallan, S.E.; Silverman, L.B. Clinical course and outcome in children with acute lymphoblastic leukemia and asparaginase-associated pancreatitis. Pediatr. Blood Cancer 2009, 53, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rank, C.U.; Wolthers, B.O.; Grell, K.; Albertsen, B.K.; Frandsen, T.L.; Overgaard, U.M.; Toft, N.; Nielsen, O.J.; Wehner, P.S.; Harila-Saari, A.; et al. Asparaginase-Associated Pancreatitis in Acute Lymphoblastic Leukemia: Results From the NOPHO ALL2008 Treatment of Patients 1–45 Years of Age. J. Clin. Oncol. 2020, 38, 145–154. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Gonska, T.; Durie, P.R.; Freedman, S.D. Genetic testing in pancreatitis. Gastroenterology 2010, 138, 2202–2206. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, W.; Devidas, M.; Cheng, C.; Pei, D.; Smith, C.; Carroll, W.L.; Raetz, E.A.; Bowman, W.P.; Larsen, E.C.; et al. Clinical and Genetic Risk Factors for Acute Pancreatitis in Patients With Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Wolthers, B.O.; Frandsen, T.L.; Abrahamsson, J.; Albertsen, B.K.; Helt, L.R.; Heyman, M.; Jónsson, Ó.G.; Kõrgvee, L.T.; Lund, B.; Raja, R.A.; et al. Asparaginase-associated pancreatitis: A study on phenotype and genotype in the NOPHO ALL2008 protocol. Leukemia 2017, 31, 325–332. [Google Scholar] [CrossRef]
- Wolthers, B.O.; Frandsen, T.L.; Patel, C.J.; Abaji, R.; Attarbaschi, A.; Barzilai, S.; Colombini, A.; Escherich, G.; Grosjean, M.; Krajinovic, M.; et al. Trypsin-encoding PRSS1-PRSS2 variations influence the risk of asparaginase-associated pancreatitis in children with acute lymphoblastic leukemia: A Ponte di Legno toxicity working group report. Haematologica 2019, 104, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Valsecchi, M.G.; Bartram, C.R.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Parasole, R.; Zimmermann, M.; Dworzak, M.; Buldini, B.; et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: Results of the AIEOP-BFM-ALL 2000 study. Blood 2011, 118, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; Te Kronnie, G.; Bourquin, J.P.; Bornhauser, B.; et al. IKZF1(plus) Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Stanulla, M.; Schaeffeler, E.; Möricke, A.; Buchmann, S.; Zimmermann, M.; Igel, S.; Schmiegelow, K.; Flotho, C.; Hartmann, H.; Illsinger, S.; et al. Hepatic sinusoidal obstruction syndrome and short-term application of 6-thioguanine in pediatric acute lymphoblastic leukemia. Leukemia 2021, 35, 2650–2657. [Google Scholar] [CrossRef]
- Meissner, B.; Bartram, T.; Eckert, C.; Trka, J.; Panzer-Grümayer, R.; Hermanova, I.; Ellinghaus, E.; Franke, A.; Möricke, A.; Schrauder, A.; et al. Frequent and sex-biased deletion of SLX4IP by illegitimate V(D)J-mediated recombination in childhood acute lymphoblastic leukemia. Hum. Mol. Genet. 2014, 23, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S. Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef]
- Ellinghaus, E.; Stanulla, M.; Richter, G.; Ellinghaus, D.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Horstmann, M.; Panzer Grümayer, R.; Cavé, H.; et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 2012, 26, 902–909. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treepongkaruna, S.; Thongpak, N.; Pakakasama, S.; Pienvichit, P.; Sirachainan, N.; Hongeng, S. Acute pancreatitis in children with acute lymphoblastic leukemia after chemotherapy. J. Pediatr. Hematol. Oncol. 2009, 31, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, S.; Dhir, S.; Slack, J.; Iyer, P.; Wade, R.; Clack, R.; Vora, A.; Goulden, N. Incidence and outcome of pancreatitis in children and young adults with acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br. J. Haematol. 2013, 162, 710–713. [Google Scholar] [CrossRef]
- Raja, R.A.; Schmiegelow, K.; Albertsen, B.K.; Prunsild, K.; Zeller, B.; Vaitkeviciene, G.; Abrahamsson, J.; Heyman, M.; Taskinen, M.; Harila-Saari, A. Asparaginase-associated pancreatitis in children with acute lymphoblastic leukaemia in the NOPHO ALL2008 protocol. Br. J. Haematol. 2014, 165, 126–133. [Google Scholar] [CrossRef]
- Borst, P.; de Wolf, C.; van de Wetering, K. Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Arch. 2007, 453, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Lee, K.; Walther, S.; Raftogianis, R.B.; Kuwano, M.; Zeng, H.; Kruh, G.D. Analysis of methotrexate and folate transport by multidrug resistance protein 4 (ABCC4): MRP4 is a component of the methotrexate efflux system. Cancer Res. 2002, 62, 3144–3150. [Google Scholar] [PubMed]
- Janke, D.; Mehralivand, S.; Strand, D.; Gödtel-Armbrust, U.; Habermeier, A.; Gradhand, U.; Fischer, C.; Toliat, M.R.; Fritz, P.; Zanger, U.M. 6-mercaptopurine and 9-(2-phosphonyl-methoxyethyl) adenine (PMEA) transport altered by two missense mutations in the drug transporter gene ABCC4. Hum. Mutat. 2008, 29, 659–669. [Google Scholar] [CrossRef]
- Russel, F.G.; Koenderink, J.B.; Masereeuw, R. Multidrug resistance protein 4 (MRP4/ABCC4): A versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol. Sci. 2008, 29, 200–207. [Google Scholar] [CrossRef]
- König, J.; Hartel, M.; Nies, A.T.; Martignoni, M.E.; Guo, J.; Büchler, M.W.; Friess, H.; Keppler, D. Expression and localization of human multidrug resistance protein (ABCC) family members in pancreatic carcinoma. Int. J. Cancer 2005, 115, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Ventimiglia, M.S.; Najenson, A.C.; Perazzo, J.C.; Carozzo, A.; Vatta, M.S.; Davio, C.A.; Bianciotti, L.G. Blockade of Multidrug Resistance-Associated Proteins Aggravates Acute Pancreatitis and Blunts Atrial Natriuretic Factor's Beneficial Effect in Rats: Role of MRP4 (ABCC4). Mol. Med. 2015, 21, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.; Yokoyama, M.; Ishiwata, T.; Friess, H.; Büchler, M.W.; Malfertheiner, P.; Korc, M. Alteration of fibroblast growth factor and receptor expression after acute pancreatitis in humans. Pancreas 1999, 18, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, M.; Beger, H.G.; Korc, M. Role of fibroblast growth factors and their receptors in pancreatic cancer and chronic pancreatitis. Pancreas 1998, 17, 169–175. [Google Scholar] [CrossRef]
- Nandy, D.; Mukhopadhyay, D. Growth factor mediated signaling in pancreatic pathogenesis. Cancers 2011, 3, 841–871. [Google Scholar] [CrossRef] [Green Version]
- Bhushan, A.; Itoh, N.; Kato, S.; Thiery, J.P.; Czernichow, P.; Bellusci, S.; Scharfmann, R. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development 2001, 128, 5109–5117. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Naito, Z.; Lu, Y.P.; Kawahara, K.; Fujii, T.; Kawamoto, Y.; Teduka, K.; Sugisaki, Y. Differential distribution of fibroblast growth factor (FGF)-7 and FGF-10 in L-arginine-induced acute pancreatitis. Exp. Mol. Pathol. 2002, 73, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Nomme, J.; Su, Y.; Konrad, M.; Lavie, A. Structures of apo and product-bound human L-asparaginase: Insights into the mechanism of autoproteolysis and substrate hydrolysis. Biochemistry 2012, 51, 6816–6826. [Google Scholar] [CrossRef] [Green Version]
- Menniti, M.; Iuliano, R.; Föller, M.; Sopjani, M.; Alesutan, I.; Mariggiò, S.; Nofziger, C.; Perri, A.M.; Amato, R.; Blazer-Yost, B. 60kDa lysophospholipase, a new Sgk1 molecular partner involved in the regulation of ENaC. Cell Physiol. Biochem. 2010, 26, 587–596. [Google Scholar] [CrossRef]
- Sugimoto, H.; Odani, S.; Yamashita, S. Cloning and expression of cDNA encoding rat liver 60-kDa lysophospholipase containing an asparaginase-like region and ankyrin repeat. J. Biol. Chem. 1998, 273, 12536–12542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.C.; Schouten, M.; Aulchenko, Y.S.; Haley, C.S.; de Koning, D.J. Rapid and robust association mapping of expression quantitative trait loci. BMC Proc. 2007, 1 (Suppl. 1), S144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorlov, I.P.; Gorlova, O.Y.; Sunyaev, S.R.; Spitz, M.R.; Amos, C.I. Shifting paradigm of association studies: Value of rare single-nucleotide polymorphisms. Am. J. Hum. Genet. 2008, 82, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Tabangin, M.E.; Woo, J.G.; Martin, L.J. The effect of minor allele frequency on the likelihood of obtaining false positives. BMC Proc. 2009, 3 (Suppl. 7), S41. [Google Scholar] [CrossRef] [Green Version]
- Cohn, J.A.; Friedman, K.J.; Noone, P.G.; Knowles, M.R.; Silverman, L.M.; Jowell, P.S. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N. Engl. J. Med. 1998, 339, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.C.; Ermentrout, G.B. A mathematical model of the pancreatic duct cell generating high bicarbonate concentrations in pancreatic juice. Pancreas 2004, 29, e30–e40. [Google Scholar] [CrossRef]
- Hegyi, P.; Maléth, J.; Venglovecz, V.; Rakonczay, Z., Jr. Pancreatic ductal bicarbonate secretion: Challenge of the acinar Acid load. Front. Physiol. 2011, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Larusch, J.; Sun, X.; Aloe, A.; Lamb, J.; Hawes, R.; Cotton, P.; Brand, R.E.; Anderson, M.A.; Money, M.E. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology 2011, 140, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Mounzer, R.; Whitcomb, D.C. Genetics of acute and chronic pancreatitis. Curr. Opin. Gastroenterol. 2013, 29, 544–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).