
RTA404, an Activator of Nrf2, Activates the Checkpoint Kinases and Induces Apoptosis through Intrinsic Apoptotic Pathway in Malignant Glioma

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Viability
2.4. Cell Cycle Analysis
2.5. Apoptosis Measurement
2.6. Evaluation of Mitochondrial Membrane Potential
2.7. Western Blotting
2.8. Mitotic Index Analysis
2.9. Data Analysis
3. Result
3.1. RTA404 Reduces the Viability of U87MG Cells by Both Time and Does Dependent Manner
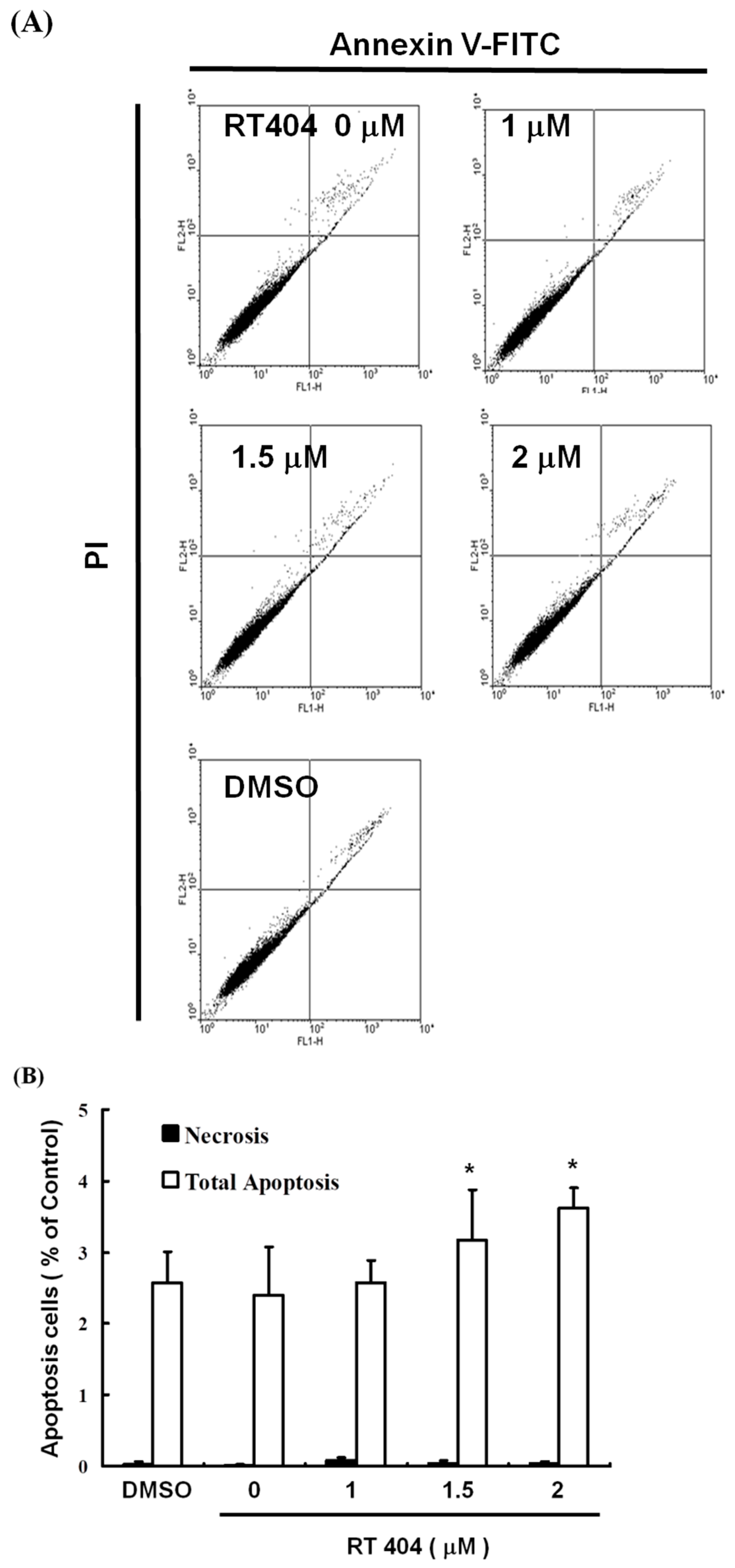
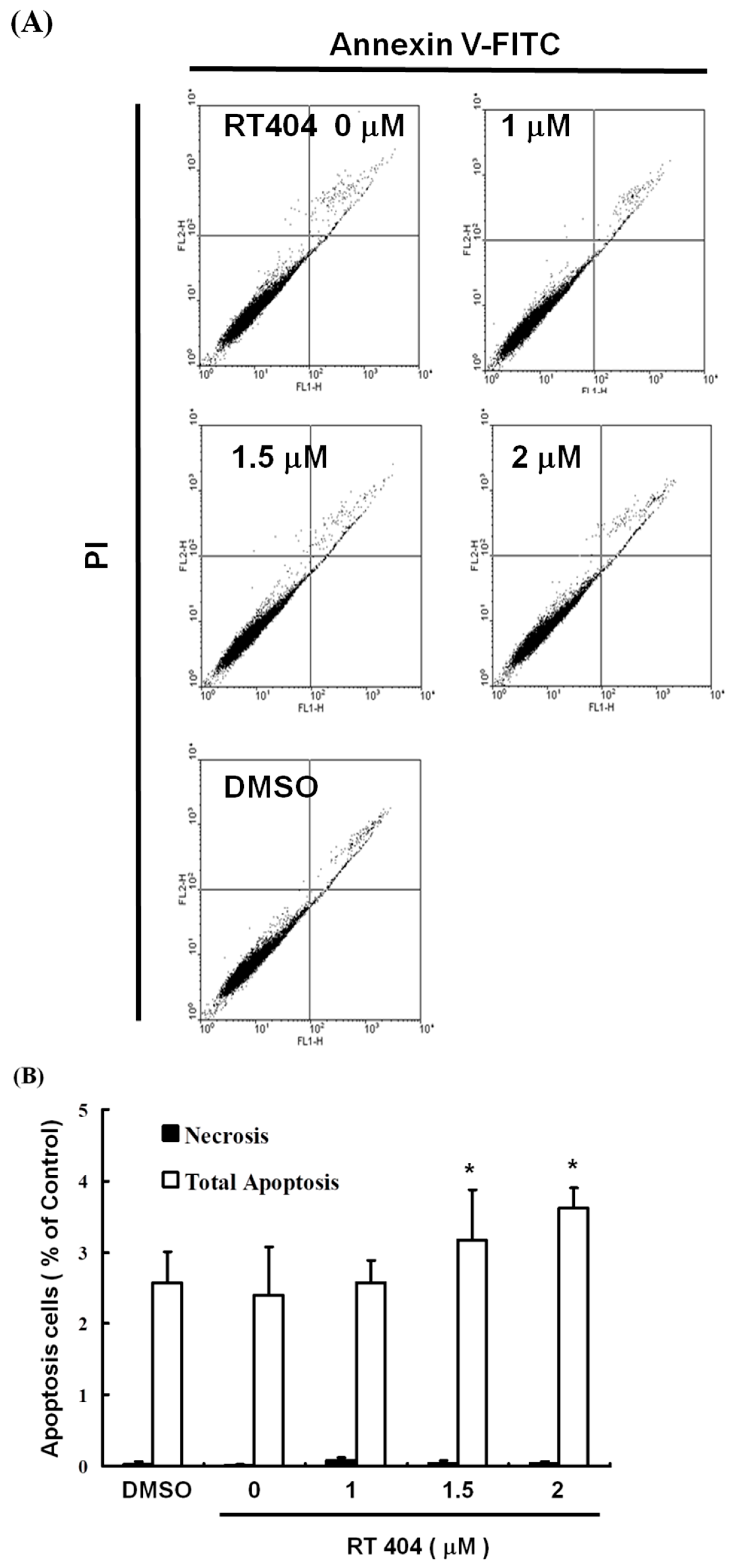
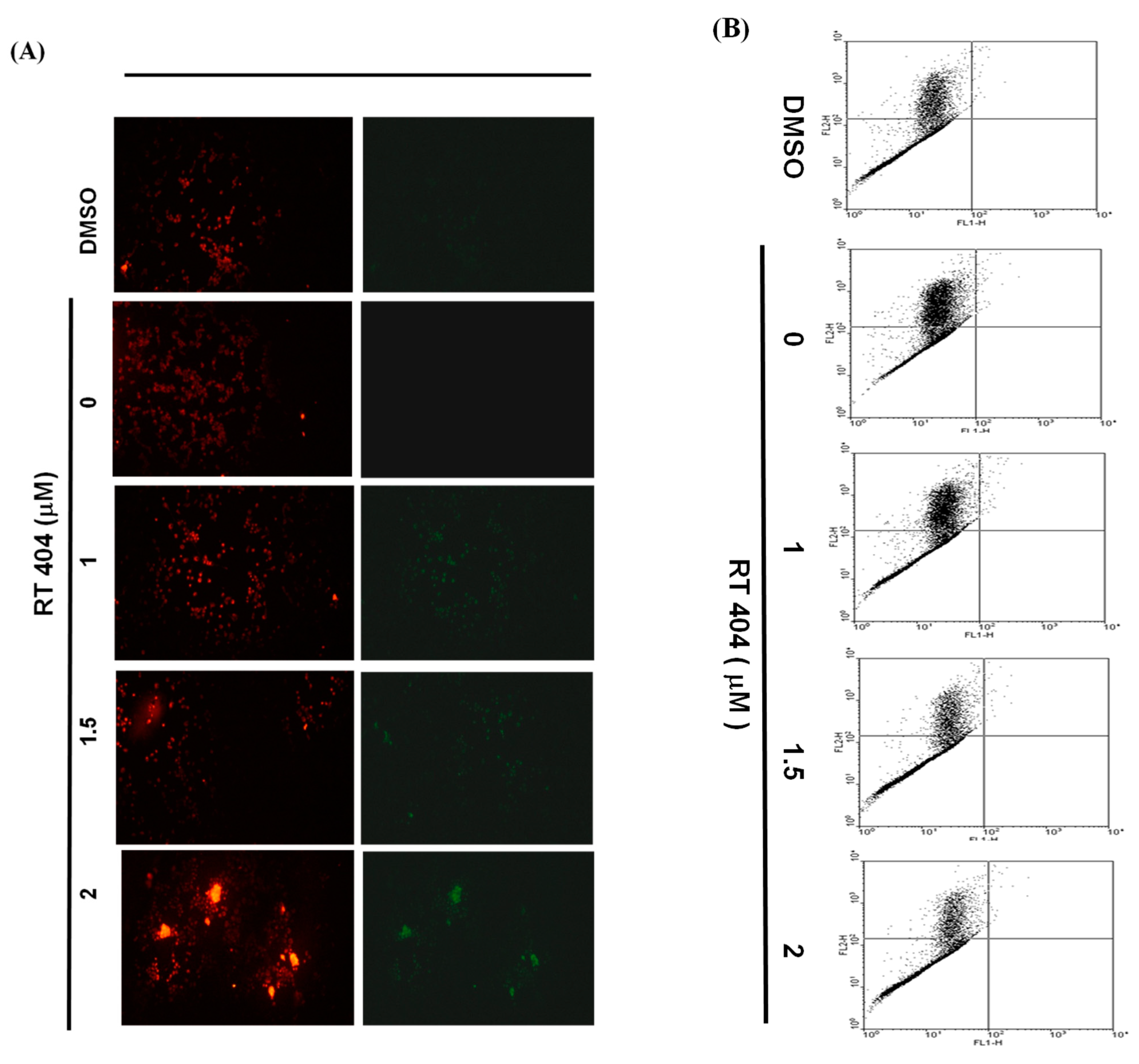
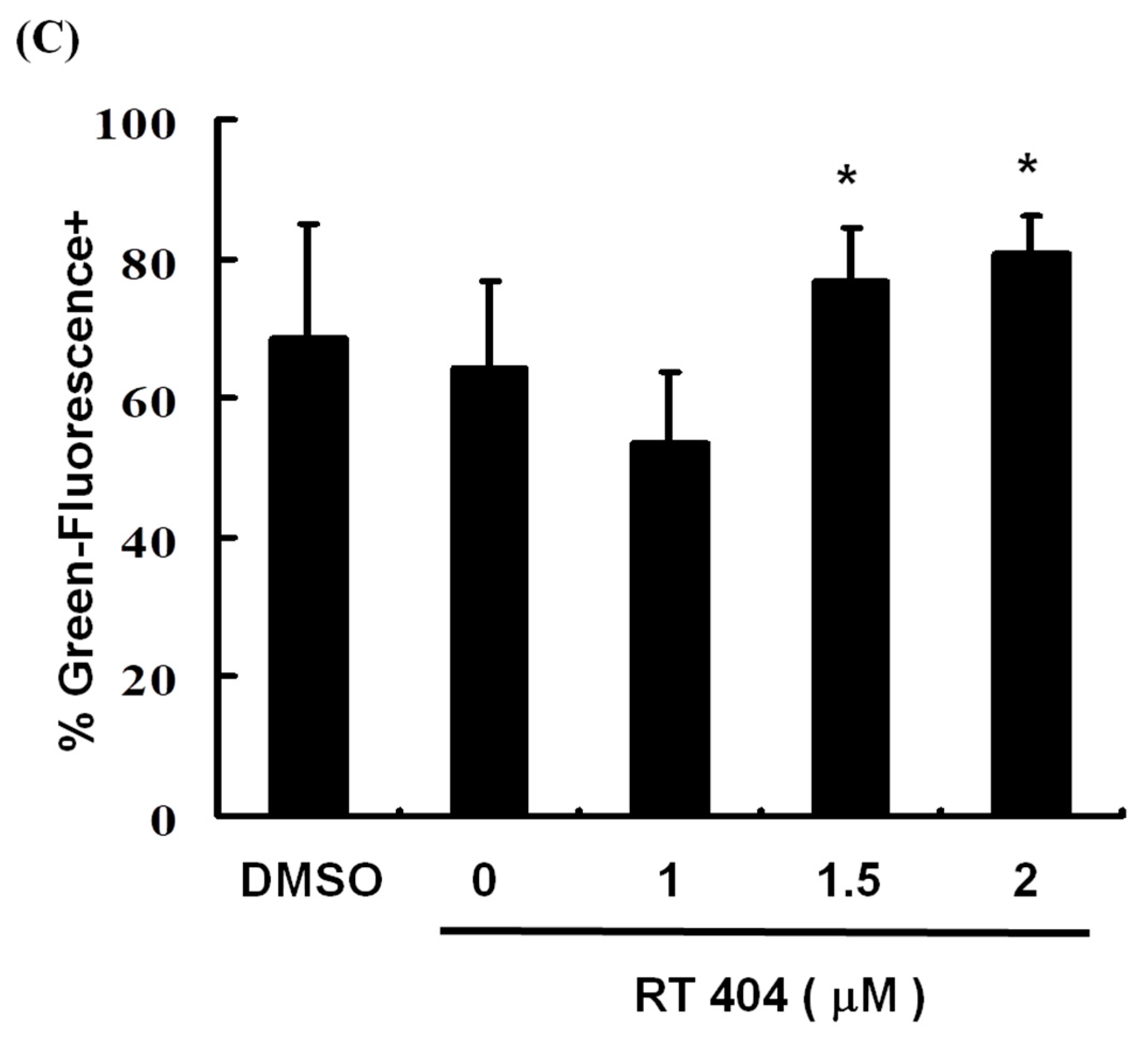
3.2. RTA404 Induced Apoptosis in U87MG Cells by Does Dependent Manner
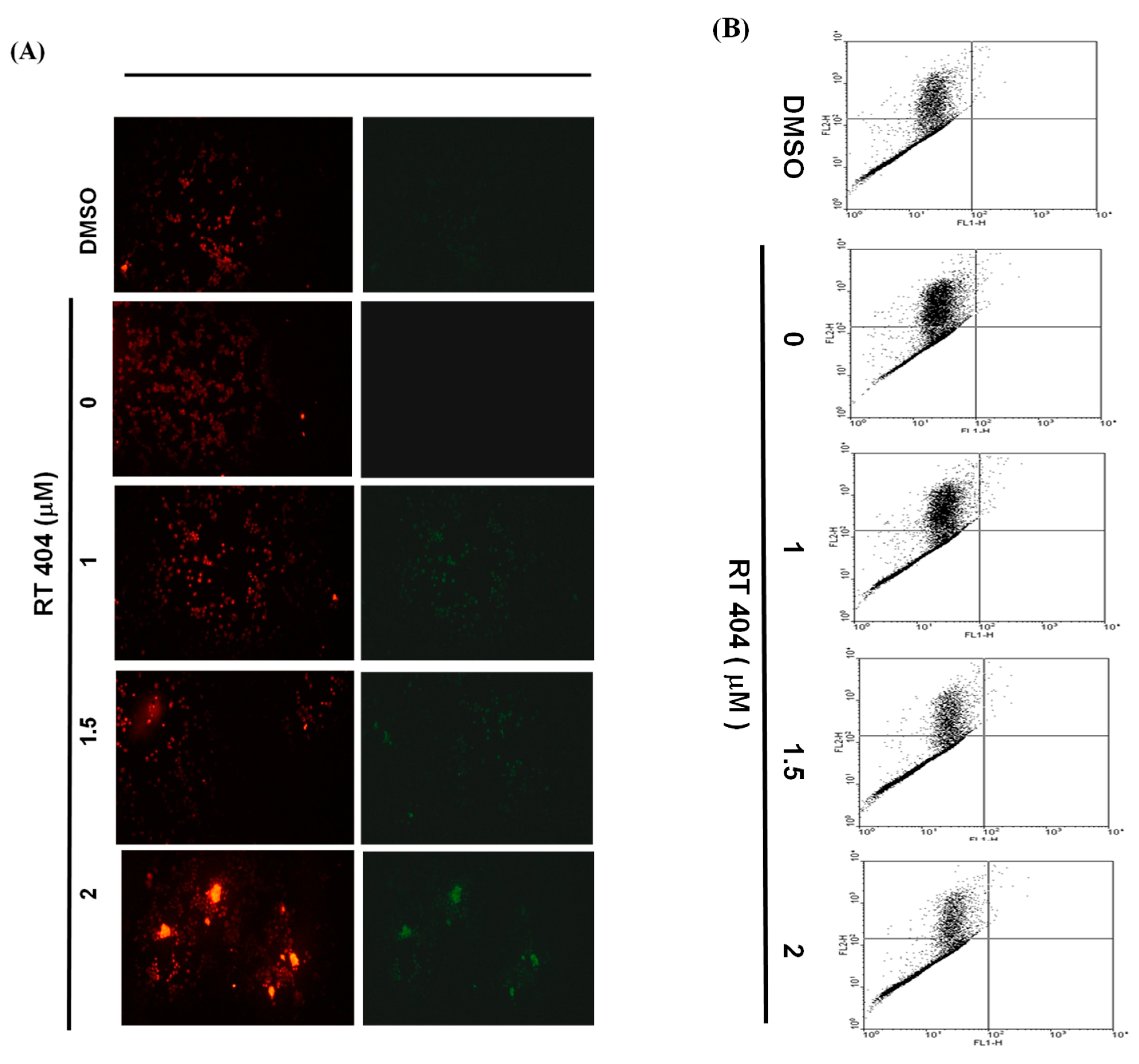
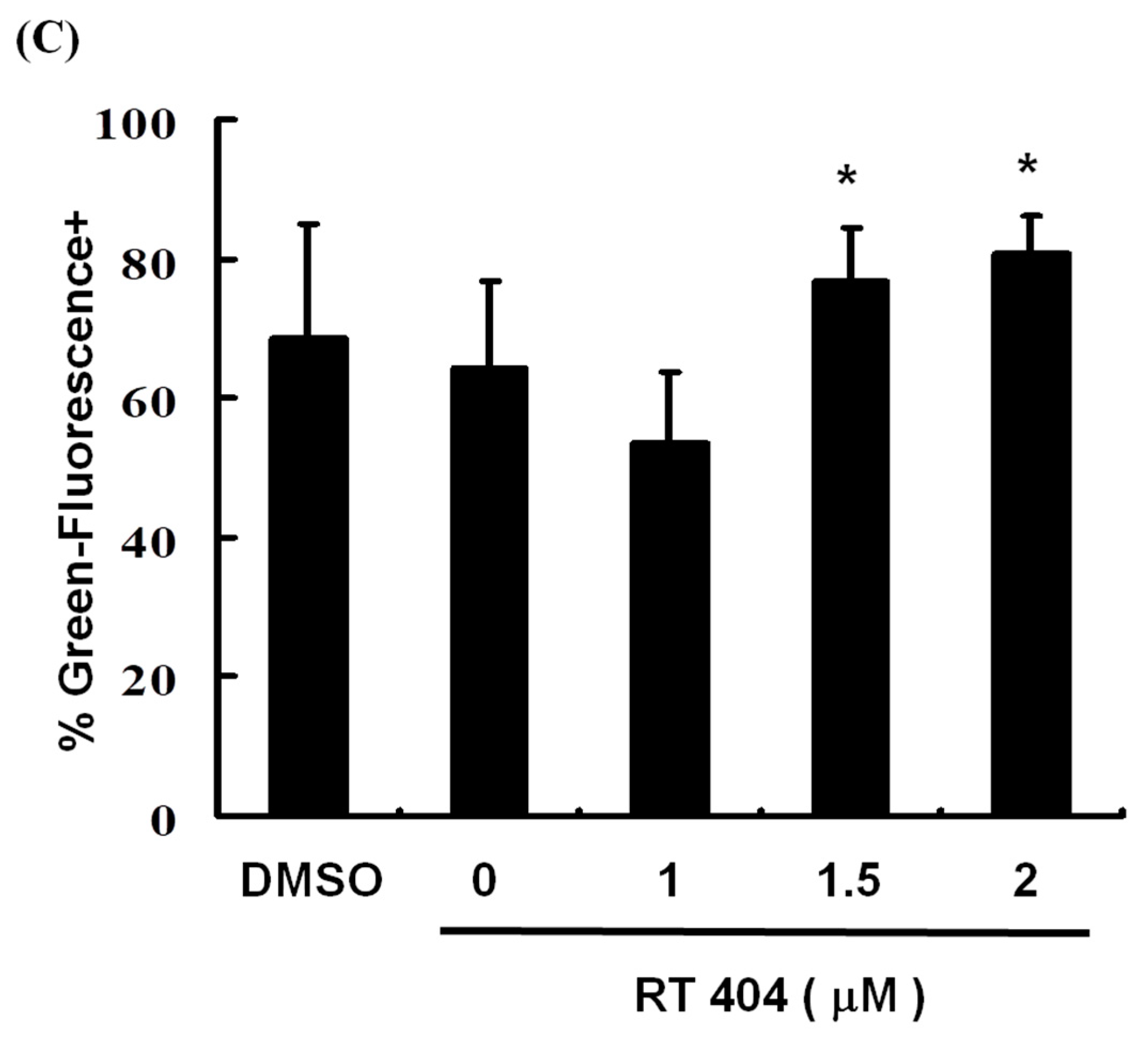
3.3. RTA404 Treatment Causes the Loss of Mitochondrial Membrane Potential
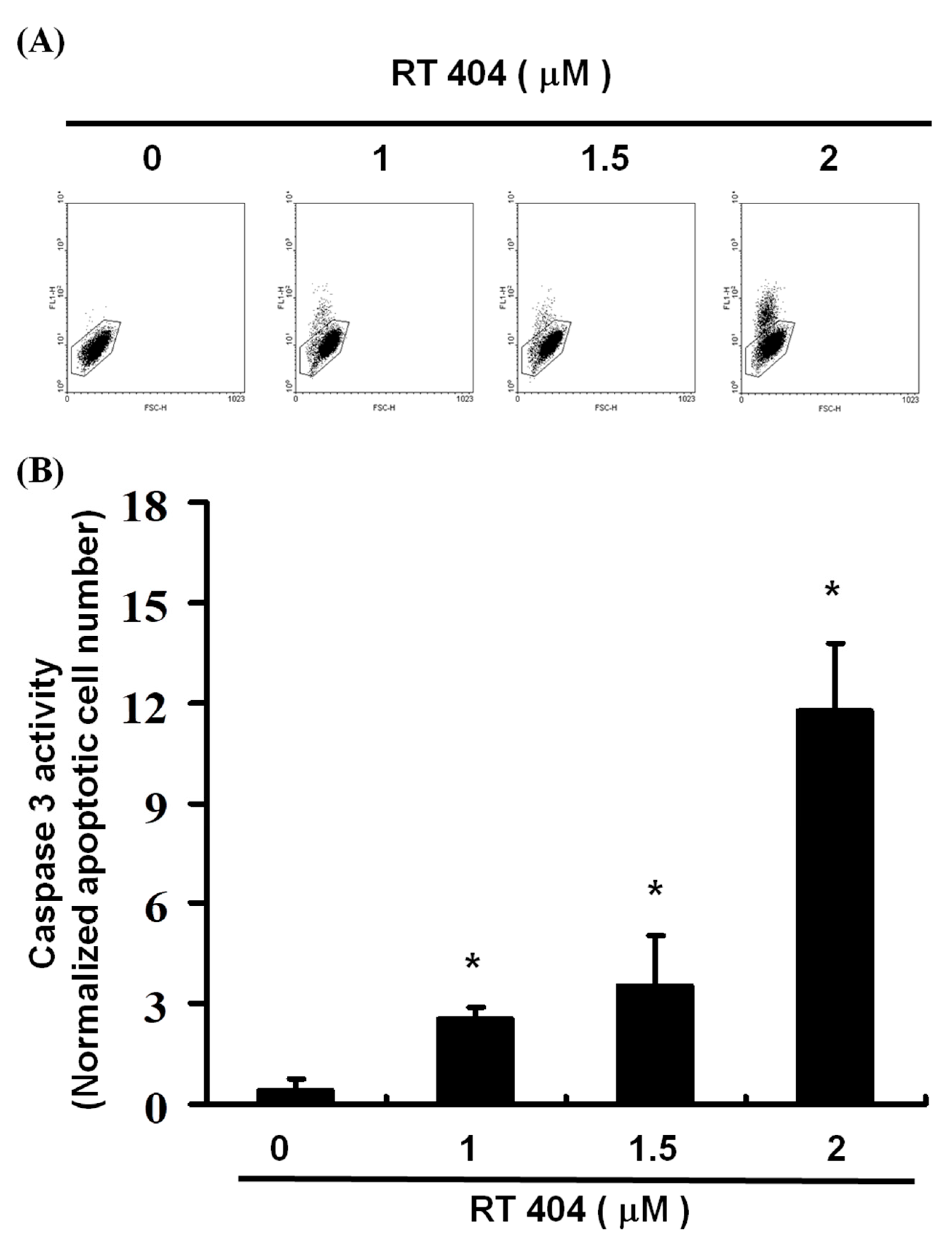
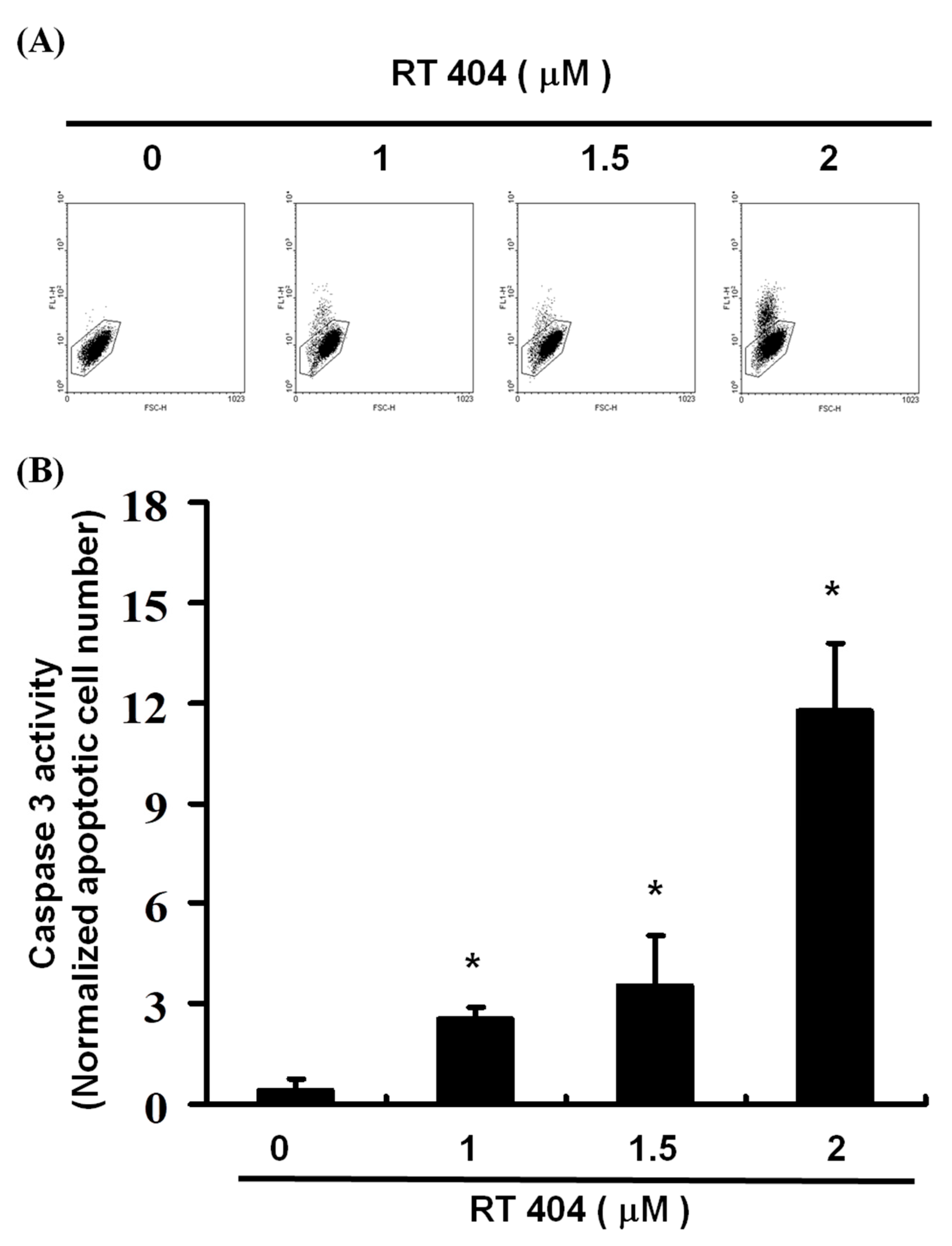
3.4. RTA404 Treatment Increases the Numbers of Active Caspase-3
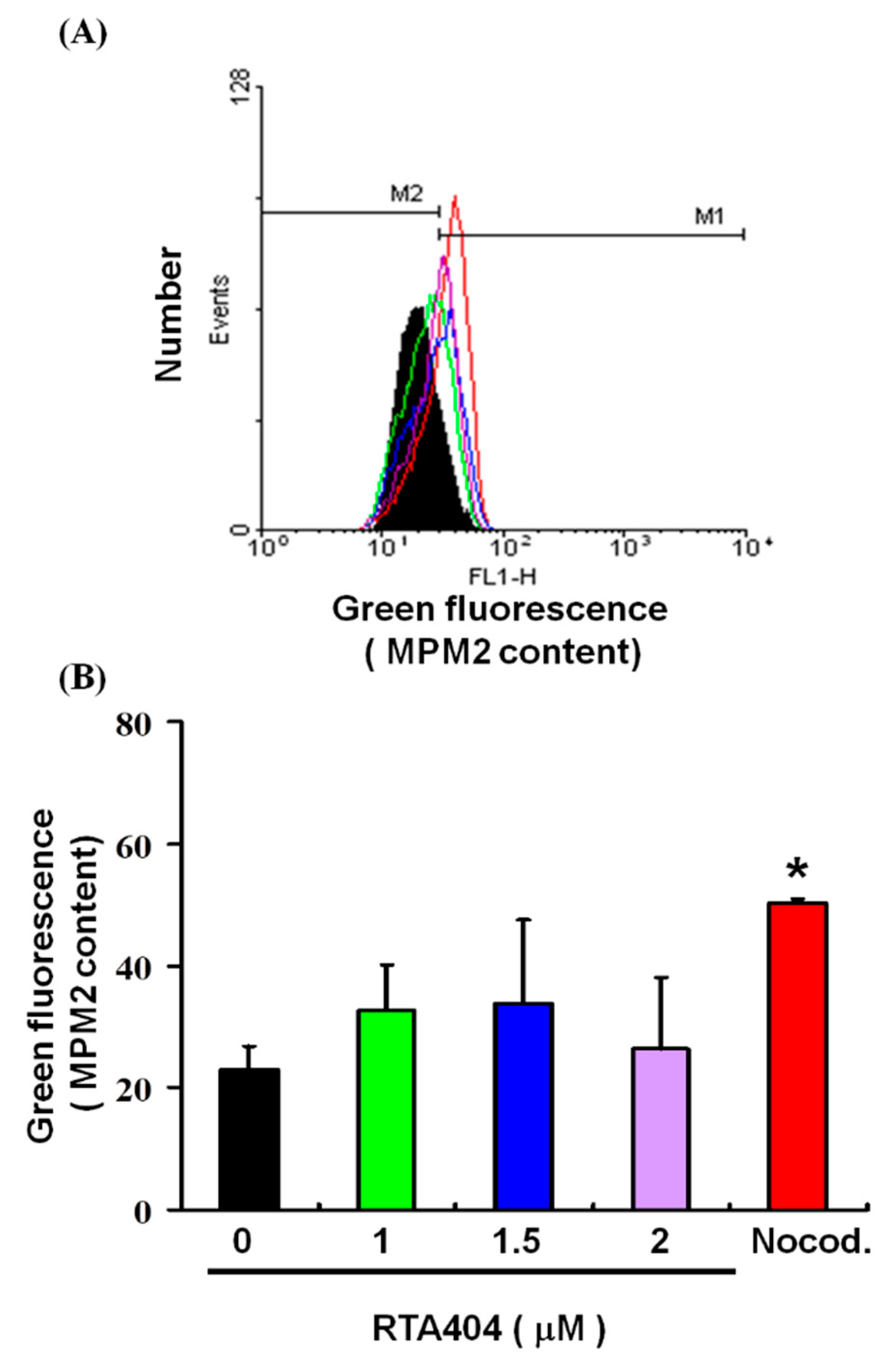
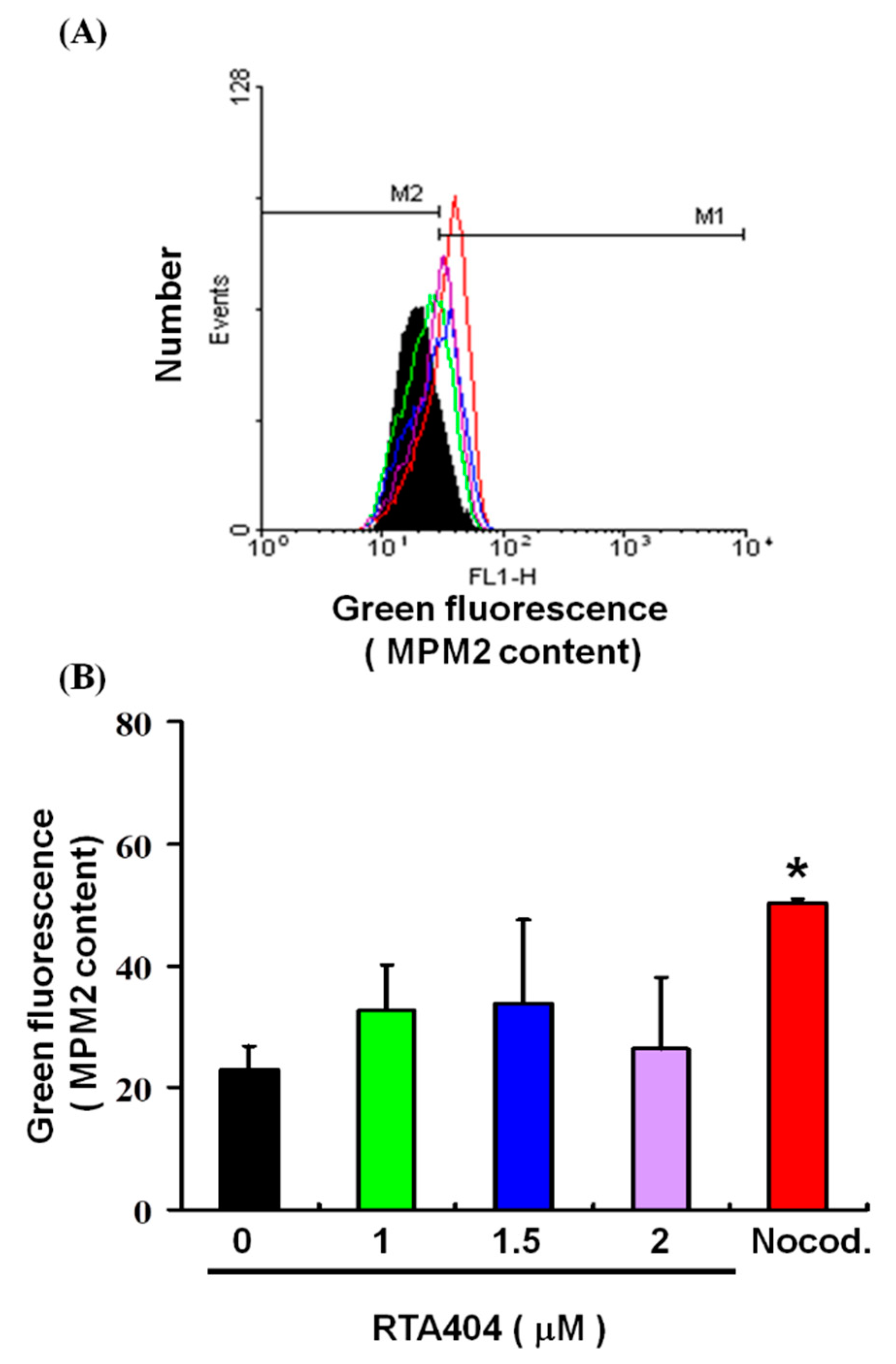
3.5. Cell Pass the G2 Checkpoint without Cell Cycle Arrest by RTA404 Treatment in U87MG Cells
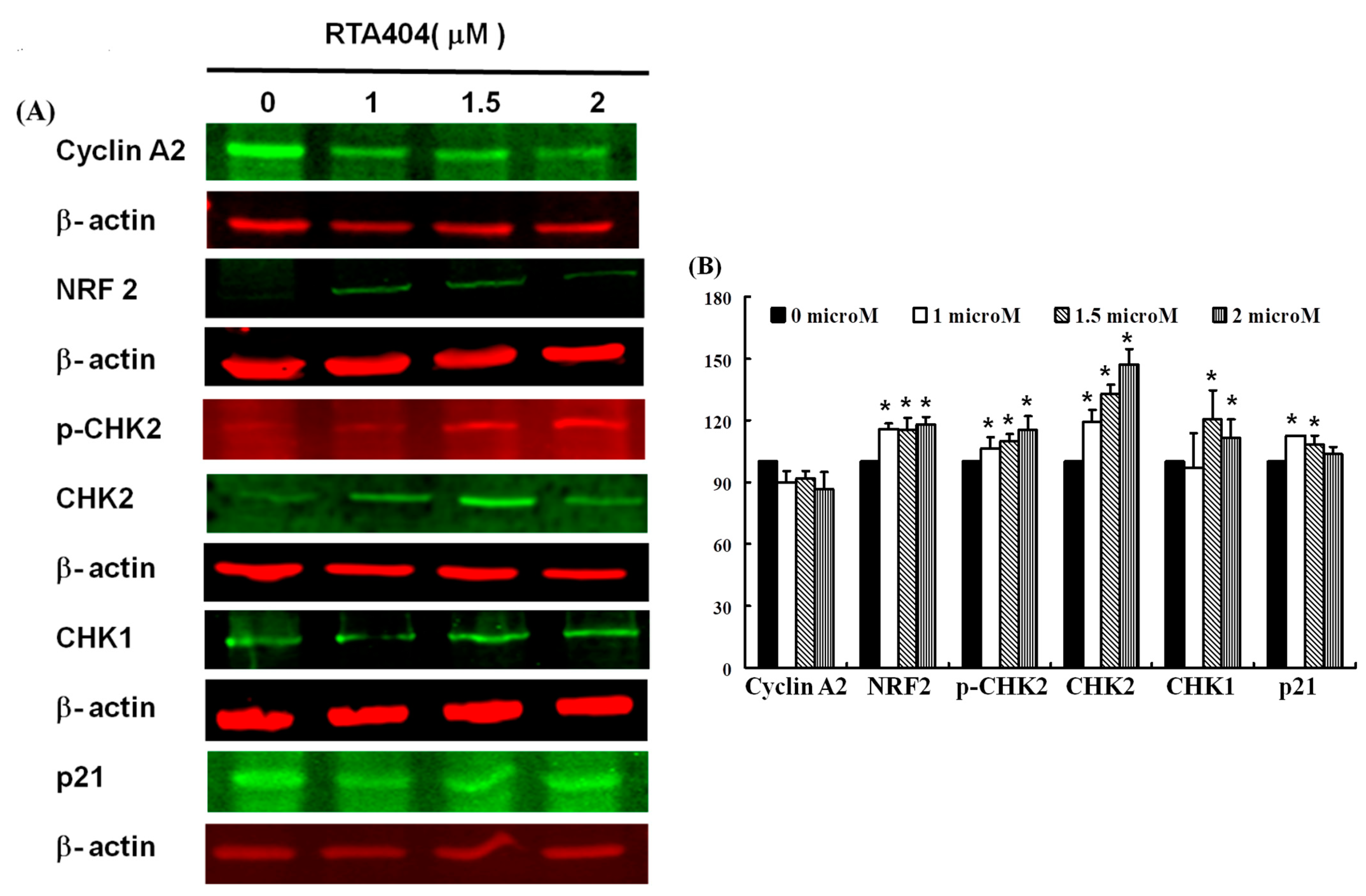
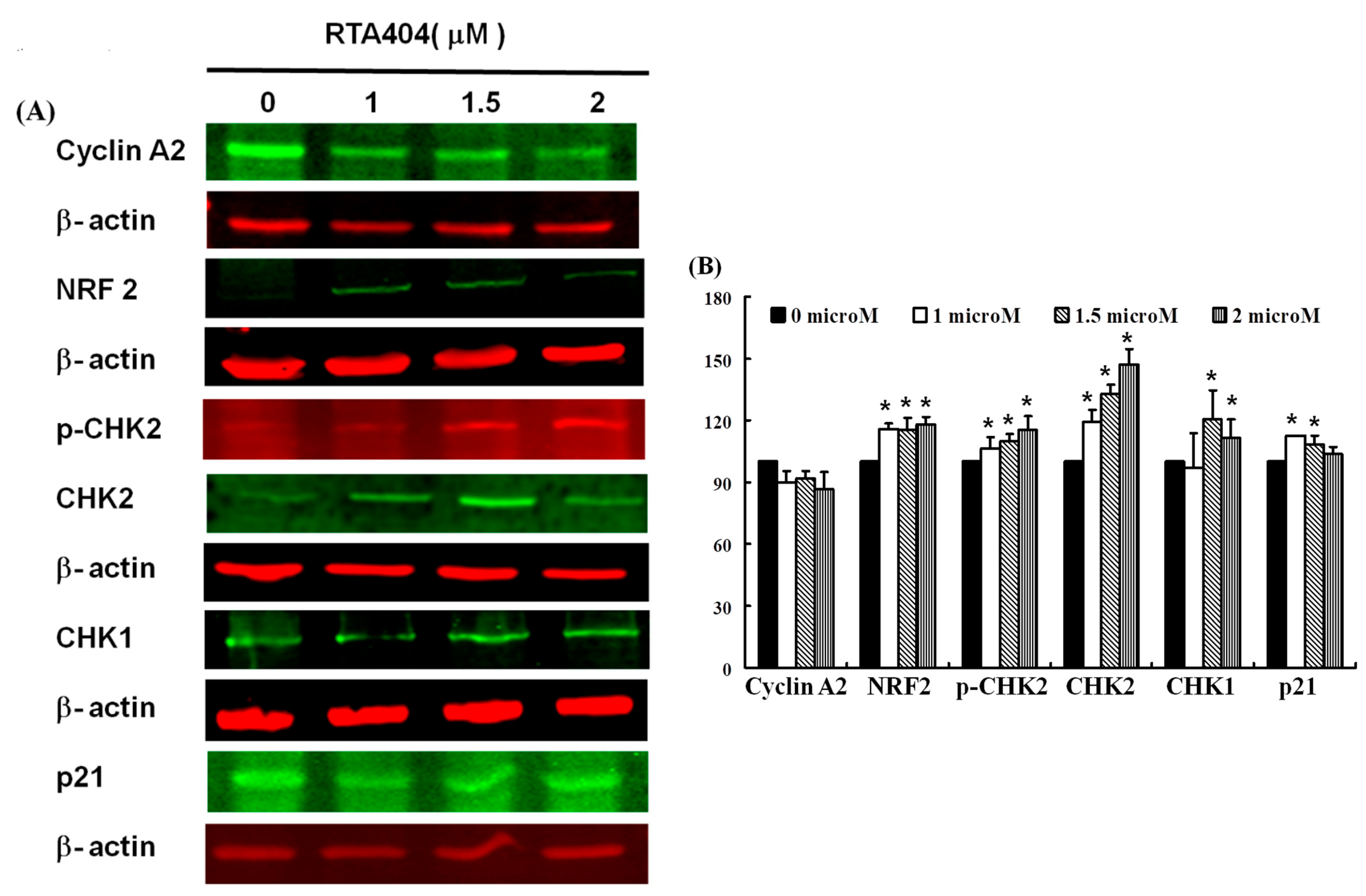
3.6. Protein of NRF2, p-CHK2, CHK1/2, p21 to Be Up-Regulated Expressions in RTA404-Treated U87MG Cells
4. Discussion
4.1. RTA404 Induced Apoptosis in Malignant Glioma
4.2. RTA404 Induced Apoptosis through Intrinsic Apoptotic Pathway
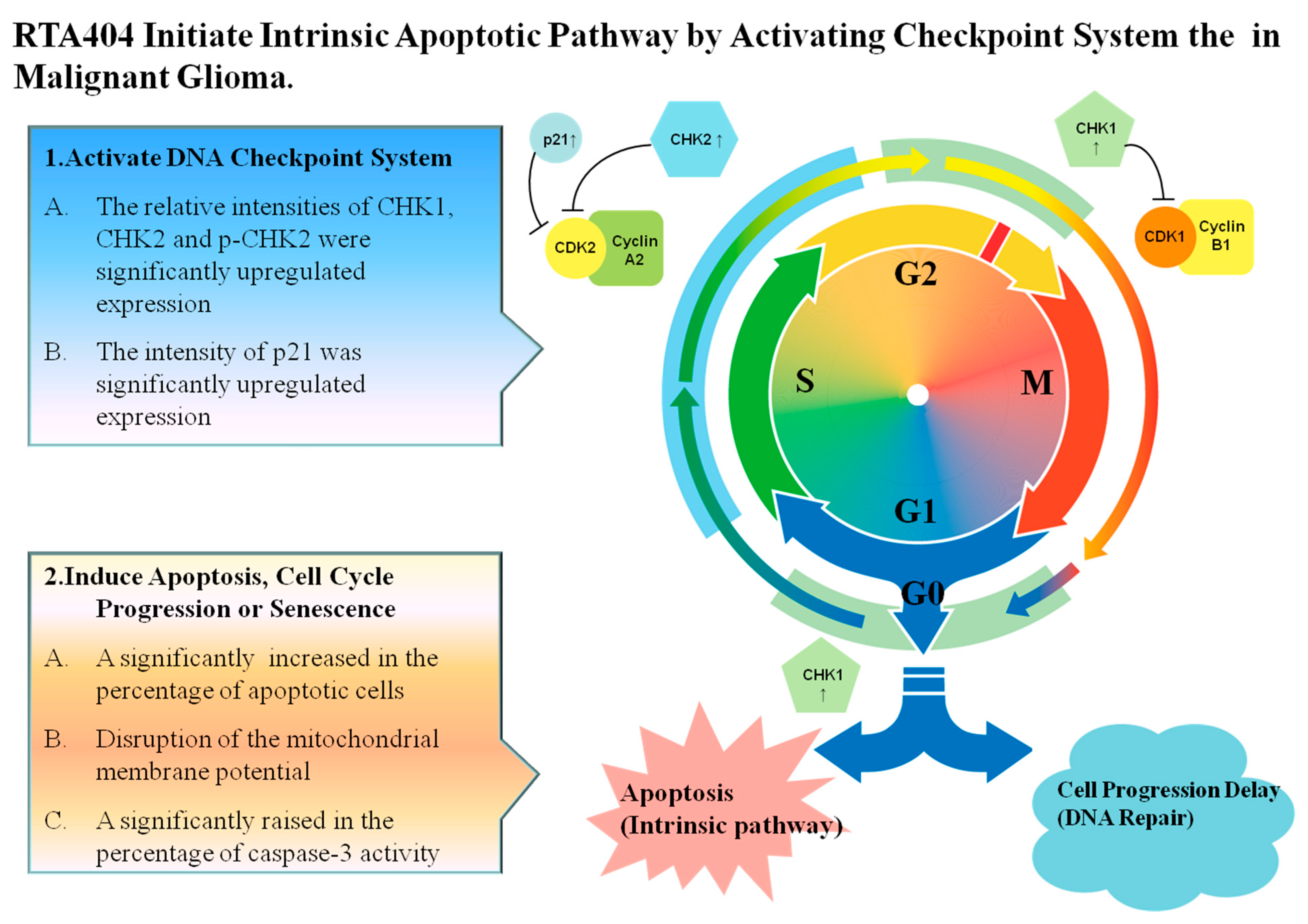
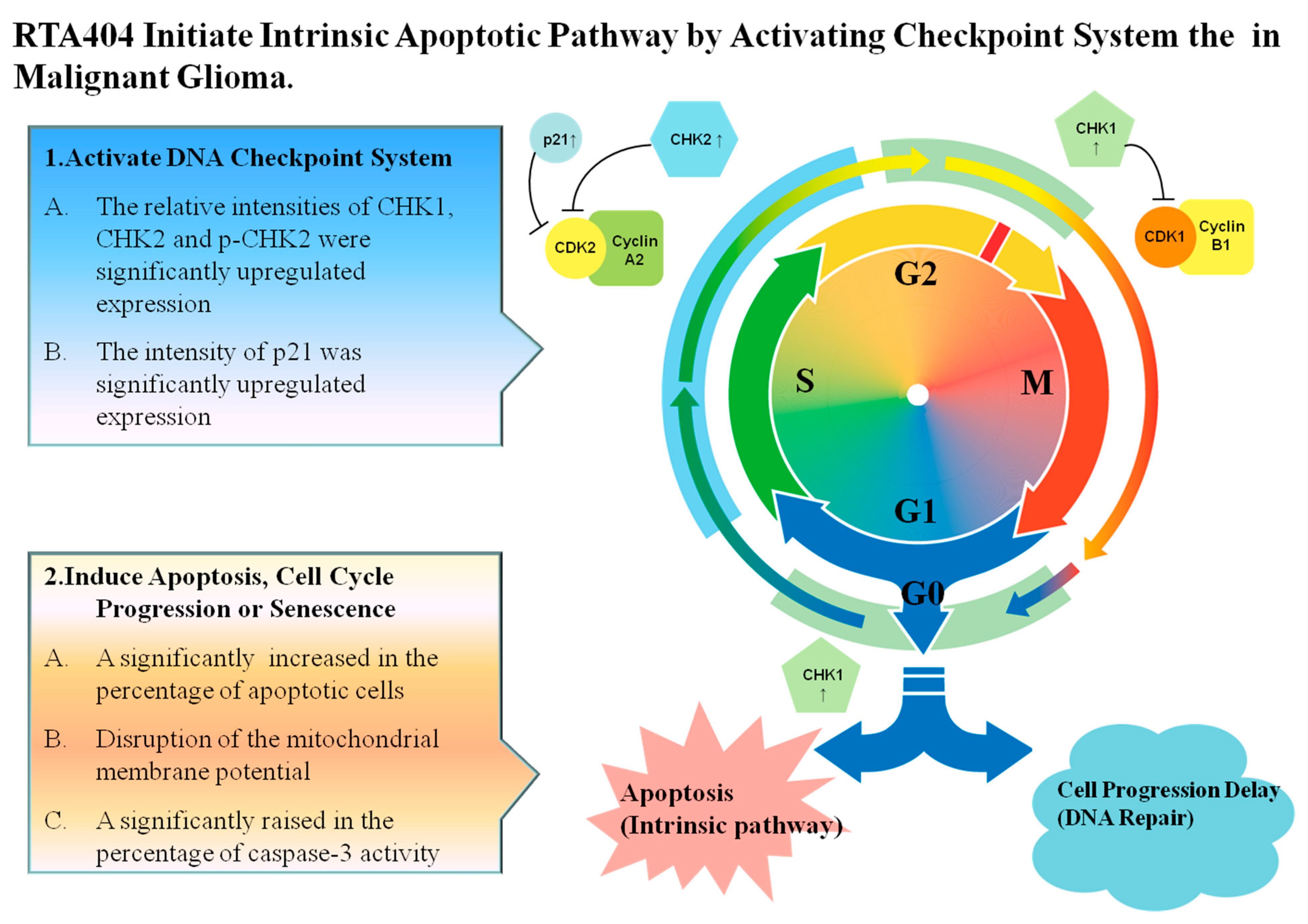
4.3. RTA404 Initiate Apoptosis by Activating DNA Damage Checkpoint System
4.4. RTA404, an Activator of Nrf2, Has Multi-Pharmacological Functions
4.5. RTA404 Induced Intrinsic Apoptotic Pathway Initiated by Checkpoint Kinases in Malignant Glioma
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Antioxidant Response Element | ARE |
| 2-cyano-3-,12-dioxoolean-1,9-dien-28-oic acid | CDDO |
| CDDO imidazolide | CDDO-Im |
| CDDO methyl ester | CDDO-Me |
| CDDOtrifluoroethyl amide | CDDO-TFEA |
| Glioblastoma multiforme | GBM |
| Malignant Glioma | MG |
| Kelch-like ECH-associated protein 1 | Keap1 |
| Nuclear factor erythroid 2-related factor 2 | Nrf2 |
| World Health Organization | WHO |
References
- Cahill, D.; Turcan, S. Origin of Gliomas. Semin. Neurol. 2018, 38, 5–10. [Google Scholar]
- Bush, N.A.; Chang, S.M.; Berger, M.S. Current and future strategies for treatment of glioma. Neurosurg. Rev. 2017, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro Oncol. 2016, 18 (Suppl. S5), v1–v75. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, M.A.; Oliveira-Costa, J.P.; Carter, B.S.; Stott, S.L.; Nahed, B.V. Blood-based biomarkers for the diagnosis and monitoring of gliomas. Neuro Oncol. 2018, 20, 1155–1161. [Google Scholar] [CrossRef] [Green Version]
- Saleem, M.; Asif, J.; Asif, M.; Saleem, U. Amygdalin from Apricot kernels induces apoptosis and causes cell cycle arrest in cancer cells: An updated review. Anti-Cancer Agents Med. Chem. 2018, 18, 1650–1655. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, N.Y.L.; Choong, C.; Lee, J.; Bellot, G.; Wong, A.L.A.; Goh, B.C.; Pervaiz, S. Targeting mitochondrial apoptosis to overcome treatment resistance in cancer. Cancers 2020, 12, 574. [Google Scholar] [CrossRef] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Zuo, Q.; Jiang, T.; Song, H.; Zhou, J. A newly synthesized oleanolic acid derivative inhibits the growth of osteosarcoma cells in vitro and in vivo by decreasing c-MYC-dependent glycolysis. J. Cell. Biochem. 2019, 120, 9264–9276. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhang, C.Y.; Ma, Y.Q.; He, Z.X.; Zhe, H.; Zhou, S.F. Therapeutic effects of C-28 methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO-Me; bardoxolone methyl) on radiation-induced lung inflammation and fibrosis in mice. Drug Des. Dev. Ther. 2015, 9, 3163–3178. [Google Scholar]
- Yang, T.; Sun, Y.; Li, Q.; Li, S.; Shi, Y.; Leak, R.K.; Chen, J.; Zhang, F. Ischemic preconditioning provides long-lasting neuroprotection against ischemic stroke: The role of Nrf2. Exp. Neurol. 2020, 325, 113142. [Google Scholar] [CrossRef]
- Gupta, K.; Patani, R.; Baxter, P.; Serio, A.; Story, D.; Tsujita, T.; Hayes, J.D.; Pedersen, R.A.; Hardingham, G.E.; Chandran, S. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ. 2012, 19, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Pareek, T.K.; Belkadi, A.; Kesavapany, S.; Zaremba, A.; Loh, S.L.; Bai, L.; Cohen, M.L.; Meyer, C.; Liby, K.T.; Miller, R.H.; et al. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci. Rep. 2011, 1, 201. [Google Scholar] [CrossRef]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabran, J.L.; Cheuk, A.; Liby, K.; Sporn, M.; Khan, J.; Letterio, J.; Leskov, K.S. Human neuroblastoma cells rapidly enter cell cycle arrest and apoptosis following exposure to C-28 derivatives of the synthetic triterpenoid CDDO. Cancer Biol. Ther. 2008, 7, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.H.; Lieu, A.S.; Wang, Y.W.; Yang, S.F.; Hsu, Y.C.; Lin, C.L. Therapeutic potential of RTA 404 in human brain malignant glioma cell lines via cell cycle arrest via p21/AKT signaling. BioMed Res. Int. 2021, 2021, 5552226. [Google Scholar] [CrossRef] [PubMed]
- Kabore, A.F.; Johnston, J.B.; Gibson, S.B. Changes in the apoptotic and survival signaling in cancer cells and their potential therapeutic implications. Curr. Cancer Drug Targets 2004, 4, 147–163. [Google Scholar] [CrossRef]
- Melet, A.; Song, K.; Bucur, O.; Jagani, Z.; Grassian, A.R.; Khosravi-Far, R. Apoptotic pathways in tumor progression and therapy. Adv. Exp. Med. Biol. 2008, 615, 47–79. [Google Scholar]
- An, W.; Lai, H.; Zhang, Y.; Liu, M.; Lin, X.; Cao, S. Apoptotic Pathway as the therapeutic target for anticancer traditional chinese medicines. Front. Pharmacol. 2019, 10, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formigli, L.; Papucci, L.; Tani, A.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Orlandini, S.Z. Aponecrosis: Morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell. Physiol. 2000, 182, 41–49. [Google Scholar] [CrossRef]
- Sperandio, S.; de Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnaiyan, A.M. The apoptosome: Heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Boldin, M.P.; Goncharov, T.M.; Goltsev, Y.V.; Wallach, D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996, 85, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [Green Version]
- Stennicke, H.R.; Jürgensmeier, J.M.; Shin, H.; Deveraux, Q.; Wolf, B.B.; Yang, X.; Zhou, Q.; Ellerby, H.M.; Ellerby, L.M.; Bredesen, D.; et al. Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem. 1998, 273, 27084–27090. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [Green Version]
- Beishline, K.; Azizkhan-Clifford, J. Interplay between the cell cycle and double-strand break response in mammalian cells. Methods Mol. Biol. 2014, 1170, 41–59. [Google Scholar] [PubMed]
- Lukas, C.; Bartkova, J.; Latella, L.; Falck, J.; Mailand, N.; Schroeder, T.; Sehested, M.; Lukas, J.; Bartek, J. DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res. 2001, 61, 4990–4993. [Google Scholar] [PubMed]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.B.; McNeely, S.C.; Beckmann, R.P. Achieving precision death with cell-cycle inhibitors that target DNA replication and repair. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3232–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stawinska, M.; Cygankiewicz, A.; Trzcinski, R.; Mik, M.; Dziki, A.; Krajewska, W.M. Alterations of Chk1 and Chk2 expression in colon cancer. Int. J. Colorectal Dis. 2008, 23, 1243–1249. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, T.-H.; Lieu, A.-S.; Huang, T.-Y.; Kwan, A.-L.; Lin, C.-L.; Hsu, Y.-C. RTA404, an Activator of Nrf2, Activates the Checkpoint Kinases and Induces Apoptosis through Intrinsic Apoptotic Pathway in Malignant Glioma. J. Clin. Med. 2021, 10, 4805. https://doi.org/10.3390/jcm10214805
Tsai T-H, Lieu A-S, Huang T-Y, Kwan A-L, Lin C-L, Hsu Y-C. RTA404, an Activator of Nrf2, Activates the Checkpoint Kinases and Induces Apoptosis through Intrinsic Apoptotic Pathway in Malignant Glioma. Journal of Clinical Medicine. 2021; 10(21):4805. https://doi.org/10.3390/jcm10214805
Chicago/Turabian StyleTsai, Tai-Hsin, Ann-Shung Lieu, Tzuu-Yuan Huang, Aij-Lie Kwan, Chih-Lung Lin, and Yi-Chiang Hsu. 2021. "RTA404, an Activator of Nrf2, Activates the Checkpoint Kinases and Induces Apoptosis through Intrinsic Apoptotic Pathway in Malignant Glioma" Journal of Clinical Medicine 10, no. 21: 4805. https://doi.org/10.3390/jcm10214805
APA StyleTsai, T.-H., Lieu, A.-S., Huang, T.-Y., Kwan, A.-L., Lin, C.-L., & Hsu, Y.-C. (2021). RTA404, an Activator of Nrf2, Activates the Checkpoint Kinases and Induces Apoptosis through Intrinsic Apoptotic Pathway in Malignant Glioma. Journal of Clinical Medicine, 10(21), 4805. https://doi.org/10.3390/jcm10214805