
Heart Failure in Patients with Arrhythmogenic Cardiomyopathy


Abstract
1. Introduction
2. Prevalence of HF in ACM
3. Clinical Characteristics and Classification
3.1. Clinical Characteristics
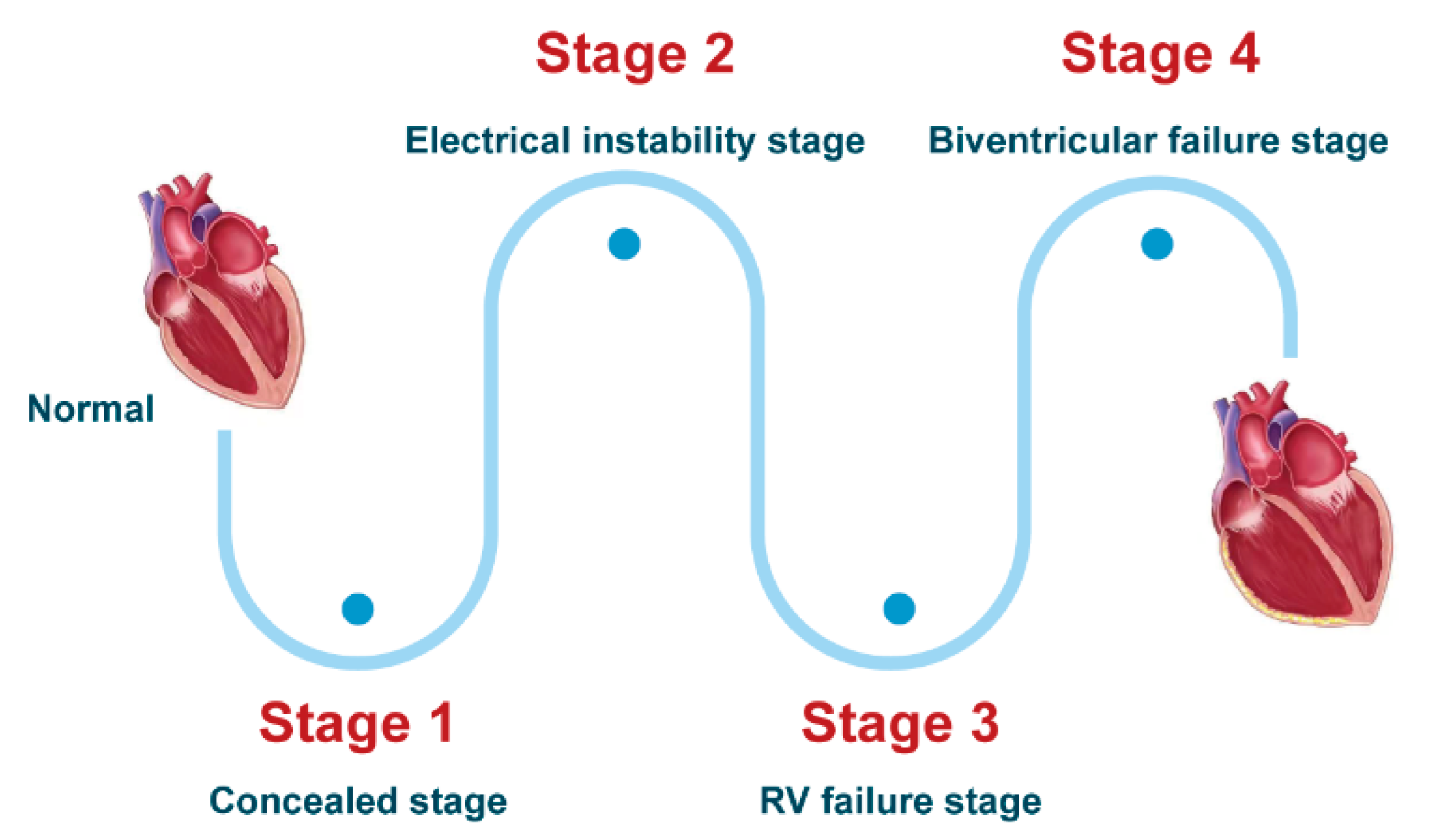
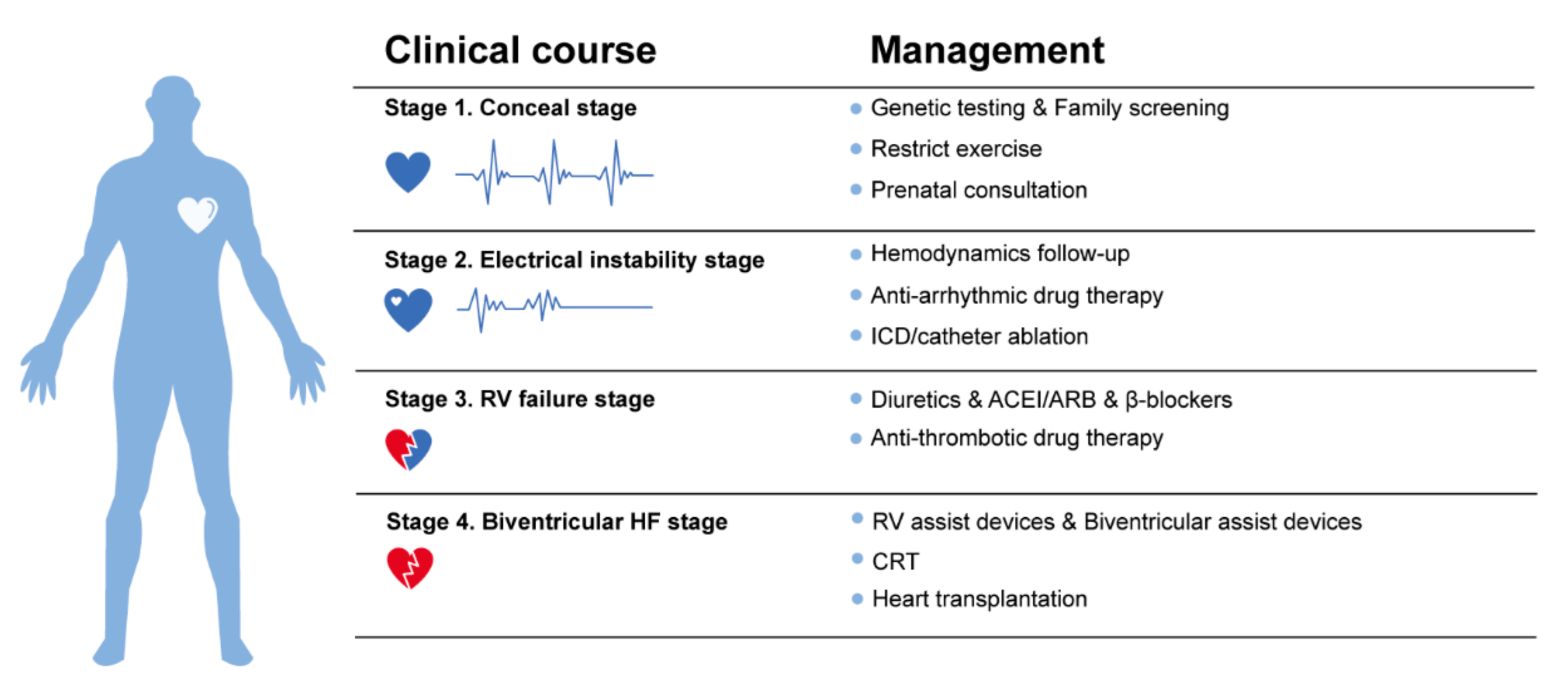
3.1.1. Clinical Course and Symptoms
- Stage 1, the concealed stage, characterized by minor structural changes in RV without obvious abnormality in electrocardiogram (ECG), echocardiogram and histological findings. At this stage, SCD and life-threatening ventricular arrythmias can be the first manifestation in young patients, especially if they are engaged in competitive sports and endurance exercise.
- Stage 2, electrical instability stage, in which RV structural remodeling and dysfunction become an overt phenotype. Recurrent RV arrhythmias such as VT frequently occurred at this stage.
- Stage 3, RV failure stage, caused by diffuse progressive fibrofatty tissue replacement of RV myocardium in RV. LV function is typically preserved. Symptoms of volume overload and congestive HF appear gradually but are still under control, if proper intervention strategies are used at this stage.
- Stage 4, biventricular HF with global dilation and LV involvement. The proportion of ACM patients to reach this phase was small, which could be influenced by survival bias.
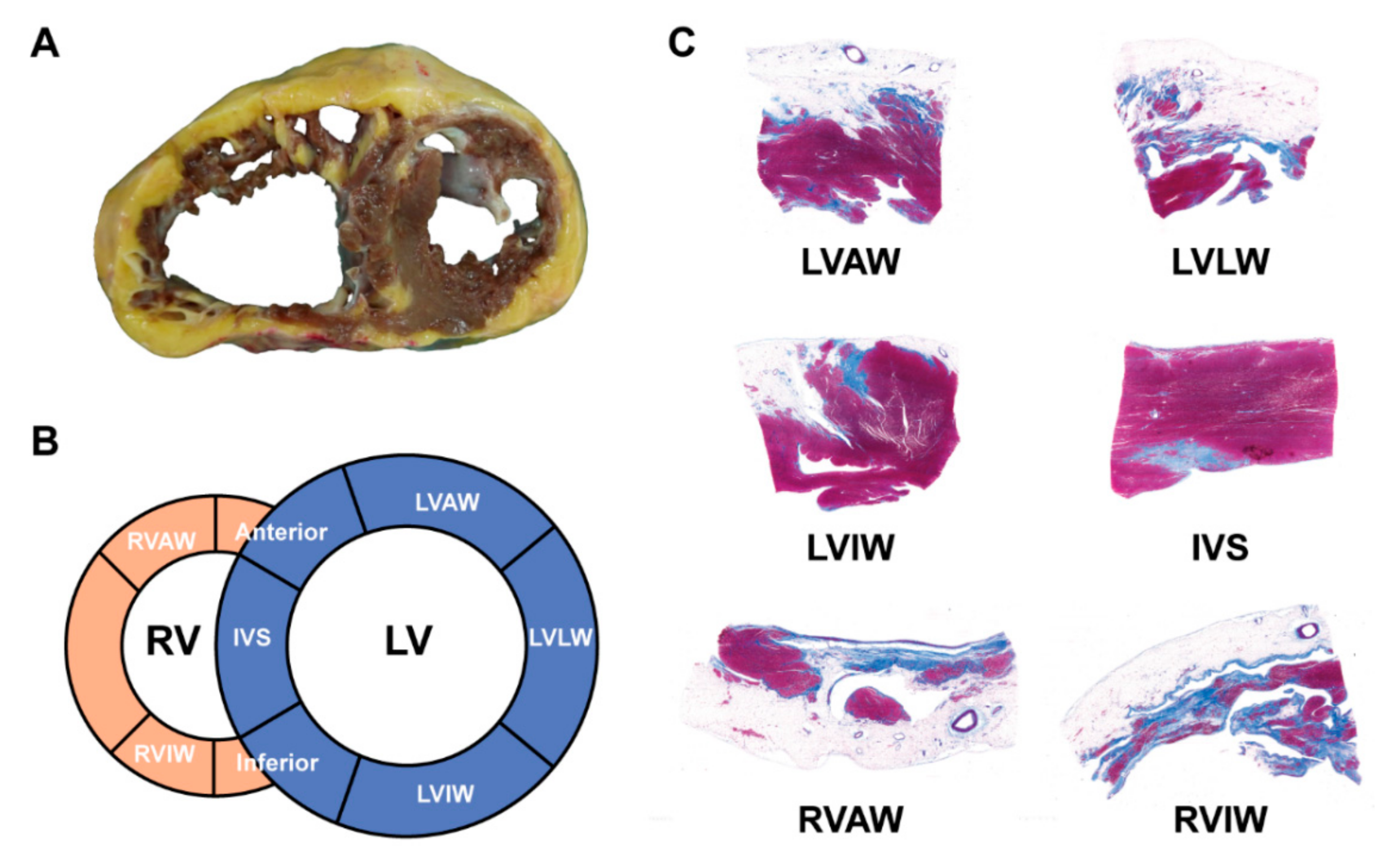
3.1.2. Imaging and ECG Phenotype
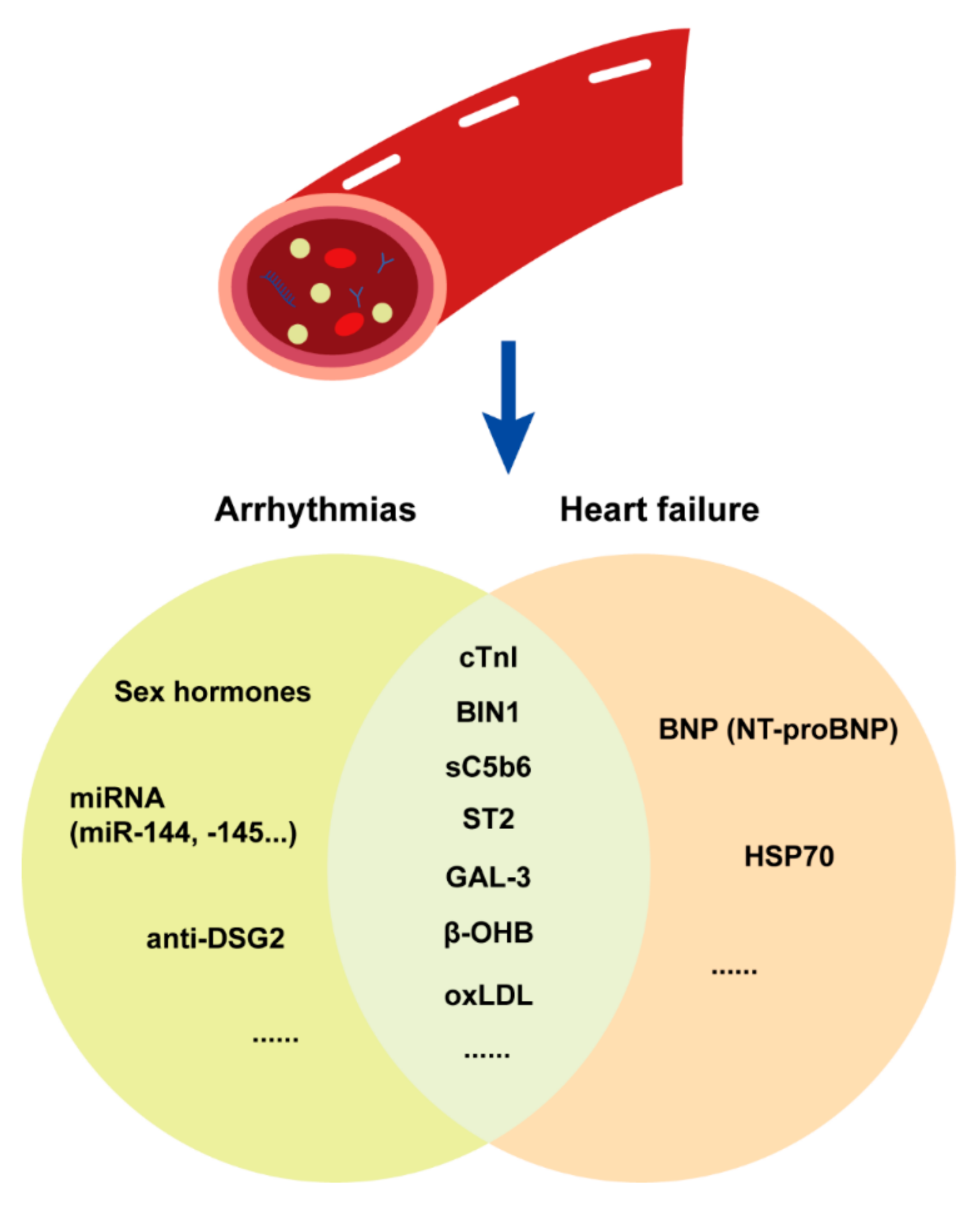
3.1.3. Plasma Biomarkers
3.1.4. Histopathological Characteristics and Endomyocardial Biopsy
3.2. Clinical Classification
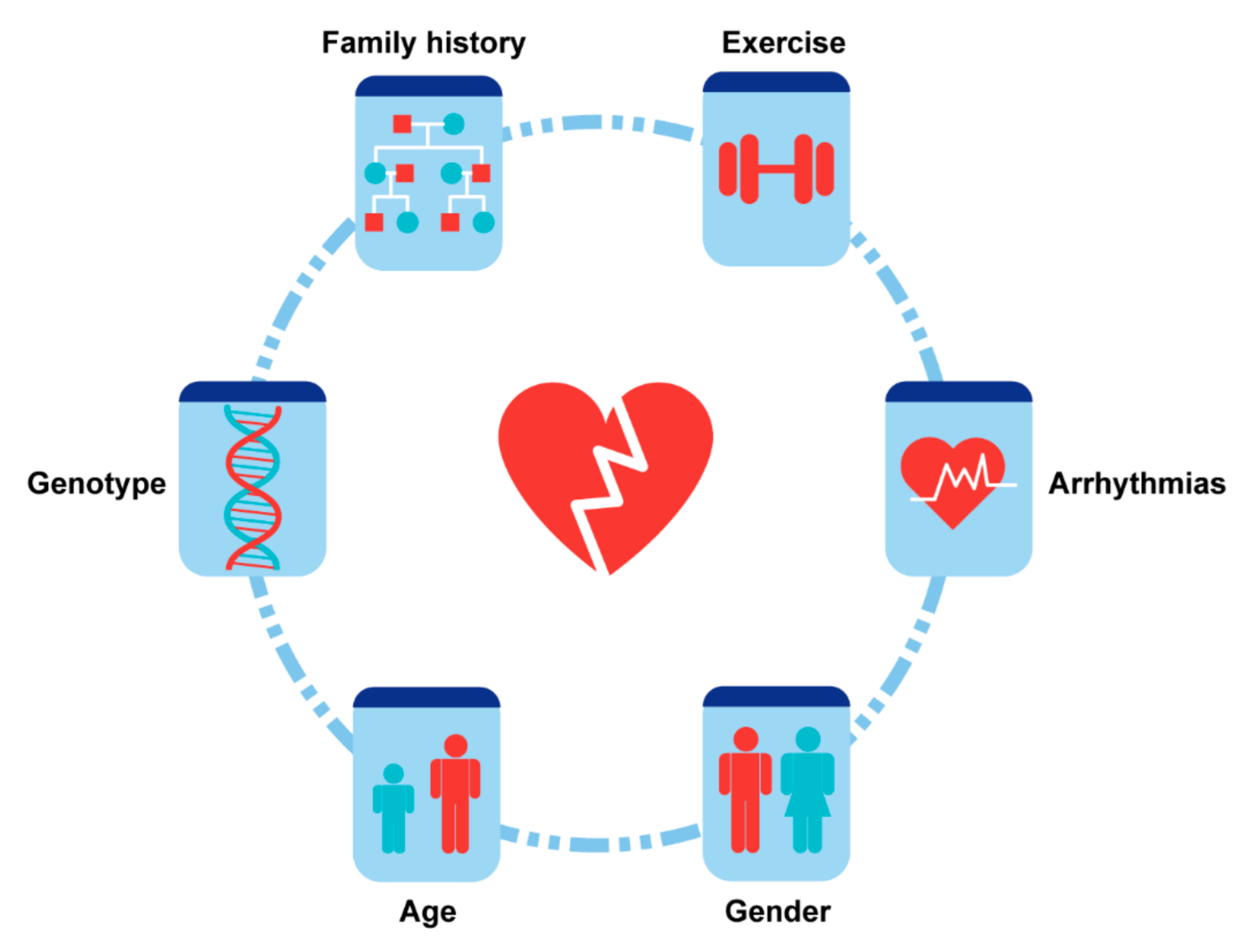
4. Risk Factors and Stratification
4.1. Demographics
4.1.1. Gender
4.1.2. Age
4.1.3. Family History and Ethnic Background
4.2. Arrhythmias and Electrophysiological Abnormalities
4.3. Genotype
4.4. Physical Activity and Exercise
5. Prevention and Management
5.1. Prevention
5.2. Drug Therapy
5.3. Defibrillator Implantation
5.4. Surgical Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Hoorntje, E.T.; Te Rijdt, W.P.T.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; Van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef]
- Bosman, L.P.; Sammani, A.; James, C.A.; Cadrin-Tourigny, J.; Calkins, H.; van Tintelen, J.P.; Hauer, R.N.W.; Asselbergs, F.W.; Te Riele, A. Predicting arrhythmic risk in arrhythmogenic right ventricular cardiomyopathy: A systematic review and meta-analysis. Heart Rhythm 2018, 15, 1097–1107. [Google Scholar] [CrossRef]
- Cadrin-Tourigny, J.; Bosman, L.P.; Nozza, A.; Wang, W.; Tadros, R.; Bhonsale, A.; Bourfiss, M.; Fortier, A.; Lie, O.H.; Saguner, A.M.; et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2019, 40, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Calkins, H.; Corrado, D.; Marcus, F. Risk Stratification in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2017, 136, 2068–2082. [Google Scholar] [CrossRef]
- Hauer, R.N.W. Prevention of Sudden Cardiac Death in Arrhythmogenic Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 769–770. [Google Scholar] [CrossRef]
- Mazzanti, A.; Ng, K.; Faragli, A.; Maragna, R.; Chiodaroli, E.; Orphanou, N.; Monteforte, N.; Memmi, M.; Gambelli, P.; Novelli, V.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Course and Predictors of Arrhythmic Risk. J. Am. Coll. Cardiol. 2016, 68, 2540–2550. [Google Scholar] [CrossRef] [PubMed]
- Vischer, A.S.; Castelletti, S.; Syrris, P.; McKenna, W.J.; Pantazis, A. Heart failure in patients with arrhythmogenic right ventricular cardiomyopathy: Genetic characteristics. Int. J. Cardiol. 2019, 286, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Gilotra, N.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.J.; Murray, B.; Tichnell, C.; Sawant, A.; Ong, C.S.; Judge, D.P.; Russell, S.D.; et al. Heart Failure Is Common and Under-Recognized in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; Te Riele, A.S.J.M.; Van Den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Komura, M.; Suzuki, J.-I.; Adachi, S.; Takahashi, A.; Otomo, K.; Nitta, J.; Nishizaki, M.; Obayashi, T.; Nogami, A.; Satoh, Y.; et al. Clinical Course of Arrhythmogenic Right Ventricular Cardiomyopathy in the Era of Implantable Cardioverter-Defibrillators and Radiofrequency Catheter Ablation. Int. Heart J. 2010, 51, 34–40. [Google Scholar] [CrossRef][Green Version]
- DeWitt, E.S.; Chandler, S.F.; Hylind, R.J.; Beausejour Ladouceur, V.; Blume, E.D.; VanderPluym, C.; Powell, A.J.; Fynn-Thompson, F.; Roberts, A.E.; Sanders, S.P.; et al. Phenotypic Manifestations of Arrhythmogenic Cardiomyopathy in Children and Adolescents. J. Am. Coll. Cardiol. 2019, 74, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Peters, S. Long-term follow-up and risk assessment of arrhythmogenic right ventricular dysplasia/cardiomyopathy: Personal experience from different primary and tertiary centres. J. Cardiovasc. Med. 2007, 8, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Trümmel, M.; Meyners, W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol. 2004, 97, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Peters, H.; Thierfelder, L. Heart failure in arrhythmogenic right ventricular dysplasia-cardiomyopathy. Int. J. Cardiol. 1999, 71, 251–256. [Google Scholar] [CrossRef]
- Hulot, J.-S.; Jouven, X.; Empana, J.-P.; Frank, R.; Fontaine, G. Natural History and Risk Stratification of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation 2004, 110, 1879–1884. [Google Scholar] [CrossRef]
- Lemola, K.; Brunckhorst, C.; Helfenstein, U.; Oechslin, E.; Jenni, R.; Duru, F. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: Long term experience of a tertiary care centre. Heart 2005, 91, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Dalal, D.; Nasir, K.; Bomma, C.; Prakasa, K.; Tandri, H.; Piccini, J.; Roguin, A.; Tichnell, C.; James, C.; Russell, S.D.; et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation 2005, 112, 3823–3832. [Google Scholar] [CrossRef]
- Watkins, D.A.; Hendricks, N.; Shaboodien, G.; Mbele, M.; Parker, M.; Vezi, B.Z.; Latib, A.; Chin, A.; Little, F.; Badri, M.; et al. Clinical features, survival experience, and profile of plakophylin-2 gene mutations in participants of the Arrhythmogenic Right Ventricular Cardiomyopathy Registry of South Africa. Heart Rhythm 2009, 6, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Pinamonti, B.; Dragos, A.M.; Pyxaras, S.A.; Merlo, M.; Pivetta, A.; Barbati, G.; Di Lenarda, A.; Morgera, T.; Mestroni, L.; Sinagra, G. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: Results from a 10-year registry. Eur. Heart J. 2011, 32, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Saguner, A.M.; Medeiros-Domingo, A.; Schwyzer, M.A.; On, C.-J.; Haegeli, L.M.; Wolber, T.; Hürlimann, D.; Steffel, J.; Krasniqi, N.; Rüeger, S.; et al. Usefulness of Inducible Ventricular Tachycardia to Predict Long-Term Adverse Outcomes in Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2013, 111, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Saguner, A.M.; Vecchiati, A.; Baldinger, S.H.; Rüeger, S.; Medeiros-Domingo, A.; Mueller-Burri, A.S.; Haegeli, L.M.; Biaggi, P.; Manka, R.; Lüscher, T.F.; et al. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ. Cardiovasc. Imaging 2014, 7, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Saberniak, J.; Hasselberg, N.E.; Borgquist, R.; Platonov, P.G.; Sarvari, S.I.; Smith, H.; Ribe, M.; Holst, A.G.; Edvardsen, T.; Haugaa, K.H. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur. J. Heart Fail. 2014, 16, 1337–1344. [Google Scholar] [CrossRef]
- Te Riele, A.S.J.M.; James, C.A.; Sawant, A.C.; Bhonsale, A.; Groeneweg, J.A.; Mast, T.P.; Murray, B.; Tichnell, C.; Dooijes, D.; Van Tintelen, J.P.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in the Pediatric Population Clinical Characterization and Comparison with Adult-Onset Disease. JACC Clin. Electrophysiol. 2015, 1, 551–560. [Google Scholar] [CrossRef]
- Gallo, C.; Blandino, A.; Giustetto, C.; Anselmino, M.; Castagno, D.; Richiardi, E.; Gaita, F. Arrhythmogenic right ventricular cardiomyopathy: ECG progression over time and correlation with long-term follow-up. J. Cardiovasc. Med. 2016, 17, 418–424. [Google Scholar] [CrossRef]
- Kimura, Y.; Noda, T.; Otsuka, Y.; Wada, M.; Nakajima, I.; Ishibashi, K.; Miyamoto, K.; Okamura, H.; Aiba, T.; Kamakura, S.; et al. Potentially Lethal Ventricular Arrhythmias and Heart Failure in Arrhythmogenic Right Ventricular Cardiomyopathy: What Are the Differences Between Men and Women? JACC Clin. Electrophysiol. 2016, 2, 546–555. [Google Scholar] [CrossRef]
- Bhonsale, A.; Te Riele, A.S.; Sawant, A.C.; Groeneweg, J.A.; James, C.A.; Murray, B.; Tichnell, C.; Mast, T.P.; van der Pols, M.J.; Cramer, M.J.; et al. Cardiac phenotype and long-term prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia patients with late presentation. Heart Rhythm 2017, 14, 883–891. [Google Scholar] [CrossRef]
- Gilljam, T.; Haugaa, K.H.; Jensen, H.K.; Svensson, A.; Bundgaard, H.; Hansen, J.; Dellgren, G.; Gustafsson, F.; Eiskjær, H.; Andreassen, A.K.; et al. Heart transplantation in arrhythmogenic right ventricular cardiomyopathy—Experience from the Nordic ARVC Registry. Int. J. Cardiol. 2018, 250, 201–206. [Google Scholar] [CrossRef]
- Orgeron, G.M.; James, C.A.; Riele, A.T.; Tichnell, C.; Murray, B.; Bhonsale, A.; Kamel, I.R.; Zimmerman, S.L.; Judge, D.P.; Crosson, J.; et al. Implantable Cardioverter-Defibrillator Therapy in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: Predictors of Appropriate Therapy, Outcomes, and Complications. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Mahida, S.; Venlet, J.; Saguner, A.M.; Kumar, S.; Baldinger, S.H.; AbdelWahab, A.; Tedrow, U.B.; Castelletti, S.; Pantazis, A.; John, R.M.; et al. Ablation compared with drug therapy for recurrent ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy: Results from a multicenter study. Heart Rhythm 2019, 16, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Liang, E.; Wu, L.; Fan, S.; Li, X.; Hu, F.; Zheng, L.; Fan, X.; Chen, G.; Ding, L.; Yao, Y. Bradyarrhythmias in Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2019, 123, 1690–1695. [Google Scholar] [CrossRef]
- Mathew, S.; Saguner, A.M.; Schenker, N.; Kaiser, L.; Zhang, P.; Yashuiro, Y.; Lemes, C.; Fink, T.; Maurer, T.; Santoro, F.; et al. Catheter Ablation of Ventricular Tachycardia in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: A Sequential Approach. J. Am. Heart Assoc. 2019, 8, e010365. [Google Scholar] [CrossRef] [PubMed]
- Laredo, M.; Da Silva, L.O.; Extramiana, F.; Lellouche, N.; Varlet, É.; Amet, D.; Algalarrondo, V.; Waintraub, X.; Duthoit, G.; Badenco, N.; et al. Catheter ablation of electrical storm in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2020, 17, 41–48. [Google Scholar] [CrossRef]
- Hermida, A.; Fressart, V.; Hidden-Lucet, F.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; et al. High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Heart Fail. 2019, 21, 792–800. [Google Scholar] [CrossRef]
- Bosman, L.P.; Verstraelen, T.E.; van Lint, F.H.M.; Cox, M.; Groeneweg, J.A.; Mast, T.P.; van der Zwaag, P.A.; Volders, P.G.A.; Evertz, R.; Wong, L.; et al. The Netherlands Arrhythmogenic Cardiomyopathy Registry: Design and status update. Neth. Heart J. 2019, 27, 480–486. [Google Scholar] [CrossRef]
- Kimura, Y.; Noda, T.; Matsuyama, T.-A.; Otsuka, Y.; Kamakura, T.; Wada, M.; Ishibashi, K.; Inoue, Y.; Miyamoto, K.; Okamura, H.; et al. Heart failure in patients with arrhythmogenic right ventricular cardiomyopathy: What are the risk factors? Int. J. Cardiol. 2017, 241, 288–294. [Google Scholar] [CrossRef]
- Camm, C.F.; James, C.A.; Tichnell, C.; Murray, B.; Bhonsale, A.; Te Riele, A.; Judge, D.P.; Tandri, H.; Calkins, H. Prevalence of atrial arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm 2013, 10, 1661–1668. [Google Scholar] [CrossRef]
- Girard, F.; Fontaine, G.; Fontaliran, F.; Zenati, O.; Gajdos, P. Catastrophic global heart failure in a patient with non-arrhythmogenic right ventricular dysplasia. Heart Vessel. 1997, 12, 152–154. [Google Scholar] [CrossRef]
- Corrado, D.; Fontaine, G.; Marcus, F.I.; McKenna, W.J.; Nava, A.; Thiene, G.; Wichter, T. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation 2000, 101, E101–E106. [Google Scholar]
- Chen, L.; Song, J.; Chen, X.; Chen, K.; Ren, J.; Zhang, N.; Rao, M.; Hu, Z.; Zhang, Y.; Gu, M.; et al. A novel genotype-based clinicopathology classification of arrhythmogenic cardiomyopathy provides novel insights into disease progression. Eur. Heart J. 2019, 40, 1690–1703. [Google Scholar] [CrossRef]
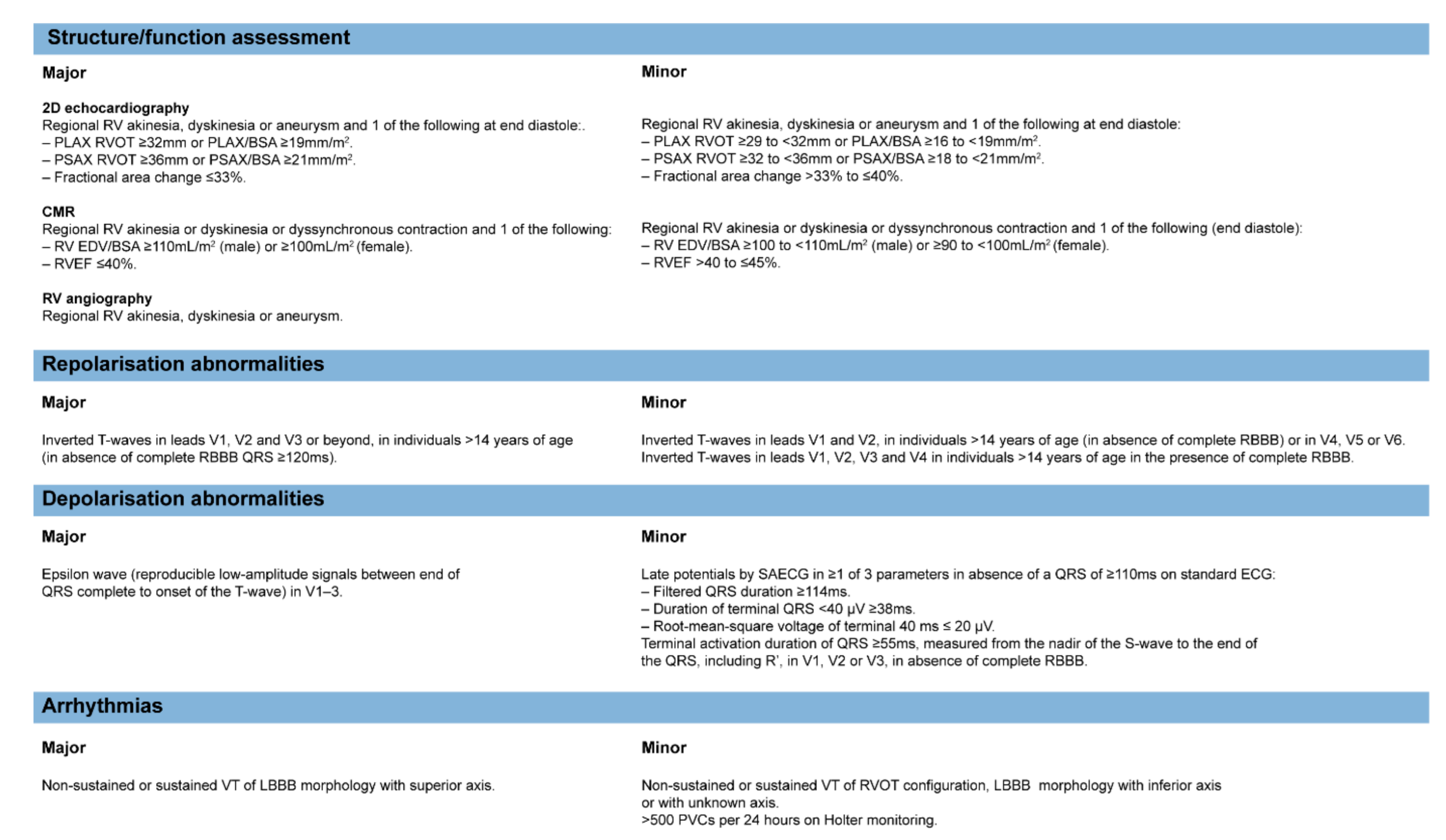
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed Modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [PubMed]
- Bernard, Y.; Meneveau, N.; Boucher, S.; Magnin, D.; Anguenot, T.; Schiele, F.; Vuillemenot, A.; Bassand, J.-P. Lack of agreement between left ventricular volumes and ejection fraction determined by two-dimensional echocardiography and contrast cineangiography in postinfarction patients. Echocardiography 2001, 18, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, L.; Cheng, H.; Song, Y.; Ji, K.; Chen, L.; Han, T.; Lu, M.; Zhao, S. Early Left Ventricular Involvement Detected by Cardiovascular Magnetic Resonance Feature Tracking in Arrhythmogenic Right Ventricular Cardiomyopathy: The Effects of Left Ventricular Late Gadolinium Enhancement and Right Ventricular Dysfunction. J. Am. Heart Assoc. 2019, 8, e012989. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef]
- Saguner, A.M.; Ganahl, S.; Kraus, A.; Baldinger, S.H.; Akdis, D.; Saguner, A.R.; Wolber, T.; Haegeli, L.M.; Steffel, J.; Krasniqi, N.; et al. Electrocardiographic features of disease progression in arrhythmogenic right ventricular cardiomyopathy/dysplasia. BMC Cardiovasc. Disord. 2015, 15, 4. [Google Scholar] [CrossRef]
- Roberts, W.C.; Kondapalli, N.; Hall, S.A. Usefulness of Total 12-Lead QRS Voltage for Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy in Patients With Heart Failure Severe Enough to Warrant Orthotopic Heart Transplantation and Morphologic Illustration of Its Cardiac Diversity. Am. J. Cardiol. 2018, 122, 1051–1061. [Google Scholar] [CrossRef]
- Saguner, A.M.; Ganahl, S.; Baldinger, S.H.; Kraus, A.; Medeiros-Domingo, A.; Nordbeck, S.; Saguner, A.R.; Mueller-Burri, A.S.; Haegeli, L.M.; Wolber, T.; et al. Usefulness of Electrocardiographic Parameters for Risk Prediction in Arrhythmogenic Right Ventricular Dysplasia. Am. J. Cardiol. 2014, 113, 1728–1734. [Google Scholar] [CrossRef]
- Cheng, H.; Lu, M.; Hou, C.; Chen, X.; Wang, J.; Yin, G.; Chu, J.; Zhang, S.; Prasad, S.K.; Pu, J.; et al. Relation Between N-Terminal Pro-Brain Natriuretic Peptide and Cardiac Remodeling and Function Assessed by Cardiovascular Magnetic Resonance Imaging in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2015, 115, 341–347. [Google Scholar] [CrossRef]
- Matsuo, K.; Nishikimi, T.; Yutani, C.; Kurita, T.; Shimizu, W.; Taguchi, A.; Suyama, K.; Aihara, N.; Kamakura, S.; Kangawa, K.; et al. Diagnostic Value of Plasma Levels of Brain Natriuretic Peptide in Arrhythmogenic Right Ventricular Dysplasia. Circulation 1998, 98, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.-J.; Huang, Y.-X.; Shen, Y.; Cui, C.-J.; Zhang, X.-L.; Zhang, H.; Hu, S.-S. Proteomic analysis reveals significant elevation of heat shock protein 70 in patients with chronic heart failure due to arrhythmogenic right ventricular cardiomyopathy. Mol. Cell. Biochem. 2009, 332, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A. BIN1: A new biomarker to track ARVC? Heart Rhythm 2012, 9, 968–969. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.-T.; Cogswell, R.; James, C.A.; Kang, G.; Pullinger, C.R.; Malloy, M.J.; Kane, J.P.; Wojciak, J.; Calkins, H.; Scheinman, M.M.; et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2012, 9, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Broch, K.; Leren, I.S.; Saberniak, J.; Ueland, T.; Edvardsen, T.; Gullestad, L.; Haugaa, K.H. Soluble ST2 is associated with disease severity in arrhythmogenic right ventricular cardiomyopathy. Biomarkers 2017, 22, 367–371. [Google Scholar] [CrossRef]
- Oz, F.; Onur, I.; Elitok, A.; Ademoglu, E.; Altun, I.; Bilge, A.K.; Adalet, K. Galectin-3 correlates with arrhythmogenic right ventricular cardiomyopathy and predicts the risk of ventricular arrhythmias in patients with implantable defibrillators. Acta Cardiol. 2017, 72, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Tsilafakis, K.; Chen, L.; Lekkos, K.; Kostavasili, I.; Varela, A.; Cokkinos, D.V.; Davos, C.H.; Sun, X.; Song, J.; et al. Crosstalk between coagulation and complement activation promotes cardiac dysfunction in arrhythmogenic right ventricular cardiomyopathy. Theranostics 2021, 11, 5939–5954. [Google Scholar] [CrossRef]
- van der Voorn, S.M.; Te Riele, A.; Basso, C.; Calkins, H.; Remme, C.A.; van Veen, T.A.B. Arrhythmogenic cardiomyopathy: Pathogenesis, pro-arrhythmic remodelling, and novel approaches for risk stratification and therapy. Cardiovasc. Res. 2020, 116, 1571–1584. [Google Scholar] [CrossRef]
- Casella, M.; Bergonti, M.; Dello Russo, A.; Maragna, R.; Gasperetti, A.; Compagnucci, P.; Catto, V.; Trombara, F.; Frappampina, A.; Conte, E.; et al. Endomyocardial Biopsy: The Forgotten Piece in the Arrhythmogenic Cardiomyopathy Puzzle. J. Am. Heart Assoc. 2021, 10, e021370. [Google Scholar] [CrossRef]
- Lutokhina, Y.; Blagova, O.; Nedostup, A.; Alexandrova, S.; Shestak, A.; Zaklyazminskaya, E. Clinical Classification of Arrhythmogenic Right Ventricular Cardiomyopathy. Pulse 2020, 8, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Duru, F.; Hauer, R.N.W. Multiple facets of arrhythmogenic cardiomyopathy: The Fuwai classification of a unique disease based on clinical features, histopathology, and genotype. Eur. Heart J. 2019, 40, 1704–1706. [Google Scholar] [CrossRef]
- Bauce, B.; Frigo, G.; Marcus, F.I.; Basso, C.; Rampazzo, A.; Maddalena, F.; Corrado, D.; Winnicki, M.; Daliento, L.; Rigato, I.; et al. Comparison of Clinical Features of Arrhythmogenic Right Ventricular Cardiomyopathy in Men Versus Women. Am. J. Cardiol. 2008, 102, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.R., 3rd; Frederick, J.R.; Hsu, V.M.; Kozin, E.D.; O’Hara, M.L.; Howell, E.; Dougherty, D.; McCormick, R.C.; Laporte, C.A.; Cohen, J.E.; et al. Risk score derived from pre-operative data analysis predicts the need for biventricular mechanical circulatory support. J. Heart Lung Transplant. 2008, 27, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, Y.; McCarthy, P.M.; Smedira, N.G.; Banbury, M.K.; Navia, J.L.; Feng, J.; Hsu, A.P.; Yeager, M.L.; Buda, T.; Hoercher, K.J.; et al. Predictors of severe right ventricular failure after implantable left ventricular assist device insertion: Analysis of 245 patients. Circulation 2002, 106, I-198–I-202. [Google Scholar]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies With Disease Severity and Family History. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy According to Revised 2010 Task Force Criteria With Inclusion of Non-Desmosomal Phospholamban Mutation Carriers. Am. J. Cardiol. 2013, 112, 1197–1206. [Google Scholar] [CrossRef]
- Link, M.S.; Laidlaw, D.; Polonsky, B.; Zareba, W.; McNitt, S.; Gear, K.; Marcus, F.; Estes, N.A., 3rd. Ventricular arrhythmias in the North American multidisciplinary study of ARVC: Predictors, characteristics, and treatment. J. Am. Coll. Cardiol. 2014, 64, 119–125. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Koehler, B. Special features of right bundle branch block in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Int. J. Cardiol. 2012, 157, 102–103. [Google Scholar] [CrossRef]
- Fressart, V.; Duthoit, G.; Donal, E.; Probst, V.; Deharo, J.-C.; Chevalier, P.; Klug, D.; Dubourg, O.; Delacretaz, E.; Cosnay, P.; et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. Europace 2010, 12, 861–868. [Google Scholar] [CrossRef]
- Chen, L.; Rao, M.; Chen, X.; Chen, K.; Ren, J.; Zhang, N.; Zhao, Q.; Yu, W.; Yuan, B.; Song, J. A founder homozygous DSG2 variant in East Asia results in ARVC with full penetrance and heart failure phenotype. Int. J. Cardiol. 2019, 274, 263–270. [Google Scholar] [CrossRef]
- Castelletti, S.; Vischer, A.S.; Syrris, P.; Crotti, L.; Spazzolini, C.; Ghidoni, A.; Parati, G.; Jenkins, S.; Kotta, M.-C.; McKenna, W.J.; et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. Int. J. Cardiol. 2017, 249, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Bauce, B.; Rampazzo, A.; Basso, C.; Mazzotti, E.; Rigato, I.; Steriotis, A.; Beffagna, G.; Lorenzon, A.; De Bortoli, M.; Pilichou, K.; et al. Clinical phenotype and diagnosis of arrhythmogenic right ventricular cardiomyopathy in pediatric patients carrying desmosomal gene mutations. Heart Rhythm 2011, 8, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and Genetic Characterization of Families With Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Provides Novel Insights Into Patterns of Disease Expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef]
- Norman, M.; Simpson, M.; Mogensen, J.; Shaw, A.; Hughes, S.; Syrris, P.; Sen-Chowdhry, S.; Rowland, E.; Crosby, A.; McKenna, W.J. Novel Mutation in Desmoplakin Causes Arrhythmogenic Left Ventricular Cardiomyopathy. Circulation 2005, 112, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- Fish, M.; Shaboodien, G.; Kraus, S.; Sliwa, K.; Seidman, C.E.; Burke, M.A.; Crotti, L.; Schwartz, P.J.; Mayosi, B.M. Mutation analysis of the phospholamban gene in 315 South Africans with dilated, hypertrophic, peripartum and arrhythmogenic right ventricular cardiomyopathies. Sci. Rep. 2016, 6, 22235. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; van der Zwaag, P.A.; Jongbloed, J.D.; Cox, M.G.; Vreeker, A.; de Boer, R.A.; van der Heijden, J.F.; van Veen, T.A.; McKenna, W.J.; van Tintelen, J.P.; et al. Left-dominant arrhythmogenic cardiomyopathy in a large family: Associated desmosomal or nondesmosomal genotype? Heart Rhythm 2013, 10, 548–559. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.; de Ruiter, R.; Nannenberg, E.A.; Groeneweg, J.A.; Post, J.G.; Hauer, R.N.; van Gelder, I.C.; van den Berg, M.P.; van der Harst, P.; et al. Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth. Heart J. 2013, 21, 286–293. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.; van der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.; Zwinderman, A.H.; Pinto, Y.M.; Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers: Results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef]
- Brun, F.; Barnes, C.V.; Sinagra, G.; Slavov, D.; Barbati, G.; Zhu, X.; Graw, S.L.; Spezzacatene, A.; Pinamonti, B.; Merlo, M.; et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2014, 51, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Song, J.; Chen, L.; Zhu, C.; Cai, H.; Sun, M.; Stern, A.; Mozdziak, P.; Ge, Y.; Means, W.J.; et al. Characterization of TTN Novex Splicing Variants across Species and the Role of RBM20 in Novex-Specific Exon Splicing. Genes 2018, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Song, J.; Wang, Z.; Rao, M.; Chen, L.; Hu, S. Absence of a primary role for TTN missense variants in arrhythmogenic cardiomyopathy: From a clinical and pathological perspective. Clin. Cardiol. 2018, 41, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Guo, G.; Li, M.; Chen, S.; Chen, K.; Chen, X.; Song, J.; Hu, S. The homozygous variant c.245G > A/p.G82D in PNPLA2 is associated with arrhythmogenic cardiomyopathy phenotypic manifestations. Clin. Genet. 2019, 96, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Krusche, C.A.; Holthöfer, B.; Hofe, V.; van de Sandt, A.M.; Eshkind, L.; Bockamp, E.; Merx, M.W.; Kant, S.; Windoffer, R.; Leube, R.E. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res. Cardiol. 2011, 106, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.; Finocchiaro, G.; Papadakis, M.; Gray, B.; Westaby, J.; Ensam, B.; Basu, J.; Parry-Williams, G.; Papatheodorou, E.; Paterson, C.; et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation 2019, 139, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.C.; Laksman, Z.W.; Mellor, G.; Sanatani, S.; Krahn, A.D. Exercise and Inherited Arrhythmias. Can. J. Cardiol. 2016, 32, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Cadrin-Tourigny, J.; Bosman, L.P.; Tadros, R.; Talajic, M.; Rivard, L.; James, C.A.; Khairy, P. Risk stratification for ventricular arrhythmias and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy: An update. Expert Rev. Cardiovasc. Ther. 2019, 17, 645–651. [Google Scholar] [CrossRef]
- Costa, S.; Gasperetti, A.; Medeiros-Domingo, A.; Akdis, D.; Brunckhorst, C.; Saguner, A.M.; Duru, F. Familial Arrhythmogenic Cardiomyopathy: Clinical Determinants of Phenotype Discordance and the Impact of Endurance Sports. J. Clin. Med. 2020, 9, 3781. [Google Scholar] [CrossRef]
- Akdis, D.; Saguner, A.M.; Burri, H.; Medeiros-Domingo, A.; Matter, C.M.; Ruschitzka, F.; Tanner, F.C.; Brunckhorst, C.; Duru, F. Clinical predictors of left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy. Am. Heart J. 2020, 223, 34–43. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of Clinicopathologic Manifestations of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: A Multicenter Study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Maron, B.J.; Chaitman, B.R.; Ackerman, M.J.; Bayes de Luna, A.; Corrado, D.; Crosson, J.E.; Deal, B.J.; Driscoll, D.J.; Estes, N.A., 3rd; Araujo, C.G.; et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004, 109, 2807–2816. [Google Scholar] [CrossRef]
- Ruwald, A.C.; Marcus, F.; Estes, N.A., 3rd; Link, M.; McNitt, S.; Polonsky, B.; Calkins, H.; Towbin, J.A.; Moss, A.J.; Zareba, W. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: Results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2015, 36, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Bundgaard, H.; Edvardsen, T.; Eschen, O.; Gilljam, T.; Hansen, J.; Jensen, H.K.; Platonov, P.G.; Svensson, A.; Svendsen, J.H. Management of patients with Arrhythmogenic Right Ventricular Cardiomyopathy in the Nordic countries. Scand. Cardiovasc. J. 2015, 49, 299–307. [Google Scholar] [CrossRef]
- Nagendran, J.; Archer, S.L.; Soliman, D.; Gurtu, V.; Moudgil, R.; Haromy, A.; St Aubin, C.; Webster, L.; Rebeyka, I.M.; Ross, D.B.; et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007, 116, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.; Calabrese, F.; Thiene, G.; Angelini, A.; Basso, C.; Nava, A.; Rossi, L. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am. J. Pathol. 1998, 152, 479–484. [Google Scholar] [PubMed]
- Corrado, D.; Wichter, T.; Link, M.S.; Hauer, R.; Marchlinski, F.; Anastasakis, A.; Bauce, B.; Basso, C.; Brunckhorst, C.; Tsatsopoulou, A.; et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: An international task force consensus statement. Eur. Heart J. 2015, 36, 3227–3237. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Chen, K.; Saguner, A.M.; Stämpfli, S.F.; Chen, X.; Chen, L.; Rao, M.; Haegeli, L.M.; Tanner, F.C.; Brunckhorst, C.; et al. Clinical Characteristics of Patients with a Right Ventricular Thrombus in Arrhythmogenic Right Ventricular Cardiomyopathy. Thromb. Haemost. 2019, 119, 1373–1378. [Google Scholar] [CrossRef]
- Chachques, J.C.; Argyriadis, P.G.; Fontaine, G.; Hebert, J.-L.; Frank, R.A.; D’Attellis, N.; Fabiani, J.-N.; Carpentier, A.F. Right ventricular cardiomyoplasty: 10-year follow-up. Ann. Thorac. Surg. 2003, 75, 1464–1468. [Google Scholar] [CrossRef]
- Zacharias, J.; Forty, J.; Doig, J.C.; Bourke, J.P.; Hilton, C.J. Right ventricular disarticulation. An 18-year single centre experience. Eur. J. Cardiothorac. Surg. 2005, 27, 1000–1004. [Google Scholar] [CrossRef]
- Bakir, I.; Brugada, P.; Sarkozy, A.; Vandepitte, C.; Wellens, F. A novel treatment strategy for therapy refractory ventricular arrhythmias in the setting of arrhythmogenic right ventricular dysplasia. Europace 2007, 9, 267–269. [Google Scholar] [CrossRef]
- Coleman, M.A.; Bos, J.M.; Johnson, J.N.; Owen, H.J.; Deschamps, C.; Moir, C.; Ackerman, M.J. Videoscopic Left Cardiac Sympathetic Denervation for Patients With Recurrent Ventricular Fibrillation/Malignant Ventricular Arrhythmia Syndromes Besides Congenital Long-QT Syndrome. Circ. Arrhythmia Electrophysiol. 2012, 5, 782–788. [Google Scholar] [CrossRef]
- McGiffin, D.; Kure, C.; McLean, J.; Marasco, S.; Bergin, P.; Hare, J.L.; Leet, A.; Patel, H.; Zimmet, A.; Rix, J.; et al. The results of a single-center experience with HeartMate 3 in a biventricular configuration. J. Heart Lung Transplant. 2021, 40, 193–200. [Google Scholar] [CrossRef]
- Minegishi, S.; Kinoshita, O.; Hoshino, Y.; Komae, H.; Kimura, M.; Shimada, S.; Yamauchi, H.; Nawata, K.; Ono, M. Long-term support by left ventricular assist device for arrhythmogenic right ventricular cardiomyopathy. Artif. Organs 2019, 43, 909–912. [Google Scholar] [CrossRef]
- Mufti, H.N.; Rajda, M.; Légaré, J.-F. Arrhythmogenic right ventricular cardiomyopathy: Use of a left ventricular assist device as a bridge to transplantation? J. Artif. Organs 2013, 16, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, D.; Toda, K.; Yoshikawa, Y.; Sawa, Y. Over 1200-day support with dual Jarvik 2000 biventricular assist device. Interact. Cardiovasc. Thorac. Surg. 2014, 19, 1083–1084. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Iacovoni, A.; Scardulla, C.; Moja, L.; Pilato, M.; Kushwaha, S.S.; Senni, M.; Clemenza, F.; Agnese, V.; Falletta, C.; et al. Prediction of right ventricular failure after ventricular assist device implant: Systematic review and meta-analysis of observational studies. Eur. J. Heart Fail. 2017, 19, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.C.; Kuo, J.Y.; Yun, C.H.; Hung, C.L.; Tsai, C.H.; Yeh, H.I. Rare case of left-dominant arrhythmogenic right ventricular cardiomyopathy with dramatic reverse remodeling after cardiac resynchronization as an adjunct to pharmacological therapy. Heart Lung 2012, 41, e39–e43. [Google Scholar] [CrossRef] [PubMed]
- Mehra, M.R.; Canter, C.E.; Hannan, M.M.; Semigran, M.J.; Uber, P.A.; Baran, D.A.; Danziger-Isakov, L.; Kirklin, J.K.; Kirk, R.; Kushwaha, S.S.; et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J. Heart Lung Transplant. 2016, 35, 1–23. [Google Scholar] [CrossRef] [PubMed]
- DePasquale, E.C.; Cheng, R.K.; Deng, M.C.; Nsair, A.; McKenna, W.J.; Fonarow, G.C.; Jacoby, D.L. Survival After Heart Transplantation in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. J. Card. Fail. 2017, 23, 107–112. [Google Scholar] [CrossRef]
- Tedford, R.J.; James, C.; Judge, D.P.; Tichnell, C.; Murray, B.; Bhonsale, A.; Philips, B.; Abraham, T.; Dalal, D.; Halushka, M.K.; et al. Cardiac Transplantation in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Am. Coll. Cardiol. 2012, 59, 289–290. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
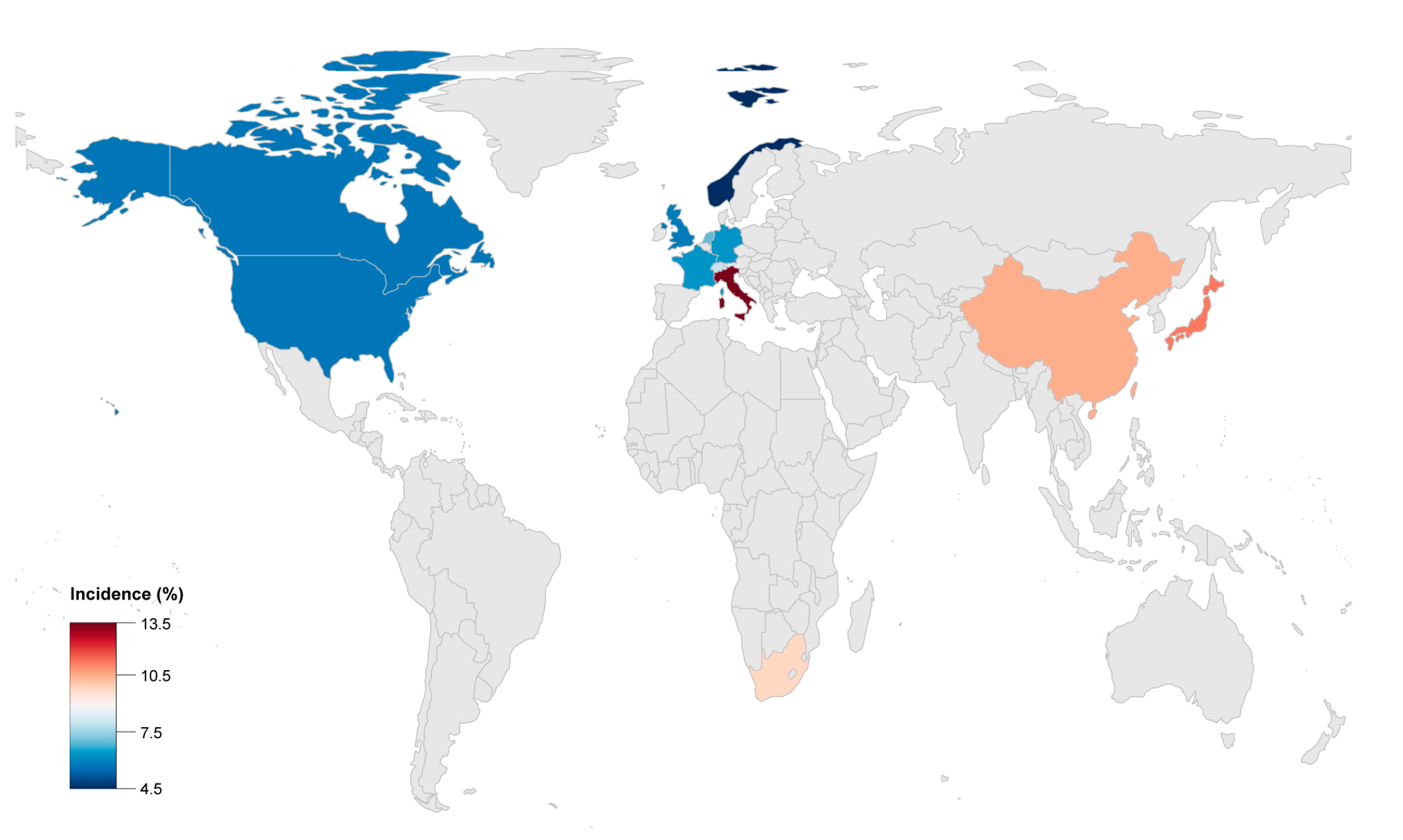
| Author | Publication Year | Study Population | Enrollment Period | Median Follow-Up (Years) | Male (%) | Endpoint | Incidence (%) | Malignant Arrythmias Events |
|---|---|---|---|---|---|---|---|---|
| Stefan Peters [17] | 1999 | 121 | 1986–1998 | 12.0 | 52.9 | 1 HTx/3 death | 3.3 | 14 RBBB |
| Jean-Sébastien Hulot [18] | 2004 | 130 | 1977–2000 | 8.1 | 76.9 | 14 death | 10.8 | 7 SCD, 17 SCA, 8 VF, 11 supraventricular arrhythmias |
| K Lemola [19] | 2005 | 61 | 4.6 | 72.1 | 5 HTx/2 death | 11.5 | 8 SCD, 8 SCA, 46 VT, 44 VT+LBBB | |
| Darshan Dalal [20] | 2005 | 100 | 6.0 | 51.0 | 2 HTx/1 death | 3 | 22 SCD, 1 SCA, 51 VT+LBBB | |
| Stefan Peters [15] | 2007 | 313 | 1986–2004 | 8.5 | 62.9 | 2 HTx/5 death | 2.2 | 5 SCD, 21 SCA, 96 SVT, 38 atrial arrhythmias |
| David A. Watkins [21] | 2009 | 50 | 2004–2009 | 4.6 | 66.0 | 2 HTx/3 death | 10 | 2 SCA, 41VT+LBBB |
| Bruno Pinamonti [22] | 2011 | 96 | 1976–2008 | 10.7 | 70.8 | 7 HTx/6 death | 13.5 | 3 SCA, 4 RBBB, 19 VT+LBBB, 4 atrial arrhythmias, 19 supraventricular arrhythmias |
| Masatoshi Komura [12] | 2010 | 35 | 4.5 | 74.3 | 4 death | 11.4 | 1 SCD | |
| Ardan M. Saguner [23] | 2013 | 62 | 7.0 | 67.7 | 3 HTx/2 death | 8.1 | 7 SCA, 15 VF, 6 supraventricular arrhythmias | |
| Ardan M. Saguner [24] | 2014 | 70 | 5.3 | 67.1 | 5 HTx | 7.1 | 2 SCA, 25 sustained VT, 7 VF | |
| Jørg Saberniak [25] | 2014 | 110 | 58.2 | 5 HTx | 4.6 | 66 VA | ||
| Anneline S.J.M. te Riele [26] | 2015 | 75 | 9.0 | 54.7 | 2 HTx/2 death | 5.3 | 11 SCD, 8 SCA, 16 sustained VT | |
| Judith A. Groeneweg [14] | 2015 | 439 | 7.0 | 64.2 | 18 HTx/6 death | 5.8 | 21 SCA, 96 sustained VT, 38 atrial arrhythmias | |
| Aditya Bhonsale [11] | 2015 | 541 | 6.0 | 58.8 | 8 HTx/12 death | 3.7 | 36 SCD, 16 SCA | |
| Cristina Gallo [27] | 2016 | 68 | 1970–2014 | 17.0 | 69.1 | 3 HTx/4 death | 10.3 | 3 SCD, 1 SCA |
| Yoshitaka Kimura [28] | 2016 | 110 | 10.0 | 75.5 | 2 HTx/8 death | 9.1 | 74 VT/VF | |
| Aditya Bhonsale [29] | 2016 | 502 | 52.6 | 19 HTx/53 death | 14.3 | 34 SCD, 24 SCA, 167 sustained VT | ||
| Nisha A. Gilotra [10] | 2017 | 289 | 1998–2014 | 50.9 | 15 HTx/7 death | 7.6 | 2 SCD, 6 SCA | |
| Thomas Gilljam [30] | 2018 | 183 | 1988–2015 | 67.2 | 28 HTx | 15.3 | 45 VT, 8 SCA, 18 atrial arrhythmias | |
| Gabriela M. Orgeron [31] | 2017 | 312 | 7.0 | 52.2 | 2 HTx/12 death | 4.5 | 158 sustained VT, 19 VF | |
| Saagar Mahida [32] | 2019 | 110 | 2000–2015 | 6.4 | 82.7 | 10 HTx/3 death | 11.8 | 3 SCD |
| Annina S. Vischer [9] | 2019 | 135 | 7.0 | 61.5 | 5 HTx/3 death | 5.9 | - | |
| Erpeng Liang [33] | 2019 | 522 | 1995–2017 | 4.3 | 71.5 | 53 HTx/62 death | 22.0 | 14 SCD, 136 sustained VT |
| Shibu Mathew [34] | 2019 | 47 | 1998–2016 | 4.2 | 100 | 1 HTx/2 death | 6.4 | 4 SCA, 18 sustained VT |
| Mikael Laredo [35] | 2019 | 23 | 2003–2015 | 3.9 | 100 | 4 HTx | 17.4 | 19 VT |
| Alexis Hermida [36] | 2019 | 118 | 2006–2013 | 5.6 | 72.9 | 9 HTx/1 death | 8.5 | 54 SVT/VF/SCA/SCD |
| Elizabeth S. DeWitt [13] | 2019 | 32 | 56.3 | 10 HTx | 31.3 | 5 SCA, 14 VT | ||
| L. P. Bosman [37] | 2019 | 850 | 9.5 | 52.1 | 7 HTx/53 death | 7.1 | - |
| Cluster 1 (Classical ACM) | Cluster 2 | Cluster 3 | Cluster 4 | |
|---|---|---|---|---|
| Clinical features | Early-onset disease, ventricular arrhythmias common, usually progressive RV (and later LV) disease, large RVEDD on echo, precordial fractionation and low voltage, MACE common during follow-up | Ventricular arrhythmias common, usually progressive disease, moderate-severe LV dysfunction, precordial fractionation and low voltage | Ventricular arrhythmias common, usually progressive disease, severe LV dysfunction, large LVEDD on echo, progression to end-stage heart failure common | Ventricular arrhythmias common, usually progressive disease, severe LV dysfunction, large LVEDD and LA diameter on echo, progression to end-stage heart failure common |
| Histopathology | Fibrofatty infiltration RV subepicardial (early), transmural (late), LV posterior wall | Fibrofatty infiltration of RV anterior wall, LV interstitial fibrosis in full thickness with only little fat | Biventricular involvement with prominent fibrofatty infiltration, LV involvement mostly of inferior wall | LV dominant involvement, mostly of inferior wall, with prominent fibrofatty infiltration |
| Genetic variants | Mostly desmosomal (PKP2, DSG2, DSC2) | Mostly non-desmosomal (LMNA, PLN, TMEM43, DES, CTNNA3) | Mostly desmosomal (DSP) or non-desmosomal (PLN, CTNNA3) | No genetic variants |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Chen, L.; Duru, F.; Hu, S. Heart Failure in Patients with Arrhythmogenic Cardiomyopathy. J. Clin. Med. 2021, 10, 4782. https://doi.org/10.3390/jcm10204782
Chen S, Chen L, Duru F, Hu S. Heart Failure in Patients with Arrhythmogenic Cardiomyopathy. Journal of Clinical Medicine. 2021; 10(20):4782. https://doi.org/10.3390/jcm10204782
Chicago/Turabian StyleChen, Shi, Liang Chen, Firat Duru, and Shengshou Hu. 2021. "Heart Failure in Patients with Arrhythmogenic Cardiomyopathy" Journal of Clinical Medicine 10, no. 20: 4782. https://doi.org/10.3390/jcm10204782
APA StyleChen, S., Chen, L., Duru, F., & Hu, S. (2021). Heart Failure in Patients with Arrhythmogenic Cardiomyopathy. Journal of Clinical Medicine, 10(20), 4782. https://doi.org/10.3390/jcm10204782