
The Predominant Role of Arrestin3 in General GPCR Desensitization in Platelets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Isolation of Mouse Platelets
2.4. Platelet Aggregation and Dense Granule Secretion
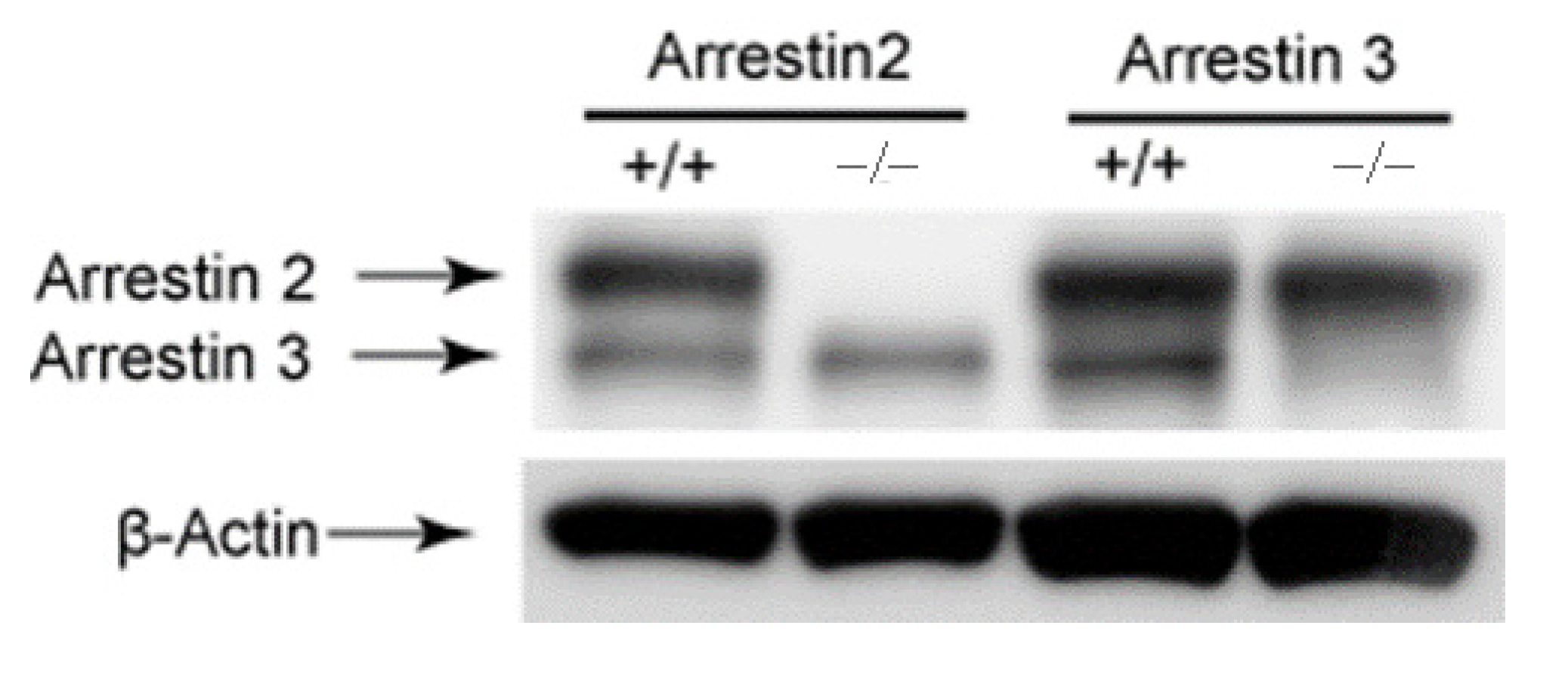
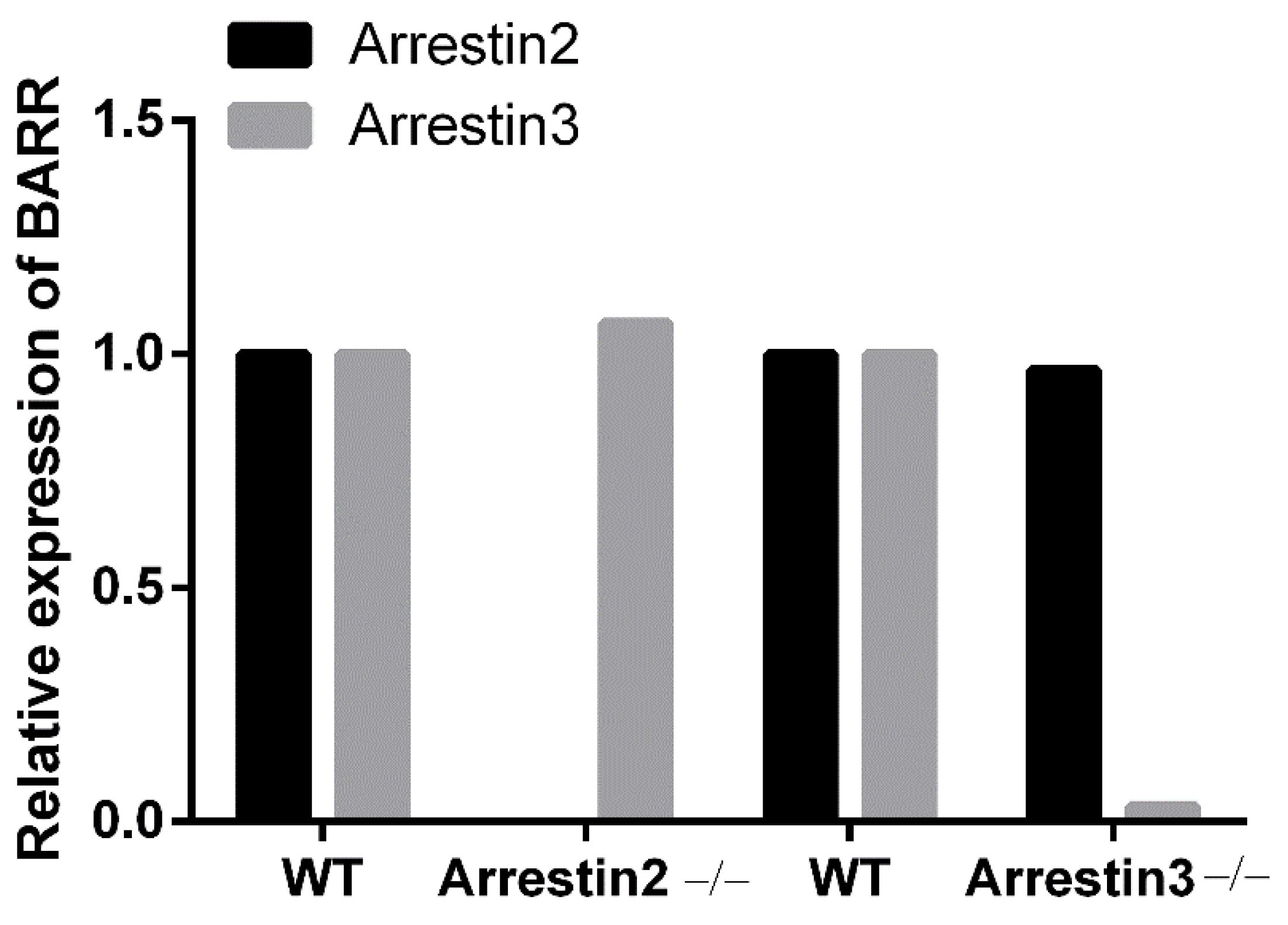
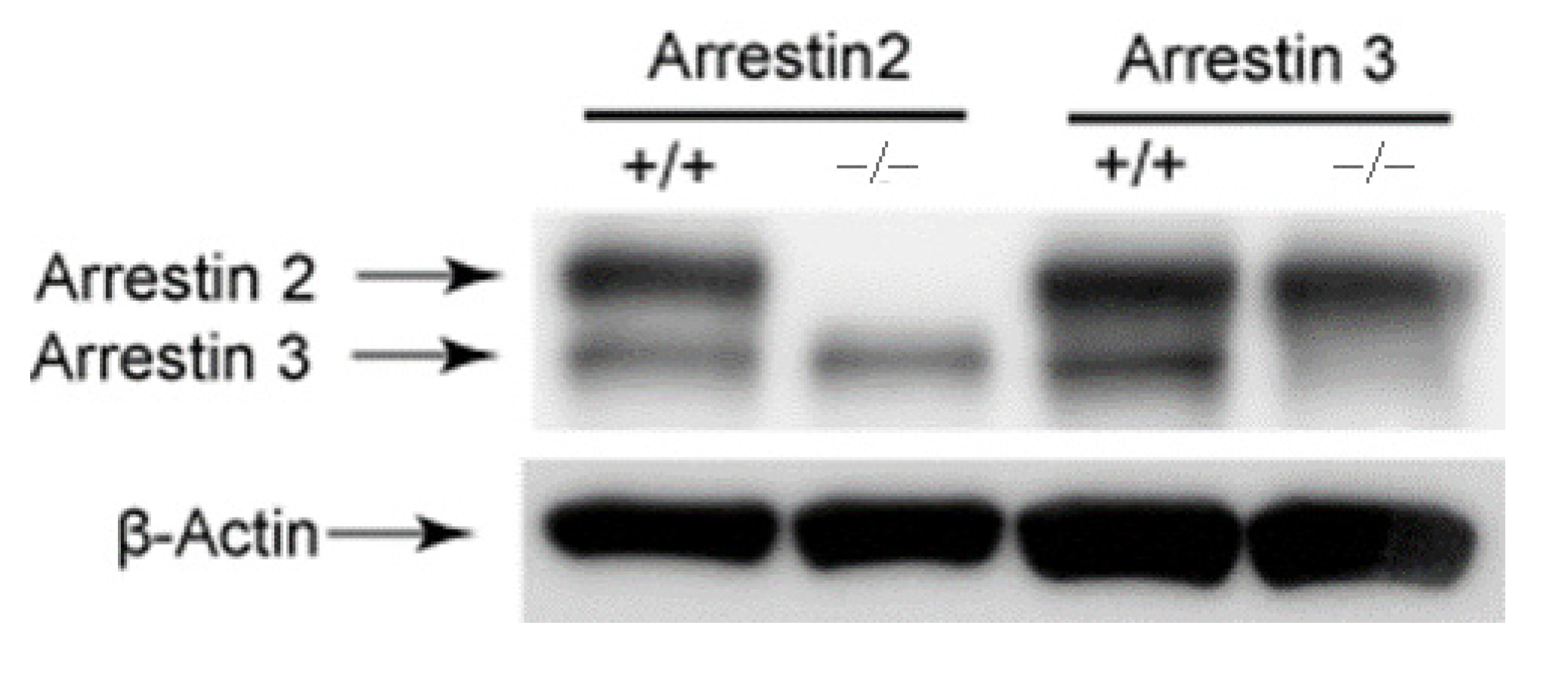
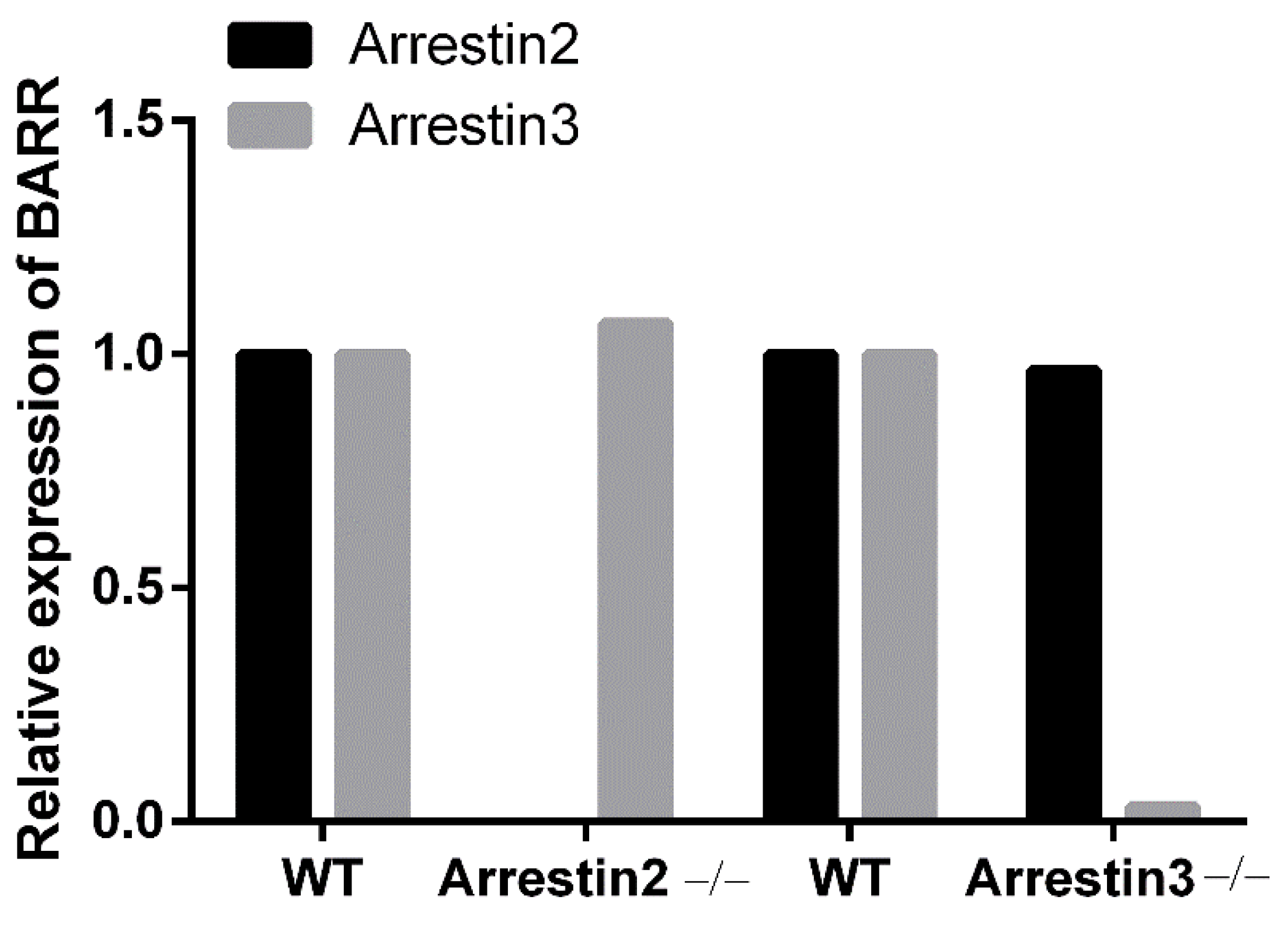
2.5. Immuno-Blotting
2.6. In Vivo Thrombosis Model Using FeCl3-Induced Carotid Artery Injury
2.7. Statistical Analysis
3. Results
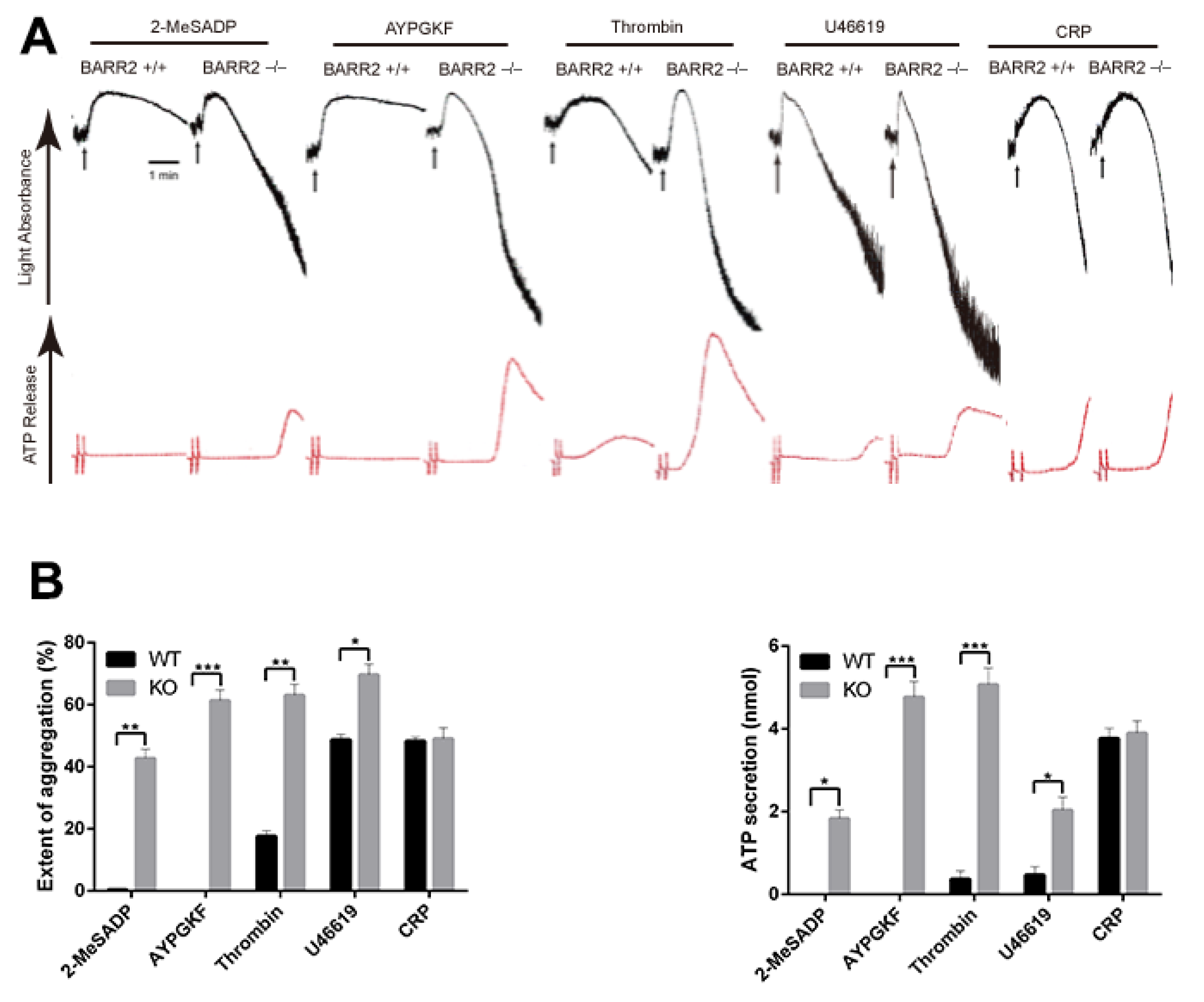
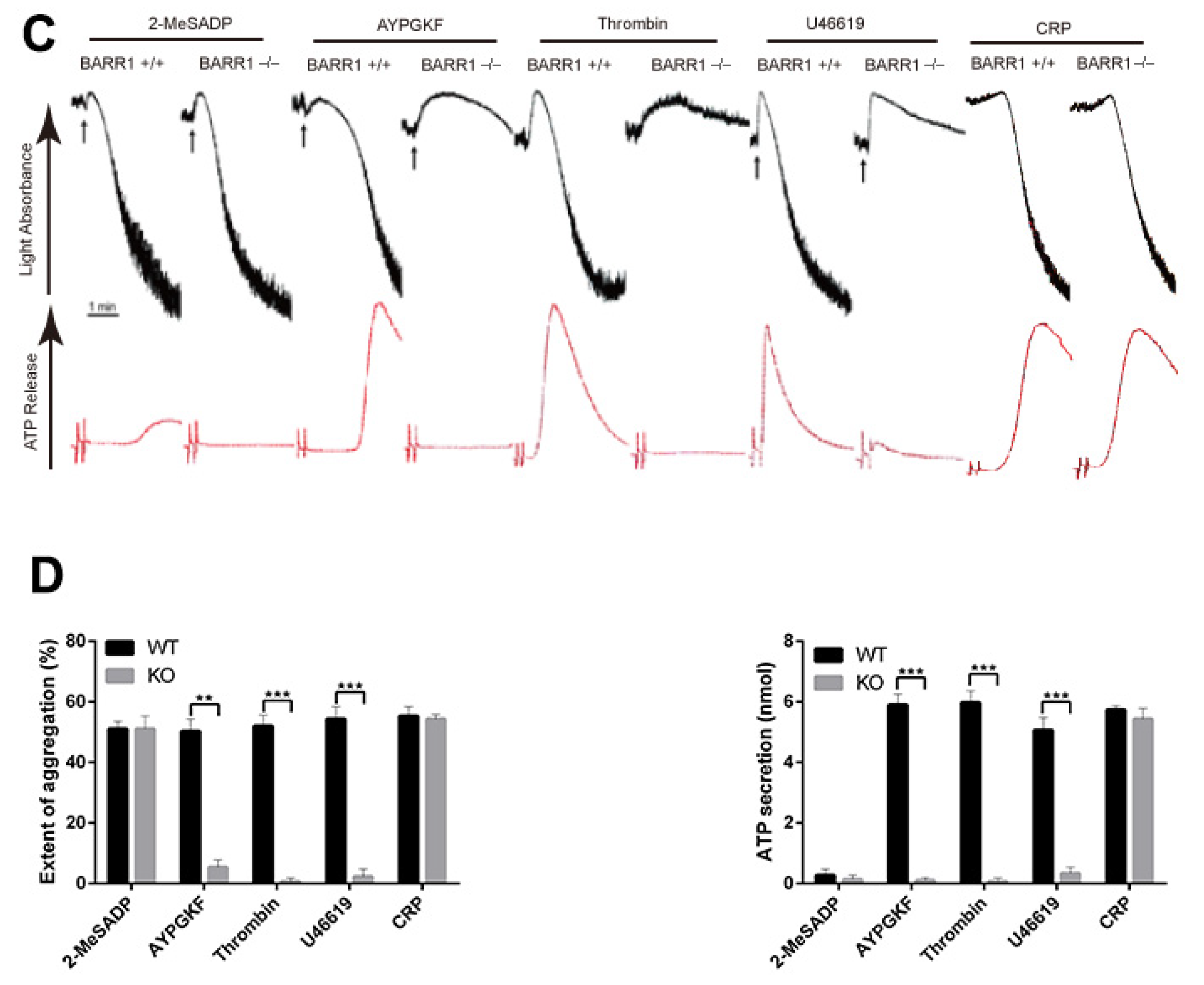
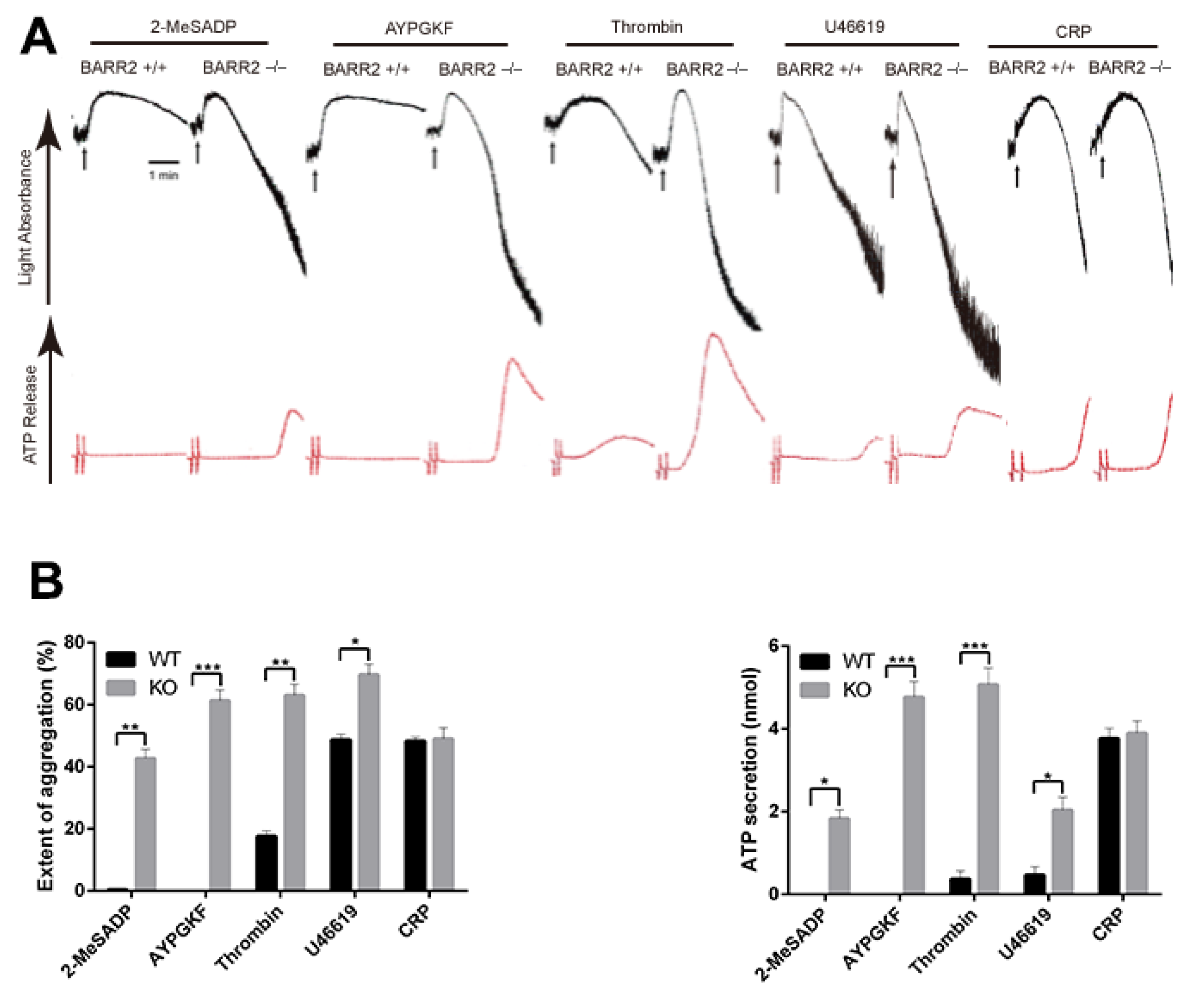
3.1. Arrestin3 Selectively Regulates Agonist-Induced Platelet Aggregation and Secretion
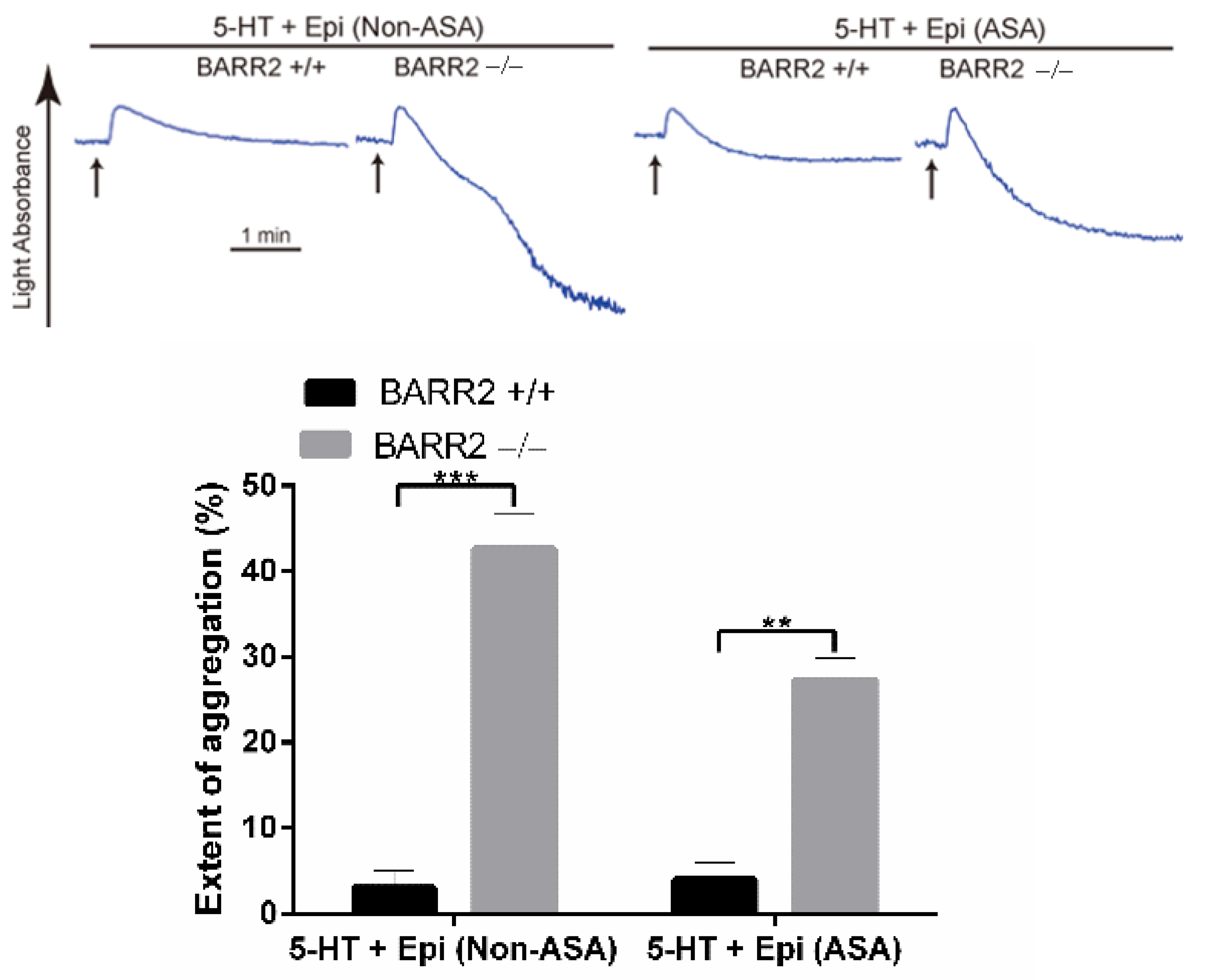
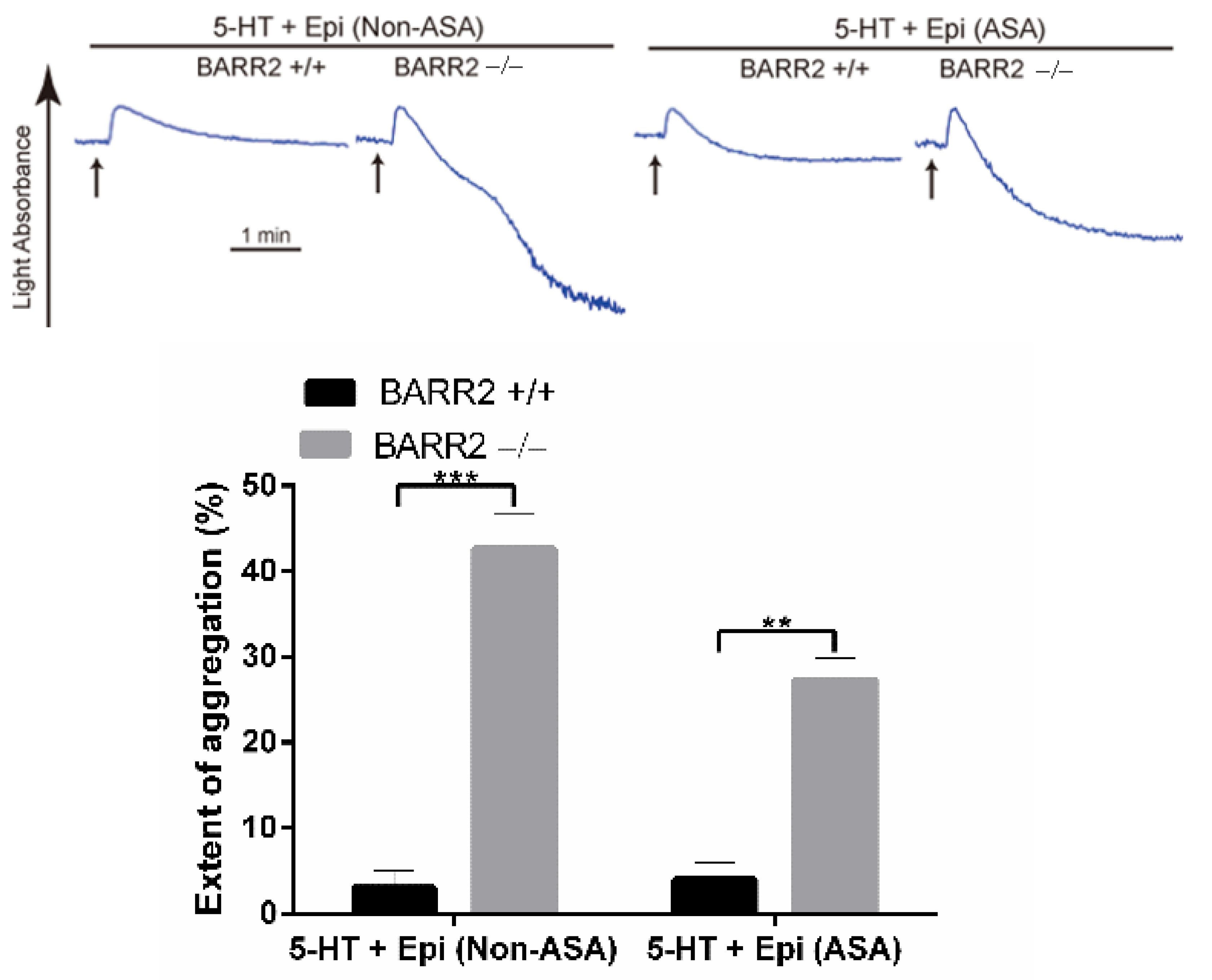
3.2. Arrestin3 Regulates Serotonin- and Epinephrine-Induced Platelet Aggregation
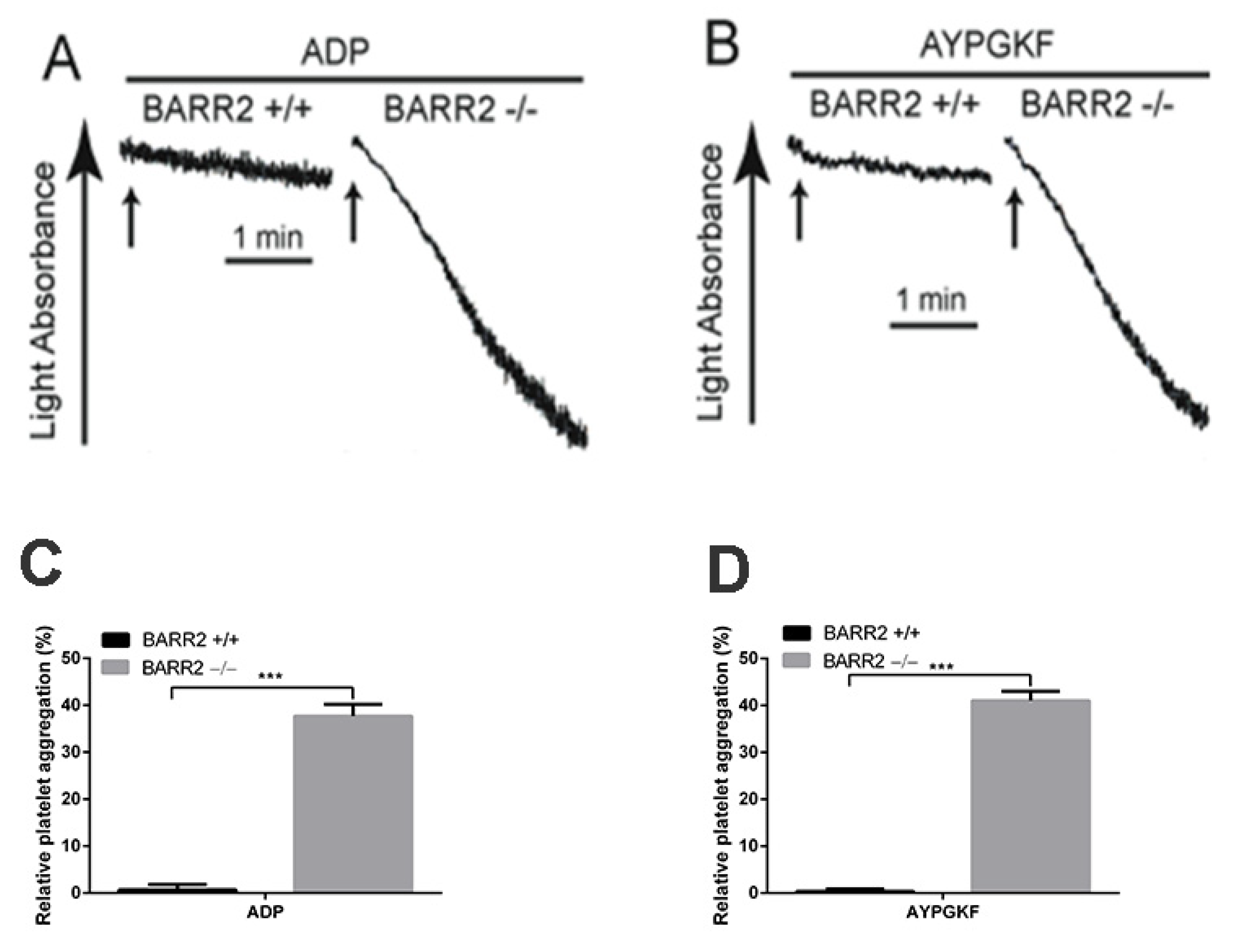
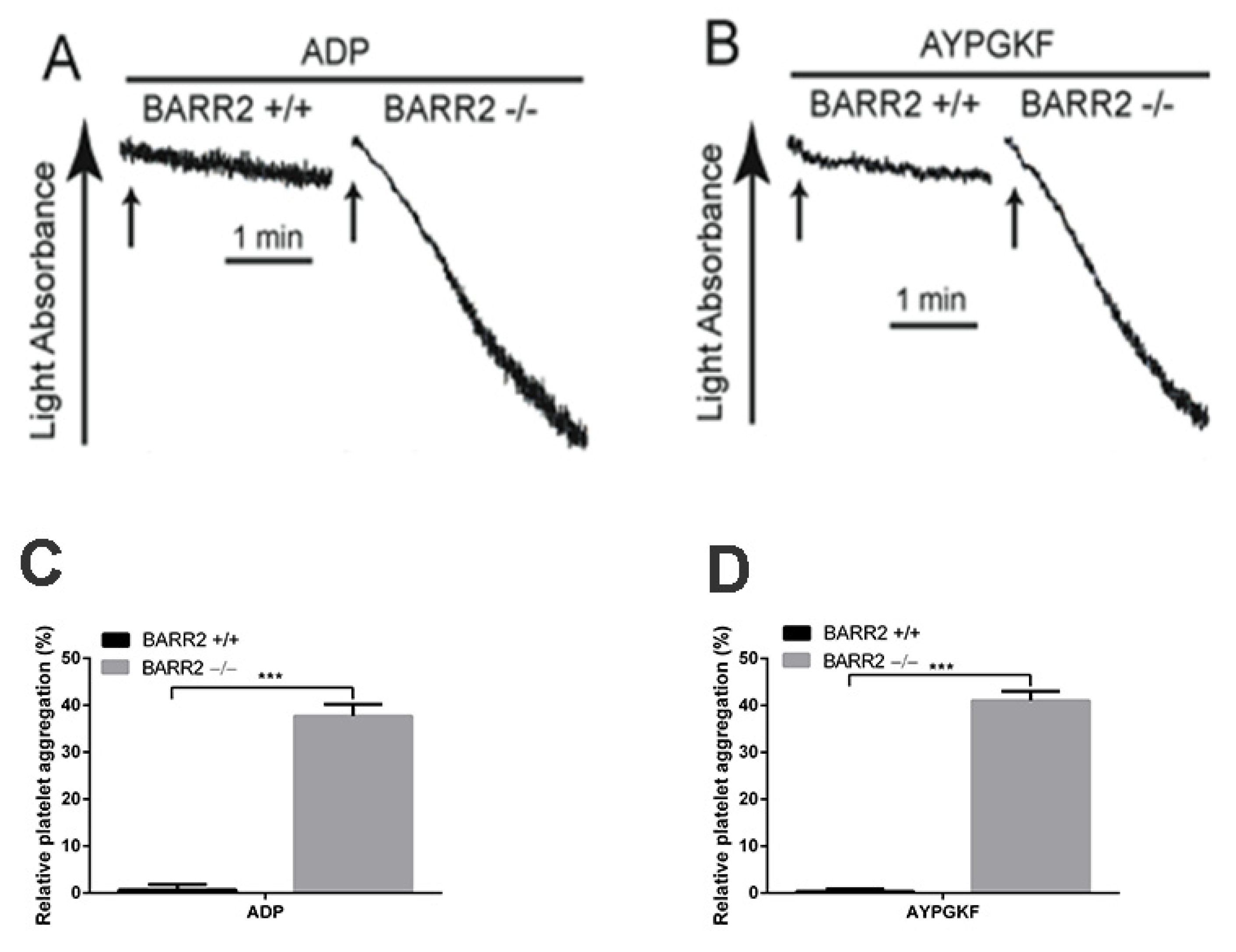
3.3. Arrestin3 Regulates ADP and PAR4 Receptor Desensitization in Platelets
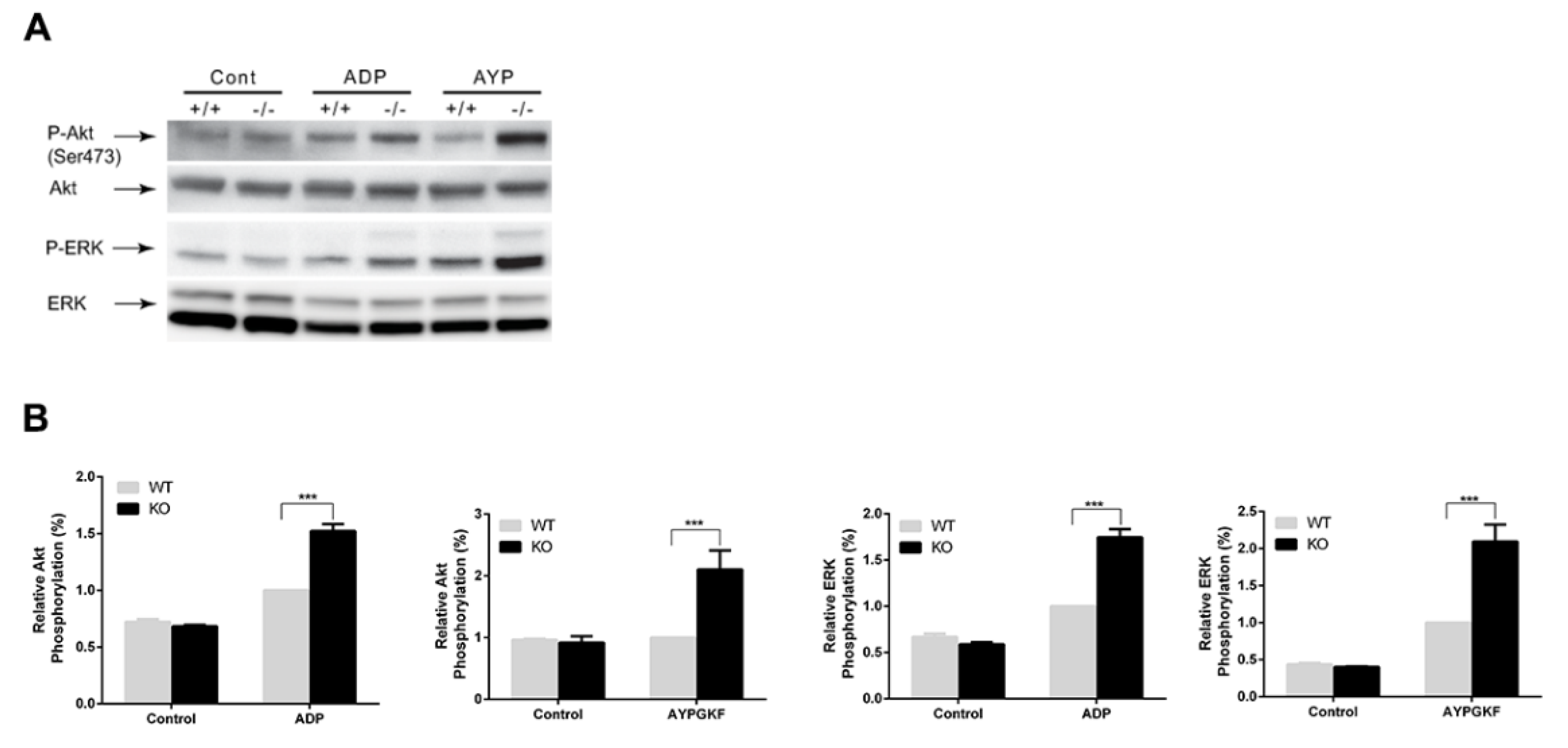
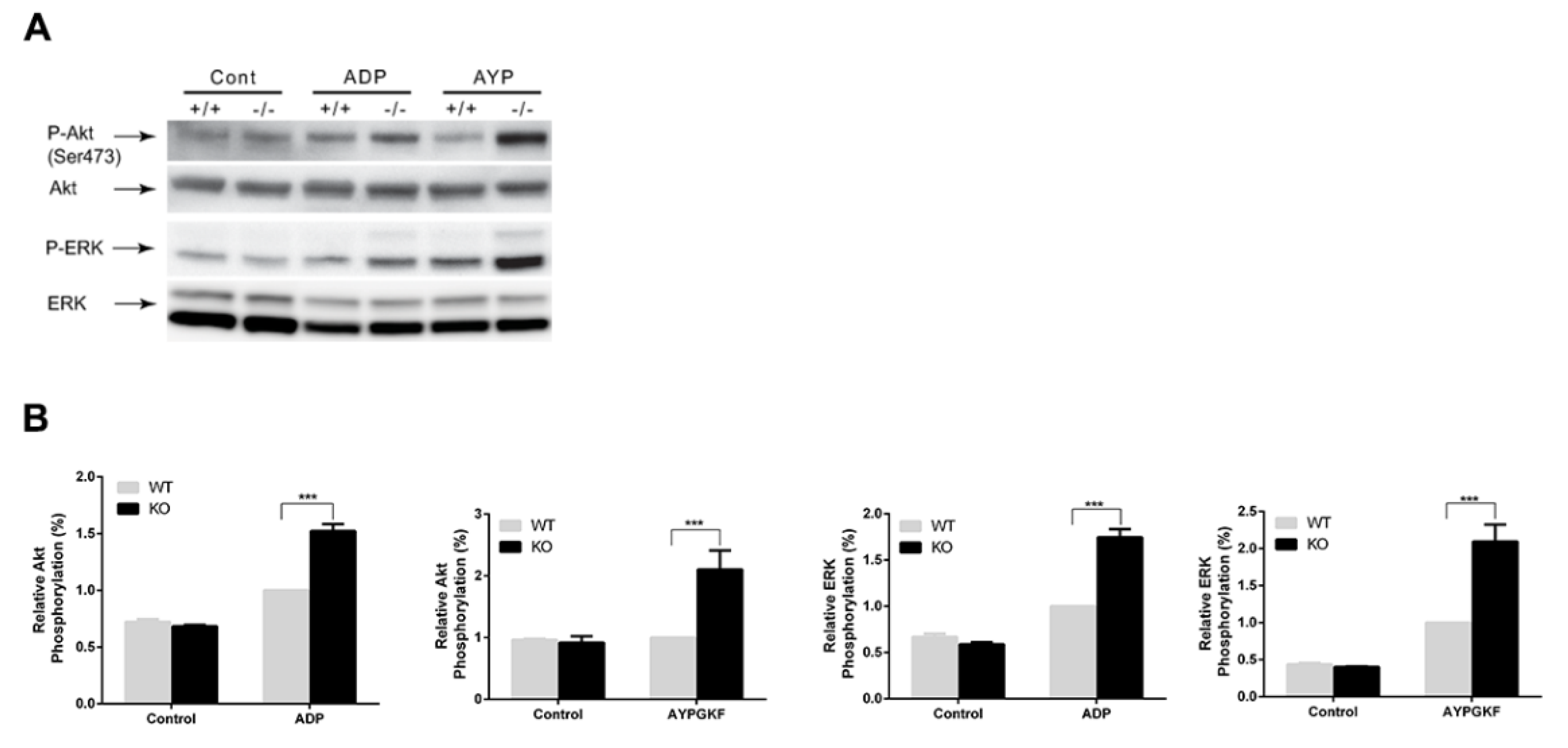
3.4. Potentiation of GPCR-mediated Signaling Events in Arrestin3-Deficient Platelets
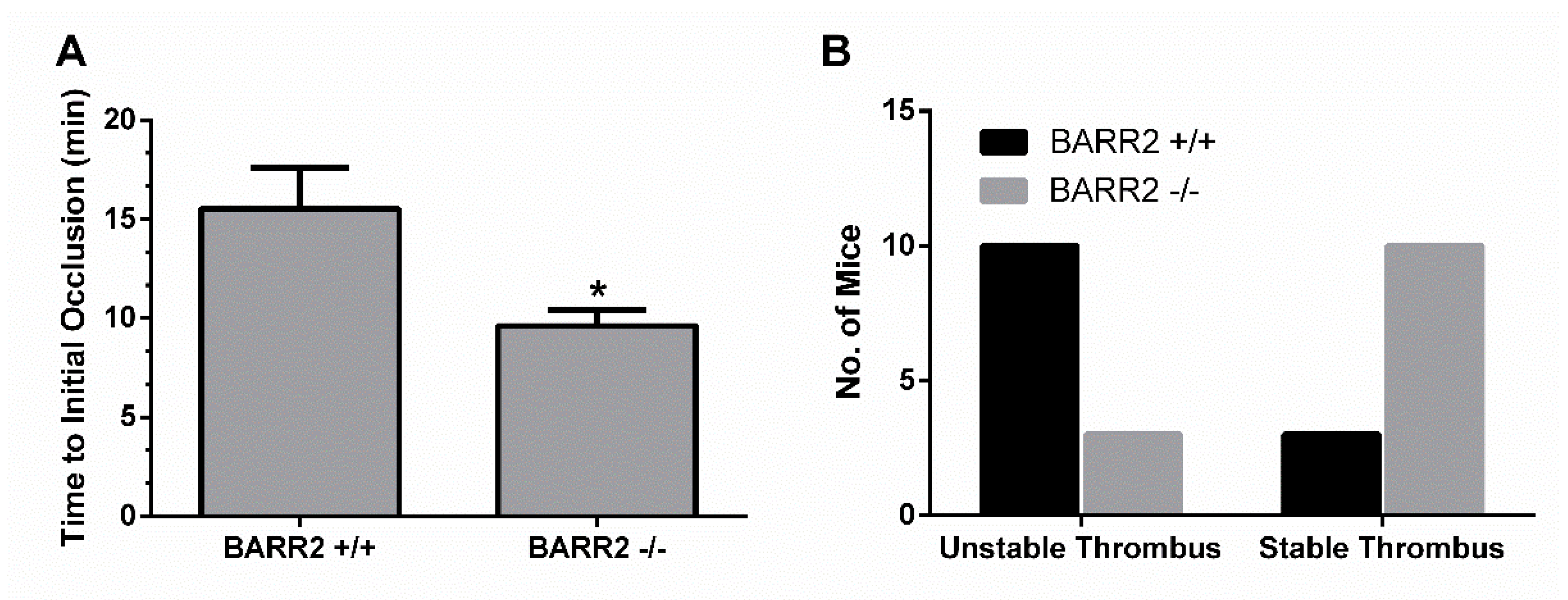
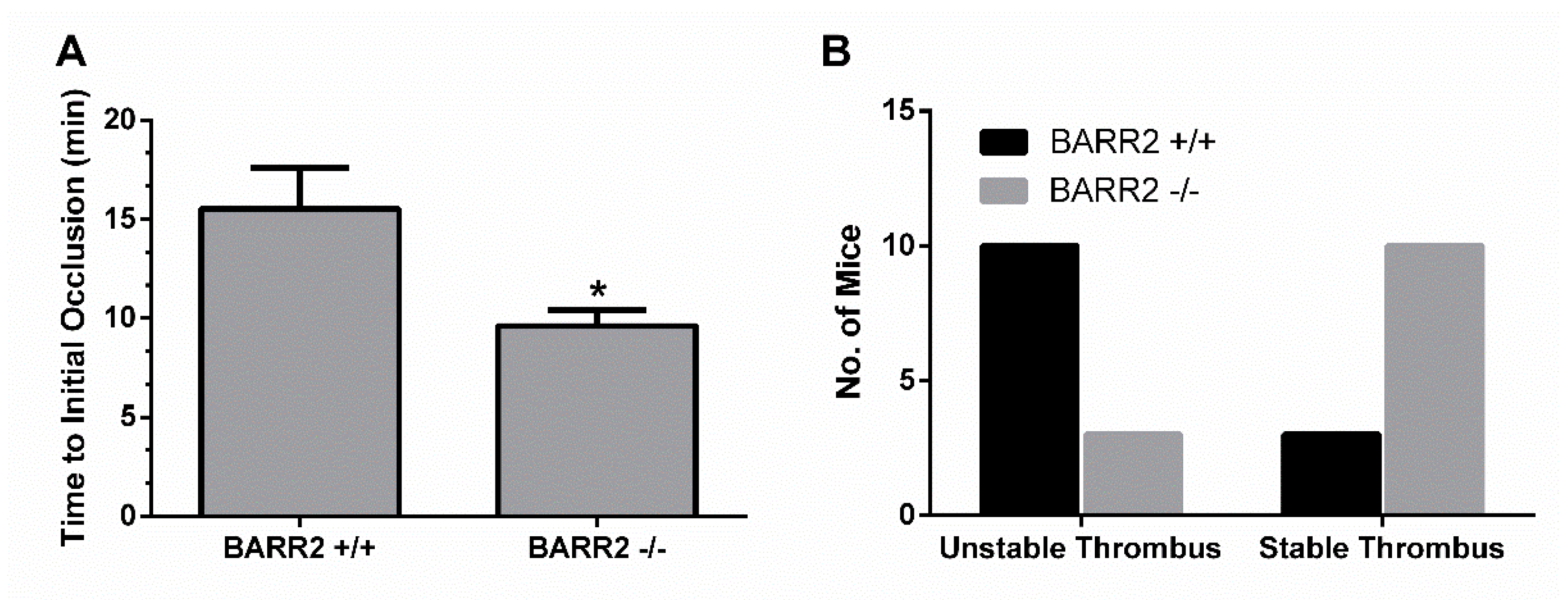
3.5. Role of Arrestin3 in Regulation of Thrombus Formation In Vivo
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Du, X. Signaling and regulation of the platelet glycoprotein Ib–IX–V complex. Curr. Opin. Hematol. 2007, 14, 262–269. [Google Scholar] [CrossRef]
- Watson, S.; Auger, J.; McCarty, O.; Pearce, A. GPVI and integrin αIIbβ3 signaling in platelets. J. Thromb. Haemost. 2005, 3, 1752–1762. [Google Scholar] [CrossRef]
- Shen, B.; Delaney, M.K.; Du, X. Inside-out, outside-in, and inside–outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr. Opin. Cell Biol. 2012, 24, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offermanns, S. Activation of platelet function through G protein–coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by ß-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar] [PubMed]
- Kohout, T.A.; Lefkowitz, R.J. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 2003, 63, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, A.; Von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Goodman, O.B.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. β-Arrestin acts as a clathrin adaptor in endocytosis of the β 2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef]
- Laporte, S.A.; Oakley, R.H.; Zhang, J.; Holt, J.A.; Ferguson, S.S.; Caron, M.G.; Barak, L.S. The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3712–3717. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S.K.; Lefkowitz, R.J. Multifaceted roles of β-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem. J. 2003, 375, 503–515. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gurevich, V.V. Arrestins: Ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Latorraca, N.R.; Wang, J.K.; Bauer, B.; Townshend, R.J.L.; Hollingsworth, S.A.; Olivieri, J.E.; Xu, H.E.; Sommer, M.E.; Dror, R.O. Molecular mechanism of GPCR-mediated arrestin activation. Nature 2018, 557, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.-T. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Conner, D.A.; Mathier, M.A.; Mortensen, R.M.; Christe, M.; Vatner, S.F.; Seidman, C.E.; Seidman, J. β-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to β-adrenergic stimulation. Circ. Res. 1997, 81, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Hara, M.R.; Davenport, C.L.; Kim, J.; Lefkowitz, R.J. Arrestin development: Emerging roles for β-arrestins in developmental signaling pathways. Dev. Cell 2009, 17, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Paing, M.M.; Stutts, A.B.; Kohout, T.A.; Lefkowitz, R.J.; Trejo, J. β-Arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J. Biol. Chem. 2002, 277, 1292–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.; Nelson, C.D.; Garrison, T.R.; Miller, W.E.; Lefkowitz, R.J. Desensitization, internalization, and signaling functions of β-arrestins demonstrated by RNA interference. Proc. Natl. Acad. Sci. USA 2003, 100, 1740–1744. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.; Wei, H.; Garrison, T.R.; Lefkowitz, R.J. Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by β-arrestins 1 and 2. J. Biol. Chem. 2004, 279, 7807–7811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundell, S.J.; Luo, J.; Benovic, J.L.; Conley, P.B.; Poole, A.W. Distinct clathrin-coated pits sort different G protein-coupled receptor cargo. Traffic 2006, 7, 1420–1431. [Google Scholar] [CrossRef]
- Hardy, A.R.; Conley, P.B.; Luo, J.; Benovic, J.L.; Poole, A.W.; Mundell, S.J. P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase-dependent mechanisms. Blood 2005, 105, 3552–3560. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.-L.; Labrecque, P.; Orsini, M.J.; Benovic, J.L. Internalization of the TXA2 receptor α and β isoforms: Role of the differentially spliced cooh terminus in agonist-promoted receptor internalization. J. Biol. Chem. 1999, 274, 8941–8948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; D′Angelo, L.; Chavez, M.; Woulfe, D.S. Arrestin-2 differentially regulates PAR4 and ADP receptor signaling in platelets. J. Biol. Chem. 2011, 286, 3805–3814. [Google Scholar] [CrossRef] [Green Version]
- Schaff, M.; Receveur, N.; Bourdon, C.; Ohlmann, P.; Lanza, F.; Gachet, C.; Mangin, P.H. β-arrestin-1 participates in thrombosis and regulates integrin aIIbβ3 signalling without affecting P2Y receptors desensitisation and function. Thromb. Haemost. 2012, 107, 735–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, J.L.; Zhao, X.; Hill, R.; Mundell, S.J. Arrestin-3 differentially regulates platelet GPCR subsets. Platelets 2020, 31, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Nisar, S.P.; Cunningham, M.; Saxena, K.; Pope, R.J.; Kelly, E.; Mundell, S.J. Arrestin scaffolds NHERF1 to the P2Y12 receptor to regulate receptor internalization. J. Biol. Chem. 2012, 287, 24505–24515. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Cipolla, L.; Guidetti, G.; Okigaki, M.; Jin, J.; Torti, M.; Kunapuli, S.P. Distinct role of Pyk2 in mediating thromboxane generation downstream of both G12/13 and integrin αIIbβ3 in platelets. J. Biol. Chem. 2013, 288, 18194–18203. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jin, J.; Kunapuli, S.P. Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood 2006, 107, 947–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bynagari-Settipalli, Y.S.; Lakhani, P.; Jin, J.; Bhavaraju, K.; Rico, M.C.; Kim, S.; Woulfe, D.; Kunapuli, S.P. Protein kinase C isoform ε negatively regulates ADP-induced calcium mobilization and thromboxane generation in platelets. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- De Chaffoy de Courcelles, D.; Roevens, P.; Van Belle, H.; De Clerck, F. The synergistic effect of serotonin and epinephrine on the human platelet at the level of signal transduction. FEBS Lett. 1987, 219, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, P.K.; Kim, S.; Jee, Y.; Lee, S.-H.; Park, K.-M.; Kim, S. Role of GRK6 in the regulation of platelet activation through selective G protein-coupled receptor (GPCR) desensitization. Int. J. Mol. Sci. 2020, 21, 3932. [Google Scholar] [CrossRef]
- Nagy, B., Jr.; Bhavaraju, K.; Getz, T.; Bynagari, Y.S.; Kim, S.; Kunapuli, S.P. Impaired activation of platelets lacking protein kinase C-θ isoform. Blood J. Am. Soc. Hematol. 2009, 113, 2557–2567. [Google Scholar] [CrossRef] [Green Version]
- Kohout, T.A.; Lin, F.-T.; Perry, S.J.; Conner, D.A.; Lefkowitz, R.J. β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. USA 2001, 98, 1601–1606. [Google Scholar] [CrossRef] [Green Version]
- Krupnick, J.G.; Benovic, J.L. The role of receptor kinases and arrestins in G protein–coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Attramadal, H.; Arriza, J.L.; Aoki, C.; Dawson, T.M.; Codina, J.; Kwatra, M.M.; Snyder, S.H.; Caron, M.G.; Lefkowitz, R.J. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem. 1992, 267, 17882–17890. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S. The GRKs reactome: Role in cell biology and pathology. Int. J. Mol. Sci. 2021, 22, 3375. [Google Scholar] [CrossRef] [PubMed]
- Shankar, H.; Garcia, A.; Prabhakar, J.; Kim, S.; Kunapuli, S. P2Y12 receptor-mediated potentiation of thrombin-induced thromboxane A2 generation in platelets occurs through regulation of Erk1/2 activation. J. Thromb. Haemost. 2006, 4, 638–647. [Google Scholar] [CrossRef]
- Kahner, B.; Shankar, H.; Murugappan, S.; Prasad, G.; Kunapuli, S. Nucleotide receptor signaling in platelets. J. Thromb. Haemost. 2006, 4, 2317–2326. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, P.K.; Kim, S.; Kim, S. The Predominant Role of Arrestin3 in General GPCR Desensitization in Platelets. J. Clin. Med. 2021, 10, 4743. https://doi.org/10.3390/jcm10204743
Chaudhary PK, Kim S, Kim S. The Predominant Role of Arrestin3 in General GPCR Desensitization in Platelets. Journal of Clinical Medicine. 2021; 10(20):4743. https://doi.org/10.3390/jcm10204743
Chicago/Turabian StyleChaudhary, Preeti Kumari, Sanggu Kim, and Soochong Kim. 2021. "The Predominant Role of Arrestin3 in General GPCR Desensitization in Platelets" Journal of Clinical Medicine 10, no. 20: 4743. https://doi.org/10.3390/jcm10204743
APA StyleChaudhary, P. K., Kim, S., & Kim, S. (2021). The Predominant Role of Arrestin3 in General GPCR Desensitization in Platelets. Journal of Clinical Medicine, 10(20), 4743. https://doi.org/10.3390/jcm10204743