
Effect of Ivabradine on Cardiac Ventricular Arrhythmias: Friend or Foe?

 ,
, 
Abstract
:1. Ivabradine—Brief Summary
2. HCN Channel Family—A Primary Target for Ivabradine
3. HCN Channels in the Heart—Expression and Role
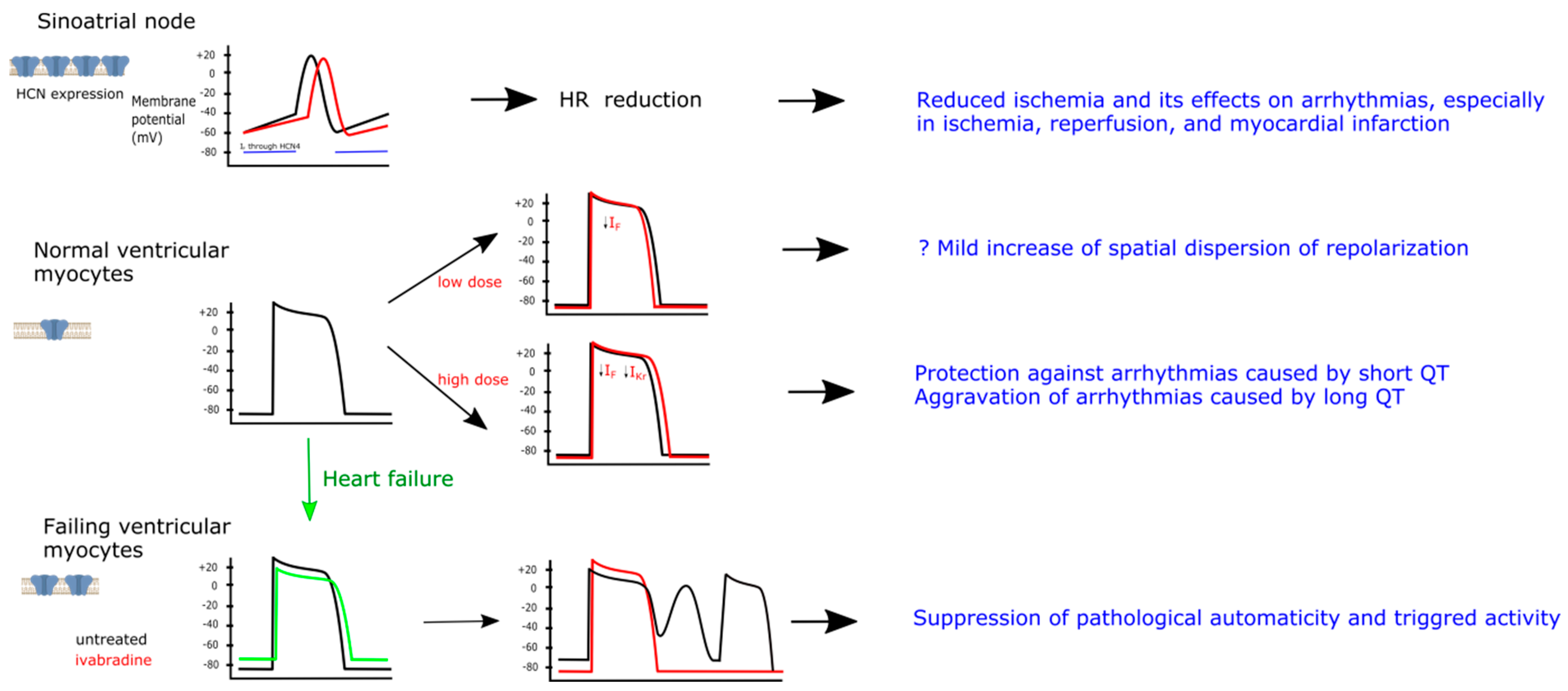
4. Effect of Ivabradine on Ion Channels
5. Pathophysiology of Ventricular Arrhythmias
6. Ivabradine and Ventricular Arrhythmias
6.1. Preclinical Evidence
6.2. Potential Mechanisms of Ivabradine Effects
6.3. Clinical Evidence
7. Possible Future Applications of Ivabradine
8. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur. Heart J. 2019, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failureThe Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Card. Fail. 2017, 23, 628–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Francesco, D.; Borer, J.S. The Funny Current. Drugs 2007, 67, 15–24. [Google Scholar] [CrossRef]
- Di Francesco, D.; Tortora, P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 1991, 351, 145–147. [Google Scholar] [CrossRef]
- Novella Romanelli, M.; Sartiani, L.; Masi, A.; Mannaioni, G.; Manetti, D.; Mugelli, A.; Cerbai, E. HCN Channels Modulators: The Need for Selectivity. Curr. Top. Med. Chem. 2016, 16, 1764–1791. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Kaupp, U.B.; Seifert, R. Molecular diversity of pacemaker ion channels. Annu. Rev. Physiol. 2001, 63, 235–257. [Google Scholar] [CrossRef]
- Wainger, B.J.; DeGennaro, M.; Santoro, B.; Siegelbaum, S.A.; Tibbs, G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 2001, 411, 805–810. [Google Scholar] [CrossRef]
- Biel, M.; Wahl-Schott, C.; Michalakis, S.; Zong, X. Hyperpolarization-activated cation channels: From genes to function. Physiol. Rev. 2009, 89, 847–885. [Google Scholar] [CrossRef] [Green Version]
- Baruscotti, M.; Bucchi, A.; Difrancesco, D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol. Ther. 2005, 107, 59–79. [Google Scholar] [CrossRef]
- Di Francesco, D. A Brief History of Pacemaking. Front. Physiol. 2020, 10, 1599. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A Coupled SYSTEM of Intracellular Ca2+ Clocks and Surface Membrane Voltage Clocks Controls the Timekeeping Mechanism of the Heart’s Pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef] [Green Version]
- Dobrzynski, H.; Nikolski, V.P.; Sambelashvili, A.T.; Greener, I.D.; Yamamoto, M.; Boyett, M.R.; Efimov, I.R. Site of Origin and Molecular Substrate of Atrioventricular Junctional Rhythm in the Rabbit Heart. Circ. Res. 2003, 93, 1102–1110. [Google Scholar] [CrossRef] [Green Version]
- Di Francesco, D. The role of the funny current in pacemaker activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, F.; Fabritz, L.; Stieber, J.; Schmitt, J.; Kirchhof, P.; Ludwig, A.; Herrmann, S. Ventricular HCN channels decrease the repolarization reserve in the hypertrophic heart. Cardiovasc. Res. 2012, 95, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl-Schott, C.; Fenske, S.; Biel, M. HCN channels: New roles in sinoatrial node function. Curr. Opin. Pharmacol. 2014, 15, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Csepe, T.A.; Hansen, B.J.; Dobrzynski, H.; Higgins, R.S.D.; Kilic, A.; Mohler, P.J.; Janssen, P.M.L.; Rosen, M.R.; Biesiadecki, B.J.; et al. Molecular Mapping of Sinoatrial Node HCN Channel Expression in the Human Heart. Circ. Arrhythmia Electrophysiol. 2015, 8, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, P.A.; Yampolsky, P.; Malik, R.; Thomas, D.; Zehelein, J.; Katus, H.A.; Koenen, M. Transcription profiling of HCN-channel isotypes throughout mouse cardiac development. Basic Res. Cardiol. 2009, 104, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Yasui, K.; Liu, W.; Opthof, T.; Kada, K.; Lee, J.K.; Kamiya, K.; Kodama, I. I(f) current and spontaneous activity in mouse embryonic ventricular myocytes. Circ. Res. 2001, 88, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, P.A.; Schröter, J.; Greiner, S.; Haas, J.; Yampolsky, P.; Mereles, D.; Buss, S.J.; Seyler, C.; Bruehl, C.; Draguhn, A.; et al. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J. Am. Coll. Cardiol. 2014, 64, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stillitano, F.; Lonardo, G.; Zicha, S.; Varro, A.; Cerbai, E.; Mugelli, A.; Nattel, S. Molecular basis of funny current (If) in normal and failing human heart. J. Mol. Cell Cardiol. 2008, 45, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, U.C.; Jansen, E.; Südkamp, M.; Beuckelmann, D.J. Hyperpolarization-Activated Inward Current in Ventricular Myocytes From Normal and Failing Human Hearts. Circulation 1998, 97, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Cerbai, E.; Pino, R.; Porciatti, F.; Sani, G.; Toscano, M.; Maccherini, M.; Giunti, G.; Mugelli, A. Characterization of the hyperpolarization-activated current, I(f), in ventricular myocytes from human failing heart. Circulation 1997, 95, 568–571. [Google Scholar] [CrossRef]
- Mackiewicz, U.; Gerges, J.Y.; Chu, S.; Duda, M.; Dobrzynski, H.; Lewartowski, B.; Mączewski, M. Ivabradine Protects Against Ventricular Arrhythmias in Acute Myocardial Infarction in the Rat. J. Cell. Physiol. 2014, 229, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Wang, Y.; Zhang, Y.; Deng, S.-B.; Du, J.-L.; Wang, X.-C.; She, Q. Dynamic changes in HCN2, HCN4, KCNE1, and KCNE2 expression in ventricular cells from acute myocardial infarction rat hearts. Biochem. Biophys. Res. Commun. 2010, 395, 330–335. [Google Scholar] [CrossRef]
- Suffredini, S.; Stillitano, F.; Comini, L.; Bouly, M.; Brogioni, S.; Ceconi, C.; Ferrari, R.; Mugelli, A.; Cerbai, E. Long-term treatment with ivabradine in post-myocardial infarcted rats counteracts f-channel overexpression. Br. J. Pharmacol. 2012, 165, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Velasco, M.; Goren, N.; Benito, G.; Blanco-Rivero, J.; Boscá, L.; Delgado, C. Regional distribution of hyperpolarization-activated current (If) and hyperpolarization-activated cyclic nucleotide-gated channel mRNA expression in ventricular cells from control and hypertrophied rat hearts. J. Physiol. 2003, 553, 395–405. [Google Scholar] [CrossRef]
- Cerbai, E.; Barbieri, M.; Mugelli, A. Occurrence and Properties of the Hyperpolarization-Activated Current If in Ventricular Myocytes From Normotensive and Hypertensive Rats During Aging. Circulation 1996, 94, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, Y.; Kuwahara, K.; Takano, M.; Kinoshita, H.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Igata, S.; Usami, S.; Minami, T.; et al. Increased Expression of HCN Channels in the Ventricular Myocardium Contributes to Enhanced Arrhythmicity in Mouse Failing Hearts. J. Am. Heart Assoc. 2013, 2, e000150. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Saito, Y.; Takano, M.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Takahashi, N.; Adachi, Y.; Takemura, G.; Horie, M.; et al. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 2003, 22, 6310–6321. [Google Scholar] [CrossRef]
- Del Lungo, M.; Melchiorre, M.; Guandalini, L.; Sartiani, L.; Mugelli, A.; Koncz, I.; Szel, T.; Varro, A.; Romanelli, M.N.; Cerbai, E. Novel blockers of hyperpolarization-activated current with isoform selectivity in recombinant cells and native tissue. Br. J. Pharmacol. 2012, 166, 602–616. [Google Scholar] [CrossRef] [Green Version]
- Hadova, K.; Kralova, E.; Doka, G.; Bies Pivackova, L.; Kmecova, Z.; Krenek, P.; Klimas, J. Isolated downregulation of HCN2 in ventricles of rats with streptozotocin-induced diabetic cardiomyopathy. BMC Cardiovasc. Disord. 2021, 21, 118. [Google Scholar] [CrossRef] [PubMed]
- Fenske, S.; Krause, S.; Biel, M.; Wahl-Schott, C. The role of HCN channels in ventricular repolarization. Trends Cardiovasc. Med. 2011, 21, 216–220. [Google Scholar] [CrossRef]
- Koruth, J.S.; Lala, A.; Pinney, S.; Reddy, V.Y.; Dukkipati, S.R. The Clinical Use of Ivabradine. J. Am. Coll. Cardiol. 2017, 70, 1777–1784. [Google Scholar] [CrossRef]
- Tanguay, J.; Callahan, K.M.; D’Avanzo, N. Characterization of drug binding within the HCN1 channel pore. Sci. Rep. 2019, 9, 465. [Google Scholar] [CrossRef]
- Bucchi, A.; Baruscotti, M.; Nardini, M.; Barbuti, A.; Micheloni, S.; Bolognesi, M.; DiFrancesco, D. Identification of the Molecular Site of Ivabradine Binding to HCN4 Channels. PLoS ONE 2013, 8, e53132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucchi, A.; Tognati, A.; Milanesi, R.; Baruscotti, M.; DiFrancesco, D. Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J. Physiol. 2006, 572, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Stieber, J.; Wieland, K.; Stöckl, G.; Ludwig, A.; Hofmann, F. Bradycardic and proarrhythmic properties of sinus node inhibitors. Mol. Pharmacol. 2006, 69, 1328–1337. [Google Scholar] [CrossRef] [Green Version]
- Delpón, E.; Valenzuela, C.; Pérez, O.; Franqueza, L.; Gay, P.; Snyders, D.J.; Tamargo, J. Mechanisms of block of a human cloned potassium channel by the enantiomers of a new bradycardic agent: S-16257-2 and S-16260-2. Br. J. Pharm. 1996, 117, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Haechl, N.; Ebner, J.; Hilber, K.; Todt, H.; Koenig, X. Pharmacological Profile of the Bradycardic Agent Ivabradine on Human Cardiac Ion Channels. Cell. Physiol. Biochem. 2019, 53, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Melgari, D.; Brack, K.E.; Zhang, C.; Zhang, Y.; El Harchi, A.; Mitcheson, J.S.; Dempsey, C.E.; Ng, G.A.; Hancox, J.C. hERG potassium channel blockade by the HCN channel inhibitor bradycardic agent ivabradine. J. Am. Heart Assoc. 2015, 4, e001813. [Google Scholar] [CrossRef] [Green Version]
- Lees-Miller, J.P.; Guo, J.; Wang, Y.; Perissinotti, L.L.; Noskov, S.Y.; Duff, H.J. Ivabradine prolongs phase 3 of cardiac repolarization and blocks the hERG1 (KCNH2) current over a concentration-range overlapping with that required to block HCN4. J. Mol. Cell. Cardiol. 2015, 85, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Koncz, I.; Szél, T.; Bitay, M.; Cerbai, E.; Jaeger, K.; Fülöp, F.; Jost, N.; Virág, L.; Orvos, P.; Tálosi, L.; et al. Electrophysiological effects of ivabradine in dog and human cardiac preparations: Potential antiarrhythmic actions. Eur. J. Pharm. 2011, 668, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tian, L.; Huang, Y.; Li, Y.; Xu, L. Pharmacokinetic and safety profile of ivabradine in healthy Chinese men: A phase I, randomized, open-label, increasing single- and multiple-dose study. Clin. Ther. 2013, 35, 1933–1945. [Google Scholar] [CrossRef] [PubMed]
- Frommeyer, G.; Sterneberg, M.; Dechering, D.G.; Ellermann, C.; Bögeholz, N.; Kochhäuser, S.; Pott, C.; Fehr, M.; Eckardt, L. Effective suppression of atrial fibrillation by ivabradine: Novel target for an established drug? Int. J. Cardiol. 2017, 236, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Young, G.T.; Emery, E.C.; Mooney, E.R.; Tsantoulas, C.; McNaughton, P.A. Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels. Pain 2014, 155, 1708–1719. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef]
- Lamothe, S.M.; Song, W.; Guo, J.; Li, W.; Yang, T.; Baranchuk, A.; Graham, C.H.; Zhang, S. Hypoxia reduces mature hERG channels through calpain up-regulation. FASEB J. 2017, 31, 5068–5077. [Google Scholar] [CrossRef] [Green Version]
- Cocco, G.; Jerie, P. Torsades de Pointes Induced by the Concomitant Use of Ivabradine and Azithromycin: An Unexpected Dangerous Interaction. Cardiovasc. Toxicol. 2015, 15, 104–106. [Google Scholar] [CrossRef]
- Mittal, S.R. Slow junctional rhythm, QTc prolongation and transient torsades de-pointes following combined use of Ivabradine, Diltiazem and Ranolazine. J. Assoc. Physicians India 2014, 62, 426–427. [Google Scholar]
- Sánchez Muñoz, J.J.; García-Alberola, A.; Martínez-Sánchez, J.; Peñafiel-Verdú, P.; Caro-Martínez, C.; Manzano-Fernández, S.; Valdés Chávarri, M. Premature ventricular complexes as a trigger for ventricular fibrillation. Rev. Española Cardiol. 2010, 63, 798–801. [Google Scholar] [CrossRef]
- Qu, Z.; Weiss, J.N. Mechanisms of ventricular arrhythmias: From molecular fluctuations to electrical turbulence. Annu. Rev. Physiol. 2015, 77, 29–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauss, S.; Bleyer, C.; Schüttler, D.; Tomsits, P.; Renner, S.; Klymiuk, N.; Wakili, R.; Massberg, S.; Wolf, E.; Kääb, S. Animal models of arrhythmia: Classic electrophysiology to genetically modified large animals. Nat. Rev. Cardiol. 2019, 16, 457–475. [Google Scholar] [CrossRef]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Janse, M.J.; Wit, A.L. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 1989, 69, 1049–1169. [Google Scholar] [CrossRef] [PubMed]
- Marciszek, M.; Paterek, A.; Oknińska, M.; Mackiewicz, U.; Mączewski, M. Ivabradine is as effective as metoprolol in the prevention of ventricular arrhythmias in acute non-reperfused myocardial infarction in the rat. Sci. Rep. 2020, 10, 15027. [Google Scholar] [CrossRef]
- Vaillant, F.; Timour, Q.; Descotes, J.; Manati, W.; Belhani, D.; Bui-Xuan, B.; Tabib, A.; Bricca, G.; Chevalier, P. Ivabradine Induces an Increase in Ventricular Fibrillation Threshold During Acute Myocardial Ischemia: An Experimental Study. J. Cardiovasc. Pharmacol. 2008, 52, 548–554. [Google Scholar] [CrossRef]
- Vaillant, F.; Dehina, L.; Mazzadi, A.; Descotes, J.; Chevalier, P.; Tabib, A.; Bui-Xuan, B.; Riera, C.; Belhani, D.; Timour, Q. Heart rate reduction with ivabradine increases ischaemia-induced ventricular fibrillation threshold: Role of myocyte structure and myocardial perfusion. Resuscitation 2011, 82, 1092–1099. [Google Scholar] [CrossRef]
- Ng, F.S.; Shadi, I.T.; Peters, N.S.; Lyon, A.R. Selective heart rate reduction with ivabradine slows ischaemia-induced electrophysiological changes and reduces ischaemia-reperfusion-induced ventricular arrhythmias. J. Mol. Cell. Cardiol. 2013, 59, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.-p.; Lin, J.-j.; Zhuang, T.-p.; Wang, W.-w.; Wu, H.-z.; Zhang, F.-l. Effects of ivabradine on ventricular electrophysiological remodeling after myocardial infarction in rats. Arch. Med. Sci. 2020, 16, 1–14. [Google Scholar] [CrossRef]
- Paterek, A.; Sochanowicz, B.; Oknińska, M.; Śmigielski, W.; Kruszewski, M.; Mackiewicz, U.; Mączewski, M.; Leszek, P. Ivabradine prevents deleterious effects of dopamine therapy in heart failure: No role for HCN4 overexpression. Biomed. Pharmacother. 2021, 136, 111250. [Google Scholar] [CrossRef] [PubMed]
- Milliez, P.; Messaoudi, S.; Nehme, J.; Rodriguez, C.; Samuel, J.-L.; Delcayre, C. Beneficial effects of delayed ivabradine treatment on cardiac anatomical and electrical remodeling in rat severe chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H435–H441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaillant, F.; Dehina, L.; Dizerens, N.; Bui-Xuan, B.; Tabib, A.; Lauzier, B.; Chevalier, P.; Descotes, J.; Timour, Q. Ivabradine but not propranolol delays the time to onset of ischaemia-induced ventricular fibrillation by preserving myocardial metabolic energy status. Resuscitation 2013, 84, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Yampolsky, P.; Koenen, M.; Mosqueira, M.; Geschwill, P.; Nauck, S.; Witzenberger, M.; Seyler, C.; Fink, T.; Kruska, M.; Bruehl, C.; et al. Augmentation of myocardial I(f) dysregulates calcium homeostasis and causes adverse cardiac remodeling. Nat. Commun. 2019, 10, 3295. [Google Scholar] [CrossRef]
- Frommeyer, G.; Weller, J.; Ellermann, C.; Kaese, S.; Kochhäuser, S.; Lange, P.S.; Dechering, D.G.; Eckardt, L. Antiarrhythmic properties of ivabradine in an experimental model of Short-QT- Syndrome. Clin. Exp. Pharmacol. Physiol. 2017, 44, 941–945. [Google Scholar] [CrossRef]
- Frommeyer, G.; Weller, J.; Ellermann, C.; Bögeholz, N.; Leitz, P.; Dechering, D.G.; Kochhäuser, S.; Wasmer, K.; Eckardt, L. Ivabradine Reduces Digitalis-induced Ventricular Arrhythmias. Basic Clin. Pharmacol. Toxicol. 2017, 121, 526–530. [Google Scholar] [CrossRef]
- Frommeyer, G.; Weller, J.; Ellermann, C.; Leitz, P.; Kochhäuser, S.; Lange, P.S.; Dechering, D.G.; Eckardt, L. Ivabradine Aggravates the Proarrhythmic Risk in Experimental Models of Long QT Syndrome. Cardiovasc. Toxicol. 2019, 19, 129–135. [Google Scholar] [CrossRef]
- Bueno-Levy, H.; Weisbrod, D.; Yadin, D.; Haron-Khun, S.; Peretz, A.; Hochhauser, E.; Arad, M.; Attali, B. The Hyperpolarization-Activated Cyclic-Nucleotide-Gated Channel Blocker Ivabradine Does Not Prevent Arrhythmias in Catecholaminergic Polymorphic Ventricular Tachycardia. Front. Pharmacol. 2020, 10, 1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvreur, N.; Tissier, R.; Pons, S.; Chetboul, V.; Gouni, V.; Bruneval, P.; Mandet, C.; Pouchelon, J.-L.; Berdeaux, A.; Ghaleh, B. Chronic heart rate reduction with ivabradine improves systolic function of the reperfused heart through a dual mechanism involving a direct mechanical effect and a long-term increase in FKBP12/12.6 expression. Eur. Heart J. 2010, 31, 1529–1537. [Google Scholar] [CrossRef]
- Vaillant, F.; Lauzier, B.; Ruiz, M.; Shi, Y.; Lachance, D.; Rivard, M.E.; Bolduc, V.; Thorin, E.; Tardif, J.C.; Des Rosiers, C. Ivabradine and metoprolol differentially affect cardiac glucose metabolism despite similar heart rate reduction in a mouse model of dyslipidemia. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H991–H1003. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.R.; Pereira, L.; Wang, L.; Han, G.; Ferguson, A.; Dao, K.; Copeland, R.J.; Despa, F.; Hart, G.W.; Ripplinger, C.M.; et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013, 502, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Oshita, K.; Itoh, M.; Hirashima, S.; Kuwabara, Y.; Ishihara, K.; Kuwahara, K.; Nakao, K.; Kimura, T.; Nakamura, K.; Ushijima, K.; et al. Ectopic automaticity induced in ventricular myocytes by transgenic overexpression of HCN2. J. Mol. Cell Cardiol. 2015, 80, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshita, K.; Kozasa, Y.; Nakagawa, Y.; Kuwabara, Y.; Kuwahara, K.; Nakagawa, T.; Nakashima, N.; Hiraki, T.; Takano, M. Overexpression of the HCN2 channel increases the arrhythmogenicity induced by hypokalemia. J. Physiol. Sci. 2019, 69, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Marciszek, M.; Paterek, A.; Oknińska, M.; Zambrowska, Z.; Mackiewicz, U.; Mączewski, M. Effect of ivabradine on cardiac arrhythmias: Antiarrhythmic or proarrhythmic? Heart Rhythm 2021, 18, 1230–1238. [Google Scholar] [CrossRef]
- Fox, K.; Ford, I.; Steg, P.G.; Tendera, M.; Ferrari, R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 807–816. [Google Scholar] [CrossRef]
- Tendera, M.; Talajic, M.; Robertson, M.; Tardif, J.-C.; Ferrari, R.; Ford, I.; Steg, P.G.; Fox, K. Safety of Ivabradine in Patients With Coronary Artery Disease and Left Ventricular Systolic Dysfunction (from the BEAUTIFUL Holter Substudy). Am. J. Cardiol. 2011, 107, 805–811. [Google Scholar] [CrossRef]
- Fox, K.; Ford, I.; Steg, P.G.; Tardif, J.-C.; Tendera, M.; Ferrari, R. Ivabradine in Stable Coronary Artery Disease without Clinical Heart Failure. N. Engl. J. Med. 2014, 371, 1091–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Böhm, M.; Borer, J.S.; Camm, J.; Ford, I.; Lloyd, S.M.; Komajda, M.; Tavazzi, L.; Talajic, M.; Lainscak, M.; Reil, J.-C.; et al. Twenty-four-hour heart rate lowering with ivabradine in chronic heart failure: Insights from the SHIFT Holter substudy. Eur. J. Heart Fail. 2015, 17, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Steg, P.G.; Lopez-de-Sà, E.; Schiele, F.; Hamon, M.; Meinertz, T.; Goicolea, J.; Werdan, K.; Lopez-Sendon, J.L.; investigators, V. Safety of intravenous ivabradine in acute ST-segment elevation myocardial infarction patients treated with primary percutaneous coronary intervention: A randomized, placebo-controlled, double-blind, pilot study. Eur. Heart J. Acute Cardiovasc. Care 2013, 2, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasullo, S.; Cannizzaro, S.; Maringhini, G.; Ganci, F.; Giambanco, F.; Vitale, G.; Pinto, V.; Migliore, G.; Torres, D.; Sarullo, F.M.; et al. Comparison of Ivabradine Versus Metoprolol in Early Phases of Reperfused Anterior Myocardial Infarction With Impaired Left Ventricular Function: Preliminary Findings. J. Card. Fail. 2009, 15, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Gerbaud, E.; Montaudon, M.; Chasseriaud, W.; Gilbert, S.; Cochet, H.; Pucheu, Y.; Horovitz, A.; Bonnet, J.; Douard, H.; Coste, P. Effect of ivabradine on left ventricular remodelling after reperfused myocardial infarction: A pilot study. Arch. Cardiovasc. Dis. 2014, 107, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mert, K.U.; Mert, G.; Morrad, B.; Tahmazov, S.; Mutlu, F.; Çavuşoglu, Y. Effects of ivabradine and beta-blocker therapy on dobutamine-induced ventricular arrhythmias. Kardiologia Polska 2017, 75, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Rayan, M.; Tawfik, M.; Alabd, A.; Gamal, A. Ivabradine, a novel heart rate slower: Is it a sword of double blades in patients with idiopathic dilated cardiomyopathy? Anadolu Kardiyoloji Dergisi 2011, 11, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Mughal, L.H.; Houghton, A.R.; Khoo, J. Significant suppression of premature ventricular ectopics with ivabradine in dilated cardiomyopathy. Br. J. Cardiol. 2019, 26, 36–37. [Google Scholar] [CrossRef]
- Vaksmann, G.; Klug, D. Efficacy of ivabradine to control ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Pacing Clin. Electrophysiol. PACE 2018, 41, 1378–1380. [Google Scholar] [CrossRef]
- Kohli, U.; Aziz, Z.; Beaser, A.D.; Nayak, H.M. Ventricular arrhythmia suppression with ivabradine in a patient with catecholaminergic polymorphic ventricular tachycardia refractory to nadolol, flecainide, and sympathectomy. Pacing Clin. Electrophysiol. 2020, 43, 527–533. [Google Scholar] [CrossRef]
- Rivolta, I.; Binda, A.; Masi, A.; DiFrancesco, J.C. Cardiac and neuronal HCN channelopathies. Pflug. Arch. Eur. J. Physiol. 2020, 472, 931–951. [Google Scholar] [CrossRef]
- Fontenla, A.; López-Gil, M.; Tamargo-Menéndez, J.; Matía-Francés, R.; Salgado-Aranda, R.; Rey-Blas, J.R.; Miracle-Blanco, Á.; Mejía-Martínez, E.; Pastor-Fuentes, A.; Toquero-Ramos, J.; et al. Ivabradine for chronic heart rate control in persistent atrial fibrillation. Design of the BRAKE-AF project. Rev. Española Cardiol. 2020, 73, 368–375. [Google Scholar] [CrossRef]
- Krishna, M.R.; Kunde, M.F.; Kumar, R.K.; Balaji, S. Ivabradine in Post-operative Junctional Ectopic Tachycardia (JET): Breaking New Ground. Pediatric Cardiol. 2019, 40, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Ergul, Y.; Ozturk, E.; Ozgur, S.; Ozyurt, A.; Cilsal, E.; Guzeltas, A. Ivabradine is an effective antiarrhythmic therapy for congenital junctional ectopic tachycardia-induced cardiomyopathy during infancy: Case studies. Pacing Clin. Electrophysiol. PACE 2018, 41, 1372–1377. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Species | Model | Study Design, Route and Method of IVA Administration | Effects on VA | Additional Effects | Reference |
|---|---|---|---|---|---|
| Ischemia and reperfusion, myocardial infarction | |||||
| Rat | non-reperfused MI, LAD ligation, first 24 h | IVA vs. saline, 24 h before MI, IVA in drinking water, daily dose 10 mg/kg | ↓mortalilty ↓VT/VF frequency ↓VT/VF duration ↓VPC | ↓RyR sensitivity ↓HCN4 expression in LV ↓dispersion of APD | [25] |
| Rat | non-reperfused MI, LAD ligation, first 6 h | IVA vs. metoprolol vs. saline, given immediately after MI induction, IVA 5 mg/kg oral gavage | ↓mortalilty ↓VT/VF frequency ↓VT/VF duration ↓VPC as effective as metoprolol | ↓QTc duration ↓RyR sensitivity -velocity of conduction -repolarization | [57] |
| Rat | non-reperfused MI, after 1 month | IVA vs. metoprolol vs. IVA + metoprolol vs. saline, starter immediately after MI and given for 4 weeks after MI, 10 mg/kg oral gavage | ↓VT/VF inducibility and VT/VF fatality, IVA+Metoprolol better than IVA or metoprolol alone, which were better than saline | ↓infarct size and ↑connexin 43 expression in all treatment groups | [61] |
| Rat | non-reperfused MI, months 3–5 | IVA vs. saline, started 2 months after MI and given for 3 months, 10 mg/kg in drinking water | ↓PVC by 89% | ↑HR variability by 22% -PR, QRS, QT duration | [63] |
| Pig | 6 episodes of I/R (LAD occlusion): 1 min ischemia + 15 min reperfusion | IVA vs. saline, given 5 min after second occlusion, IVA 0.25 mg/kg intravenous bolus | ↑VF threshold by 2.9-fold | prevention of ischemia-induced APD shortening ↓hypoxia ↑regional blood flow | [58,59] |
| Pig | ischemia (LAD occlusion) | IVA vs. propranolol vs. saline, given before ischemia, IVA 0.25 mg/kg intravenous bolus | ↓time to VF onset (2325s for IVA vs. 682s for propranolol vs. 401s for saline), abolished by pacing | Preserved cardiac energy status | [64] |
| Rat | isolated hearts, I/R episodes: 8 min of ischemia and 10 min of reperfusion | IVA vs. saline, 5 min before ischemia 1 µM IVA and 1ml bolus of 100 µM IVA before the first minute of reperfusion | ↓VF incidence (IVA: 20%, saline: 90%) effect abolished by pacing throughout I/R, but not during reperfusion alone No effects when IVA given at reperfusion | ↑time to conduction slowing ↑time to loss of electrical excitability | [60] |
| Chronic heart failure | |||||
| Rat | post-MI CHF | 1 month after MI: IVA 7 µg/min/kg or Dopamine 10 µg/min/kg or both by implantable osmotic pump for 14 days | ↓VT incidence and ↓PVC by IVA vs. Dopamine | [62] | |
| Mouse | transgenic mice (dnNRSF-Tg) with dilated cardiomyopathy | IVA vs. saline given for 6 months, daily dose 7 mg/kg in drinking water | ↓mortality ↓VT episodes (19/h vs. 92 h for saline) ↓VPC effects independent of HR reduction | ↓HCN2 and HCN4 expression in ventricular myocytes IVA prevented isoproterenol-induced spontaneous action potentials in failing ventricular myocytes | [30] |
| Mouse | transgenic mice (hHCN4 overexpression) with dilated cardiomyopathy | IVA vs. saline given for 2 months, daily dose 1.5 mg/kg by an osmotic pump | ↓VPC ↓non-sustained VT | ↓automaticity | [65] |
| Other models | |||||
| Rabbit | pinacidil-induced short QT syndrome | Langendorff-perfused hearts, IVA 5 µM | ↓VF inducibility | IVA prevented QT, APD and ERP shortening by pinacidil | [66] |
| Rabbit | ouabain-induced arrhythmias | Langendorff-perfused hearts, IVA 5 µM | ↓VF inducibility | IVA did not affect QT or APD, but prevented ERP shortening by ouabain | [67] |
| Rabbit | sotalol and veratradine-induced long QT syndrome | Langendorff-perfused hearts, IVA 5 µM | ↑incidence of polymorphic VT induced by sotalol/vertatridine and hypokalemia | IVA did not increase QT, APD or spatial dispersion of repolarization | [68] |
| Mouse | CPVT transgenic mice | IVA 6 mg/kg | -VA at rest or during a treadmill exercise test | no effect on DAD in the beating induced pluripotent stem cells derived cardiomyocytes from CPVT patients | [69] |
| Study Acronim, Year | Study Population | No. of Study Subjects | Follow-Up | Effect on VA |
|---|---|---|---|---|
| Coronary artery disease | ||||
| BEAUTIFUL 2008 [76] | CAD + LV systolic dysfunction, sinus rhythm ≥ 70 bpm | 10,917 | 1.6 years | |
| BEAUTIFUL 2008 Holter Substudy [77] | 840 | 2 × 24 hours at 1 and 6 months | VT: ΔIVA: 24–26%, PLA: 26–24% (P = NS) | |
| SIGNIFY 2014 [78] | CAD without CHF, sinus rhythm ≥ 70 bpm | 19,107 | 2.3 years | Severe VA: ΔIVA: 0.8%, PLA: 0.7% (P = NS) ΔQT prolongation: ΔIVA: 1.8%, PLA: 0.7% (P < 0.001) |
| Chronic heart failure | ||||
| SHIFT 2010 [79] | CHF, LVEF ≤ 35%, sinus rhythm ≥ 70 bpm | 6558 | 1.9 years | |
| SHIFT Holter Substudy 2010 [80] | 602 | 24 h at 8 months | nsVT: ΔIVA: 28%, PLA: 33% (P = NS) ΔPVC: ΔIVA: 78/h PLA: 69/h (P = NS) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oknińska, M.; Paterek, A.; Zambrowska, Z.; Mackiewicz, U.; Mączewski, M. Effect of Ivabradine on Cardiac Ventricular Arrhythmias: Friend or Foe? J. Clin. Med. 2021, 10, 4732. https://doi.org/10.3390/jcm10204732
Oknińska M, Paterek A, Zambrowska Z, Mackiewicz U, Mączewski M. Effect of Ivabradine on Cardiac Ventricular Arrhythmias: Friend or Foe? Journal of Clinical Medicine. 2021; 10(20):4732. https://doi.org/10.3390/jcm10204732
Chicago/Turabian StyleOknińska, Marta, Aleksandra Paterek, Zuzanna Zambrowska, Urszula Mackiewicz, and Michał Mączewski. 2021. "Effect of Ivabradine on Cardiac Ventricular Arrhythmias: Friend or Foe?" Journal of Clinical Medicine 10, no. 20: 4732. https://doi.org/10.3390/jcm10204732
APA StyleOknińska, M., Paterek, A., Zambrowska, Z., Mackiewicz, U., & Mączewski, M. (2021). Effect of Ivabradine on Cardiac Ventricular Arrhythmias: Friend or Foe? Journal of Clinical Medicine, 10(20), 4732. https://doi.org/10.3390/jcm10204732