
NLRC4 GOF Mutations, a Challenging Diagnosis from Neonatal Age to Adulthood

 , , ,
, , ,
Abstract
:1. Introduction
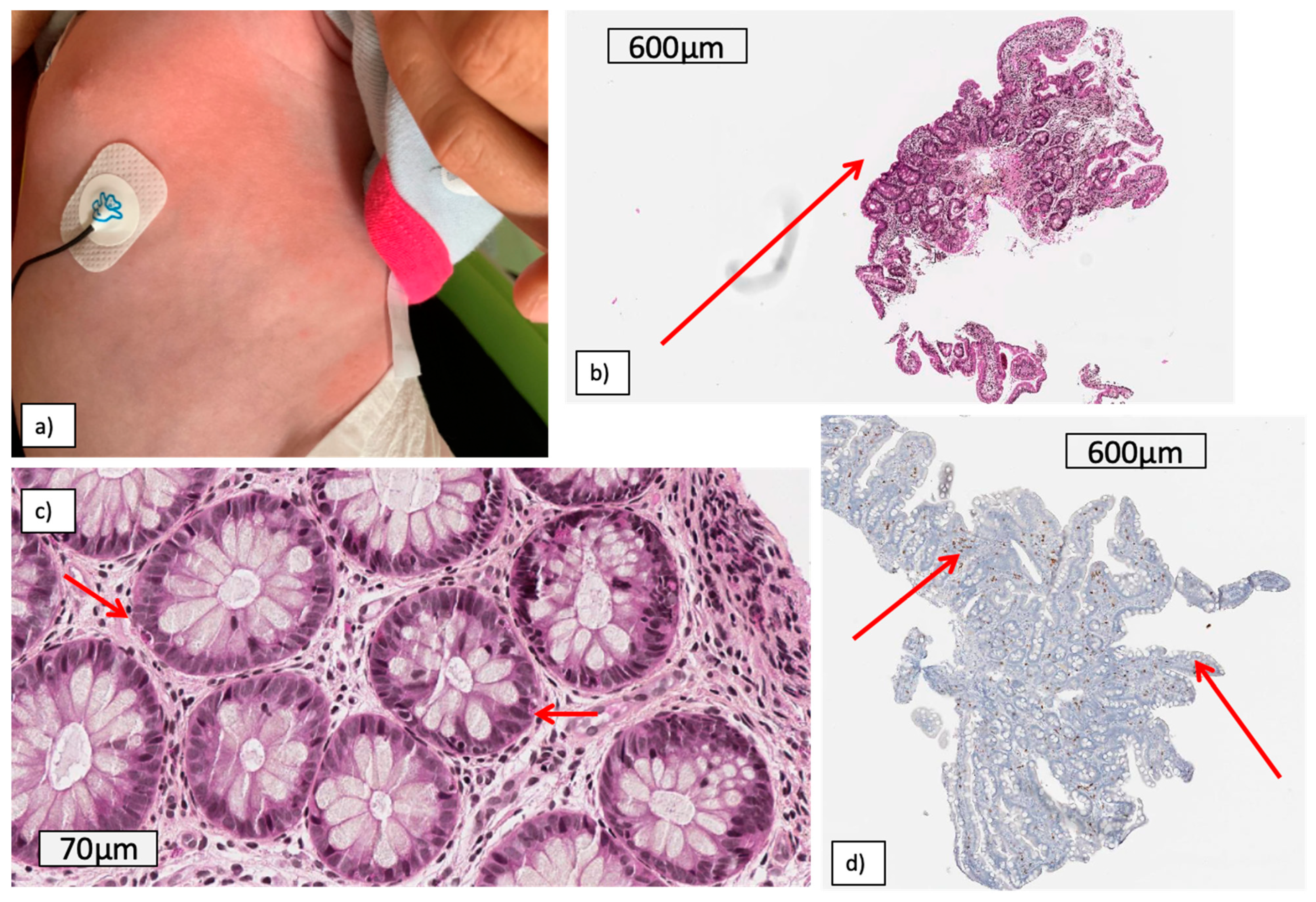
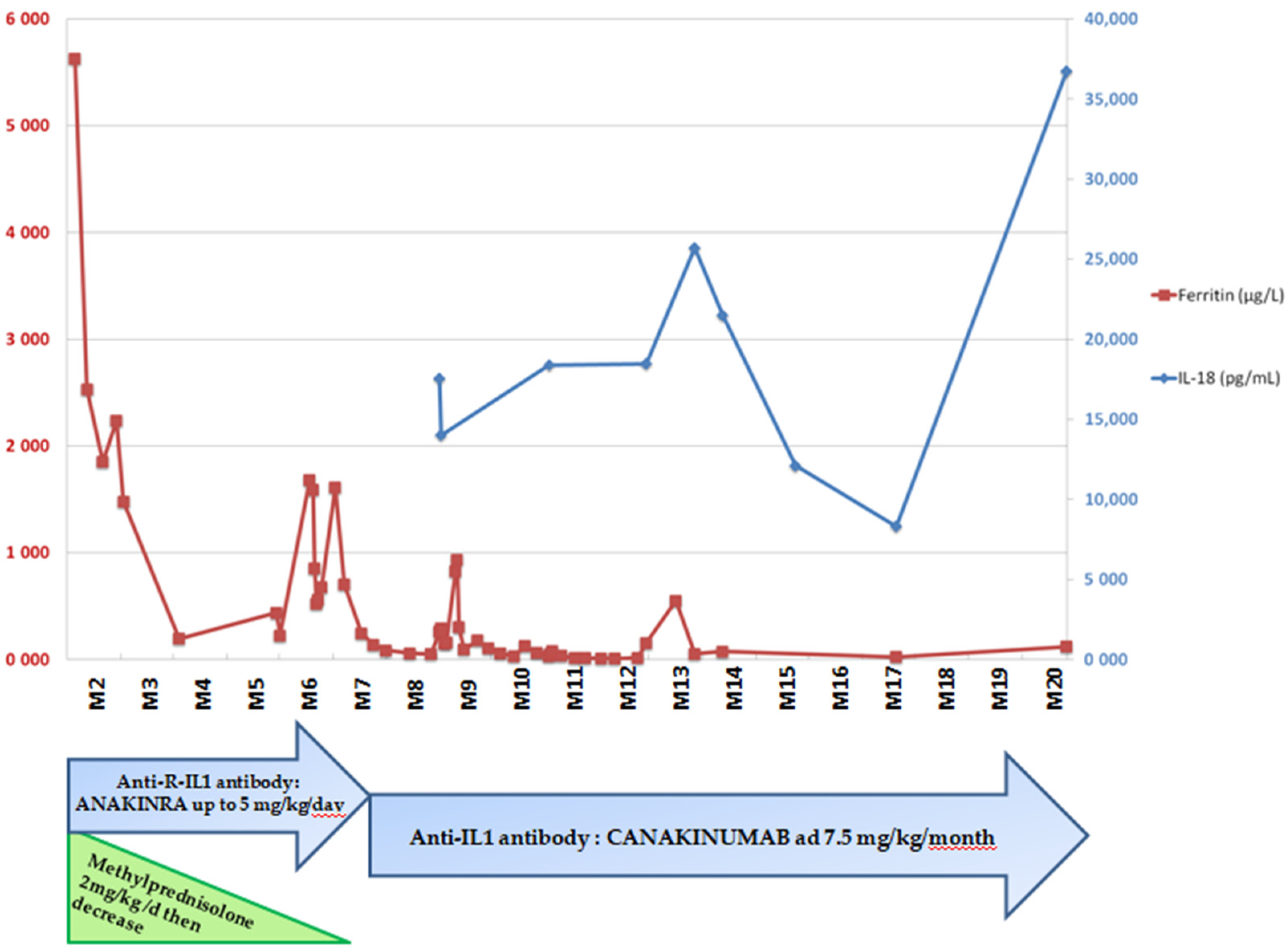
2. Clinical Case Reports
2.1. Patient 1: An Early Digestive and systemic Inflammation with Shocks
2.2. Patient 2: Recurrent Macrophagic Activation Syndrome
3. Literature Review
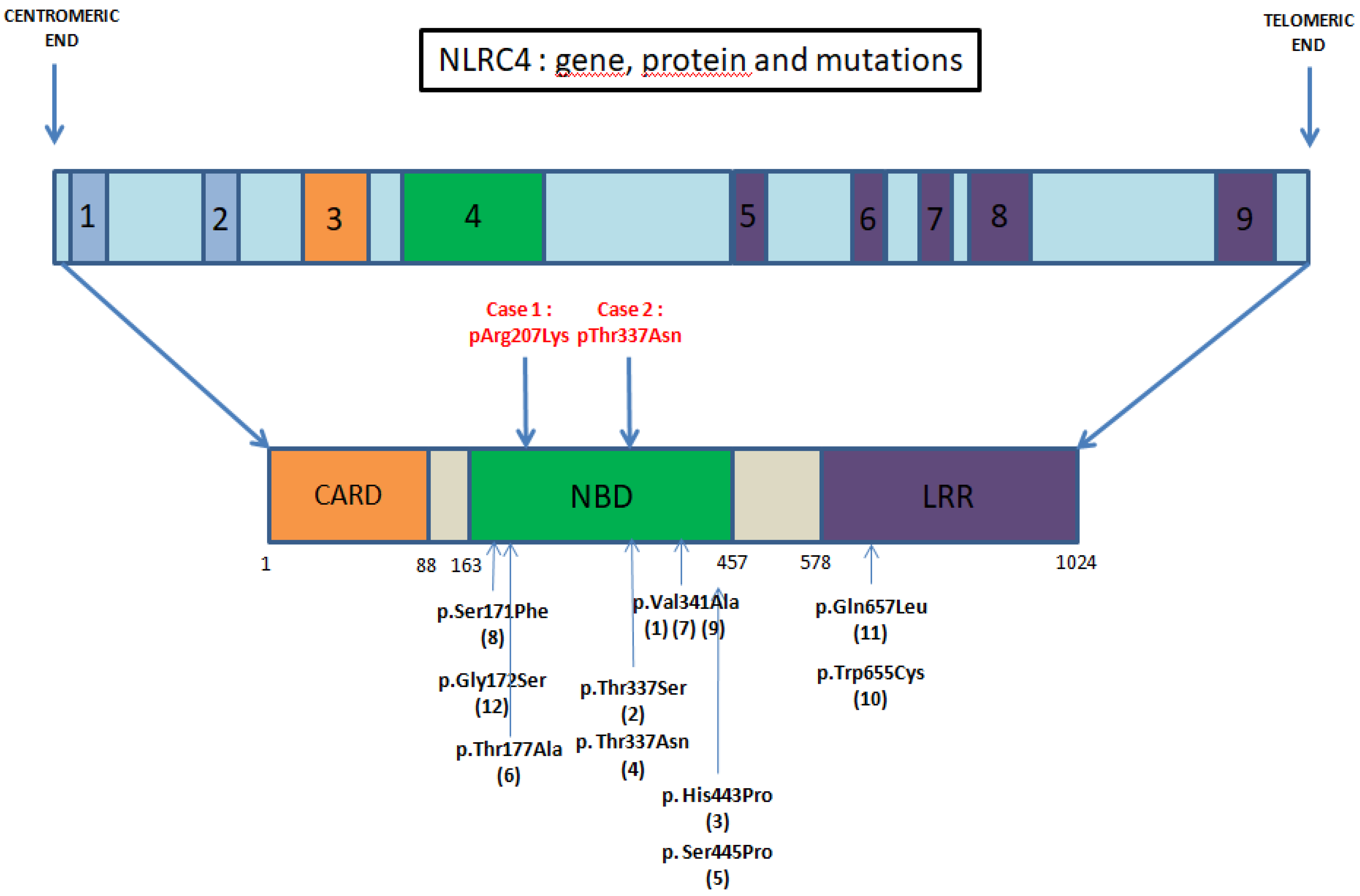
4. Discussion
- Mutations within the NBD—Kitamura et al. [11] showed that a mutation in this domain promotes the oligomerization of the inflammasome with an increased response to usual stimuli.
- Mutations in LRR domain, described as a regulator domain. Its absence results in a constitutively active NLRC4 inflammasome [3].
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poyet, J.-L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a Human Caspase-1-activating Protein Related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.A.; Canna, S.W. The NLRC4 Inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef]
- Hu, Z.; Yan, C.; Liu, P.; Huang, Z.; Ma, R.; Zhang, C.; Wang, R.; Zhang, Y.; Martinon, F.; Miao, D.; et al. Crystal Structure of NLRC4 Reveals Its Autoinhibition Mechanism. Science 2013, 341, 172–175. [Google Scholar] [CrossRef]
- Romberg, N.; Vogel, T.P.; Canna, S.W. NLRC4 inflammasomopathies. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Bracaglia, C. Anti Interferon-Gamma (IFN-γ) Monoclonal Antibody Treatment Ina Patient Carrying an NLRC4 Mutation and Severe Hemophagocytic Lymphohistiocytosis. Pediatric Rheumatol. 2015, 13 (Suppl. 1), O68. [Google Scholar] [CrossRef] [Green Version]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canna, S.W.; Girard-Guyonvarc’H, C.; Malle, L.; de Jesus, A.; Romberg, N.; Kelsen, J.; Surrey, L.F.; Russo, P.; Sleight, A.; Schiffrin, E.; et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. 2017, 139, 1698–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsalou, J.; Blincoe, A.; Fernandez, I.; Dal-Soglio, D.; Marchitto, L.; Selleri, S.; Haddad, E.; Benyoucef, A.; Touzot, F. Rapamycin as an Adjunctive Therapy for NLRC4 Associated Macrophage Activation Syndrome. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.M.; Sanchez, G.A.M.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Alfano, D.N.; Squires, J.E.; Riley, M.M.; Parks, W.T.; Kofler, J.; El-Gharbawy, A.; Madan-Kheterpal, S.; Acquaro, R.; Picarsic, J. Novel NLRC4 Mutation Causes a Syndrome of Perinatal Autoinflammation With Hemophagocytic Lymphohistiocytosis, Hepatosplenomegaly, Fetal Thrombotic Vasculopathy, and Congenital Anemia and Ascites. Pediatr. Dev. Pathol. 2017, 20, 498–505. [Google Scholar] [CrossRef]
- Kitamura, A.; Sasaki, Y.; Abe, T.; Kano, H.; Yasutomo, K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 2014, 211, 2385–2396. [Google Scholar] [CrossRef] [Green Version]
- Volker-Touw, C.M.L.; De Koning, H.D.; Giltay, J.C.; de Kovel, C.; Van Kempen, T.S.; Oberndorff, K.; Boes, M.; van Steensel, M.; Van Well, G.T.J.; Blokx, W.; et al. Erythematous nodes, urticarial rash and arthralgias in a large pedigree with NLRC 4-related autoinflammatory disease, expansion of the phenotype. Br. J. Dermatol. 2017, 176, 244–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yu, Z.; Gou, L.; Zhong, L.; Li, J.; Ma, M.; Wang, C.; Zhou, Y.; Ru, Y.; Sun, Z.; et al. Single-Center Overview of Pediatric Monogenic Autoinflammatory Diseases in the Past Decade: A Summary and Beyond. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Oda, H.; Ito, J.; Niwa, A.; Tanaka, T.; Hijikata, A.; Seki, R.; Nagahashi, A.; Osawa, M.; Asaka, I.; et al. Identification of a High-Frequency Somatic NLRC4 Mutation as a Cause of Autoinflammation by Pluripotent Cell-Based Phenotype Dissection. Arthritis Rheumatol. 2017, 69, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghaddas, F.; Zeng, P.; Zhang, Y.; Schützle, H.; Brenner, S.; Hofmann, S.R.; Berner, R.; Zhao, Y.; Lu, B.; Chen, X.; et al. Autoinflammatory mutation in NLRC4 reveals a leucine-rich repeat (LRR)–LRR oligomerization interface. J. Allergy Clin. Immunol. 2018, 142, 1956–1967.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chear, C.T.; Nallusamy, R.; Canna, S.W.; Chan, K.C.; Baharin, M.F.; Hishamshah, M.; Ghani, H.; Ripen, A.M.; Bin Mohamad, S. A novel de novo NLRC4 mutation reinforces the likely pathogenicity of specific LRR domain mutation. Clin. Immunol. 2020, 211, 108328. [Google Scholar] [CrossRef]
- Nowak-Węgrzyn, A.; Jarocka-Cyrta, E.; Moschione Castro, A. Food Protein-Induced Enterocolitis Syndrome. J. Investig. Allergol. Clin. Immunol. 2017, 27, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Takada, H.; Nomura, A.; Ohga, S.; Hara, T. Interleukin-18 in Hemophagocytic Lymphohistiocytosis. Leuk. Lymphoma 2001, 42, 21–28. [Google Scholar] [CrossRef]
- Weiss, E.S.; Girard-Guyonvarc’H, C.; Holzinger, D.; De Jesus, A.A.; Tariq, Z.; Picarsic, J.; Schiffrin, E.J.; Foell, D.; Grom, A.A.; Ammann, S.; et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018, 131, 1442–1455. [Google Scholar] [CrossRef]
- Krei, J.M.; Møller, H.J.; Larsen, J.B. The role of interleukin-18 in the diagnosis and monitoring of hemophagocytic lymphohistiocytosis/macrophage activation syndrome—A systematic review. Clin. Exp. Immunol. 2021, 203, 174–182. [Google Scholar] [CrossRef]
- Mizuta, M.; Shimizu, M.; Inoue, N.; Ikawa, Y.; Nakagishi, Y.; Yasuoka, R.; Iwata, N.; Yachie, A. Clinical significance of interleukin-18 for the diagnosis and prediction of disease course in systemic juvenile idiopathic arthritis. Rheumatology 2021, 60, 2421–2426. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| NLRC4 Gof Mutation | Symptoms | Lab Exams | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Fever | Gastrointestinal | Hemodynamical | Hematological | Rheumatologic | Dermatological | Other | Inflammation Markers Elevated (Ferritin, CRP) | Elevated IL-18 | |
| c.620G>A, p.Arg207LysCase 1 | + | + | + | + | − | + | − | + | + |
| c.1010C>A, p.Thr337AsnCase 2 | + | + | − | + | − | − | − | + | NA |
| c.1009A > T, p.Thr337Asn [5] | + | − | − | + | − | − | − | + | NA |
| c.1022T>C, p.Val341Ala [6,7] | + | + | − | + | + | + | Respiratory (acute respiratory distress syndrome, Intra alveolar hemorrhage) /Renal (Acute failure) | + | + |
| c.1021G>C, p.Val341Leu [8] | + | + | + | + | − | + | − | + | + |
| c.1009A > T, p.Thr337Ser [9] | + | + | − | + | − | − | − | + | NA |
| c.512C> T p.Ser171Phe [10] | + | + | − | + | − | − | − | NA | NA |
| c.1589A>C, p. His443Pro [11] | + | − | − | − | + | + | − | − | NA |
| c.1333T>C p.Ser445Pro [12] | − | − | − | − | + | + | − | − | NA |
| c.514G>A p.Gly172Ser [13] | + | − | − | − | − | + | − | NA | NA |
| c.529A>G, p. Thr177Asn [14] | + | + | − | - | − | + | Neurological (mental retardation, aseptic meningitis, sensorineural deafness, brain atrophy) | NA | NA |
| c.1965G>C, p. Trp655Cys [15] | + | + | − | + | − | + | − | + | + |
| c.1970A> T, p.Gln657Leu [16] | + | + | − | + | − | + | − | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bardet, J.; Laverdure, N.; Fusaro, M.; Picard, C.; Garnier, L.; Viel, S.; Collardeau-Frachon, S.; Guillebon, J.-M.D.; Durieu, I.; Casari-Thery, C.; et al. NLRC4 GOF Mutations, a Challenging Diagnosis from Neonatal Age to Adulthood. J. Clin. Med. 2021, 10, 4369. https://doi.org/10.3390/jcm10194369
Bardet J, Laverdure N, Fusaro M, Picard C, Garnier L, Viel S, Collardeau-Frachon S, Guillebon J-MD, Durieu I, Casari-Thery C, et al. NLRC4 GOF Mutations, a Challenging Diagnosis from Neonatal Age to Adulthood. Journal of Clinical Medicine. 2021; 10(19):4369. https://doi.org/10.3390/jcm10194369
Chicago/Turabian StyleBardet, Juliette, Noémie Laverdure, Mathieu Fusaro, Capucine Picard, Lorna Garnier, Sébastien Viel, Sophie Collardeau-Frachon, Jean-Marie De Guillebon, Isabelle Durieu, Clémence Casari-Thery, and et al. 2021. "NLRC4 GOF Mutations, a Challenging Diagnosis from Neonatal Age to Adulthood" Journal of Clinical Medicine 10, no. 19: 4369. https://doi.org/10.3390/jcm10194369
APA StyleBardet, J., Laverdure, N., Fusaro, M., Picard, C., Garnier, L., Viel, S., Collardeau-Frachon, S., Guillebon, J.-M. D., Durieu, I., Casari-Thery, C., Mortamet, G., Laurent, A., & Belot, A. (2021). NLRC4 GOF Mutations, a Challenging Diagnosis from Neonatal Age to Adulthood. Journal of Clinical Medicine, 10(19), 4369. https://doi.org/10.3390/jcm10194369