
Clinical Features of Familial Hypercholesterolemia in Children and Adults in EAS-FHSC Regional Center for Rare Diseases in Poland


 ,
, 
Abstract
:1. Introduction
2. Methods
Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Vallejo-Vaz, A.J.; Stevens, C.A.; Lyons, A.R.; Dharmayat, K.I.; Freiberger, T.; Hovingh, G.K.; Mata, P.; Raal, F.J.; Santos, R.D.; Soran, H.; et al. Global perspective of familial hypercholesterolaemia: A cross-sectional study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet 2021. [Google Scholar] [CrossRef]
- Wiegman, A.; Gidding, S.S.; Watts, G.F.; Chapman, M.J.; Ginsberg, H.N.; Cuchel, M.; Ose, L.; Averna, M.; Boileau, C.; Borén, J.; et al. Familial hypercholesterolaemia in children and adolescents: Gaining decades of life by optimizing detection and treatment. Eur. Hear. J. 2015, 36, 2425–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallejo-Vaz, A.J.; De Marco, M.; Stevens, C.A.; Akram, A.; Freiberger, T.; Hovingh, G.K.; Kastelein, J.J.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Overview of the current status of familial hypercholesterolaemia care in over 60 countries—The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis 2018, 277, 234–255. [Google Scholar] [CrossRef] [Green Version]
- Vallejo-Vaz, A.J.; Akram, A.; Seshasai, S.R.K.; Cole, D.; Watts, G.; Hovingh, G.K.; Kastelein, J.J.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Pooling and expanding registries of familial hypercholesterolaemia to assess gaps in care and improve disease management and outcomes: Rationale and design of the global EAS Familial Hypercholesterolaemia Studies Collaboration. Atheroscler. Suppl. 2016, 22, 1–32. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; Giacobbe, C.; Fortunato, G. Familial hypercholesterolemia: A complex genetic disease with variable phenotypes. Eur. J. Med. Genet. 2020, 63, 103831. [Google Scholar] [CrossRef]
- Bianconi, V.; Banach, M.; Pirro, M. Why patients with familial hypercholesterolemia are at high cardiovascular risk? Beyond LDL-C levels. Trends Cardiovasc. Med. 2020, 31, 205–215. [Google Scholar] [CrossRef]
- Hajighasemi, S.; Gorabi, A.M.; Bianconi, V.; Pirro, M.; Banach, M.; Tafti, H.A.; Reiner, Z.; Sahebkar, A. A review of gene- and cell-based therapies for familial hypercholesterolemia. Pharmacol. Res. 2019, 143, 119–132. [Google Scholar] [CrossRef]
- Dyrbuś, K.; Gąsior, M.; Desperak, P.; Osadnik, T.; Nowak, J.; Banach, M. The prevalence and management of familial hypercholesterolemia in patients with acute coronary syndrome in the Polish tertiary centre: Results from the TERCET registry with 19,781 individuals. Atherosclerosis 2019, 288, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Jankowski, P.; Jóźwiak, J.; Cybulska, B.; Windak, A.; Guzik, T.; Mamcarz, A.; Broncel, M.; Tomasik, T. PoLA/CFPiP/PCS Guidelines for the Management of Dyslipidaemias for Family Physicians 2016. Arch. Med. Sci. 2017, 1, 1–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podgórski, M.; Szatko, K.; Stańczyk, M.; Pawlak-Bratkowska, M.; Konopka, A.; Starostecka, E.; Tkaczyk, M.; Góreczny, S.; Rutkowska, L.; Gach, A.; et al. “Apple does not fall far from the tree”—Subclinical atherosclerosis in children with familial hypercholesterolemia. Lipids Health Dis. 2020, 19, 169. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž.; Simental-Mendía, L.E.; Ruscica, M.; Katsiki, N.; Banach, M.; Al Rasadi, K.; Jamialahmadi, T.; Sahebkar, A. Pulse wave velocity as a measure of arterial stiffness in patients with familial hypercholesterolemia: A systematic review and meta-analysis. Arch. Med. Sci. 2019, 15, 1365–1374. [Google Scholar] [CrossRef]
- Santos, R.D. Phenotype vs. genotype in severe familial hypercholesterolemia: What matters most for the clinician? Curr. Opin. Lipidol. 2017, 28, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Penson, P.E. What have we learned about lipids and cardiovascular risk from PCSK9 inhibitor outcome trials: ODYSSEY and FOURIER? Cardiovasc. Res. 2019, 115, e26–e31. [Google Scholar] [CrossRef]
- Solnica, B.; Sygitowicz, G.; Sitkiewicz, D.; Cybulska, B.; Jóźwiak, J.; Odrowąż-Sypniewska, G.; Banach, M. 2020 Guidelines of the Polish Society of Laboratory Diagnostics (PSLD) and the Polish Lipid Association (PoLA) on laboratory diagnostics of lipid metabolism disorders. Arch. Med. Sci. 2020, 16, 237–252. [Google Scholar] [CrossRef]
- Aboyans, V.; Ricco, J.B.; Bartelink, M.L.E.L.; Bjorck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the diagnosis and treatment of peripheral arterial diseases, in collaboration with the European Society for Vascular Surgery (ESVS): Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteries. Eur. Heart J. 2018, 39, 763–816. [Google Scholar] [PubMed] [Green Version]
- Available online: https://www.gov.pl/web/zdrowie/choroby-nieonkologiczne (accessed on 28 July 2021).
- Mysliwiec, M.; Walczak, M.; Małecka-Tendera, E.; Dobrzanska, A.; Cybulska, B.; Filipiak, K.; Mazur, A.; Jarosz-Chobot, P.; Szadkowska, A.; Rynkiewicz, A.; et al. Management of familial hypercholesterolemia in children and adolescents. Position paper of the Polish Lipid Expert Forum. J. Clin. Lipidol. 2014, 8, 173–180. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Colletti, A.; Bajraktari, G.; Descamps, O.; Djuric, D.M.; Ezhov, M.; Fras, Z.; Katsiki, N.; Langlois, M.; Latkovskis, G.; et al. Lipid lowering nutraceuticals in clinical practice: Position paper from an International Lipid Expert Panel. Arch. Med. Sci. 2017, 13, 965–1005. [Google Scholar] [CrossRef]
- Versmissen, J.; Oosterveer, D.; Yazdanpanah, M.; Defesche, J.C.; Basart, D.C.G.; Liem, A.H.; Heeringa, J.; Witteman, J.C.; Lansberg, P.J.; Kastelein, J.J.P.; et al. Efficacy of statins in familial hypercholesterolaemia: A long term cohort study. BMJ 2008, 337, a2423. [Google Scholar] [CrossRef] [Green Version]
- Katsiki, N.; Mikhailidis, D.P.; Bajraktari, G.; Miserez, A.R.; Cicero, A.F.G.; Bruckert, E.; Serban, M.C.; Mirrakhimov, E.; Alnouri, F.; Reiner, Ž.; et al. Statin therapy in athletes and patients performing regular intense exercise—Position paper from the International Lipid Expert Panel (ILEP). Pharmacol. Res. 2020, 155, 104719. [Google Scholar] [CrossRef]
- Vrablik, M.; Raslová, K.; Vohnout, B.; Blaha, V.; Satny, M.; Kyselak, O.; Vaclova, M.; Urbanek, R.; Maskova, J.; Soska, V.; et al. Real-life LDL-C treatment goals achievement in patients with heterozygous familial hypercholesterolemia in the Czech Republic and Slovakia: Results of the PLANET registry. Atherosclerosis 2018, 277, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [Green Version]
- Familial Hypercholesterolaemia; A Report of a WHO Consultation; WHO: Geneva, Switzerland, 1998.
- Starr, B.; Hadfield, S.G.; Hutten, B.A.; Lansberg, P.J.; Leren, T.P.; Damgaard, D.; Neil, H.A.W.; Humphries, S.E. Development of sensitive and specific age- and gender-specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin. Chem. Lab. Med. 2008, 46, 791–803. [Google Scholar] [CrossRef]
- Robinson, J.G.; Huijgen, R.; Ray, K.; Phil, M.; Persons, J.; Kastelein, J.J.P.; Pencina, M.J. Determining when to add nonstatin therapy: A quantitative approach. J. Am. Coll. Cardiol. 2016, 68, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Jayanna, M.B.; Brown, A.S.; Aspry, K.; Orringer, C.; Gill, E.A.; Goldberg, A.; Jones, L.K.; Maki, K.; Dixon, D.L.; et al. Enhancing the value of PCSK9 monoclonal antibodies by identifying patients most likely to benefit. A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.; Li, Q.H.; Bhatt, D.L.; Bittner, V.A.; Baccara-Dinet, M.T.; Goodman, S.G.; Jukema, J.W.; Kimura, T.; Parkhomenko, A.; Pordy, R.; et al. Intensity of statin treatment after acute coronary syndrome, residual risk, and its modification by alirocumab: Insights from the ODYSSEY OUTCOMES trial. Eur. J. Prev. Cardiol. 2020, 28, 33–43. [Google Scholar] [CrossRef]
- Banach, M.; Penson, P.E.; Vrablik, M.; Bunc, M.; Dyrbus, K.; Fedacko, J.; Gaita, D.; Gierlotka, M.; Jarai, Z.; Magda, S.L.; et al. Optimal use of lipid-lowering therapy after acute coronary syndromes: A Position Paper endorsed by the International Lipid Expert Panel (ILEP). Pharmacol. Res. 2021, 166, 105499. [Google Scholar] [CrossRef]
- Maliachova, O.; Stabouli, S. Familial Hypercholesterolemia in Children and Adolescents: Diagnosis and Treatment. Curr. Pharm. Des. 2018, 24, 3672–3677. [Google Scholar] [CrossRef]
- Goldberg, A.C.; Hopkins, P.N.; Toth, P.P.; Ballantyne, C.M.; Rader, D.J.; Robinson, J.G.; Daniels, S.R.; Gidding, S.S. Screening, diagnosis and management of pediatric and adult patients: Clinical guidance form the National Lipid Association Expert Panel on Familial Hypercholesteroemia. J. Clin. Lipidol. 2011, 5, 133–140. [Google Scholar] [CrossRef]
- Gaudet, G.; López-Sendón, J.L.; Averna, M.; Bigot, G.; Banach, M.; Letierce, A.; Loy, M.; Samuel, R.; Manvelian, G.; Batsu, I.; et al. Safety and efficacy of alirocumab in a real-life setting: The ODYSSEY APPRISE study. Eur. J. Prev. Cardiol. 2020, zwaa097. [Google Scholar] [CrossRef]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, P.M.; Thompson, P.D.; Cannon, C.P.; Guyton, J.R.; Bergeron, J.; Zieve, F.J.; Bruckert, E.; Jacobson, T.; Kopecky, S.L.; Baccara-Dinet, M.T.; et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J. Clin. Lipidol. 2015, 9, 758–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastelein, J.J.; Ginsberg, H.N.; Langslet, G.; Hovingh, G.K.; Ceska, R.; Dufour, R.; Blom, D.; Civeira, F.; Krempf, M.; Lorenzato, C.; et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur. Heart J. 2015, 36, 2996–3003. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, H.N.; Rader, D.J.; Raal, F.J.; Guyton, J.R.; Baccara-Dinet, M.T.; Lorenzato, C.; Pordy, R.; Stroes, E. Efficacy and Safety of Alirocumab in Patients with Heterozygous Familial Hypercholesterolemia and LDL-C of 160 mg/dl or Higher. Cardiovasc. Drugs Ther. 2016, 30, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Vaseghi, G.; Taheri, M.; Heshmat-Ghahdarijani, K.; Rayati, M.; Zarfeshani, S.; Pourmoghaddas, A.; Khosravi, A.; Zarepour, E.; Keshavarzrad, P.; Arabi, S.; et al. Familial Hypercholesterolemia (FH) in Iran: Findings from the Four-Year FH Registry. J. Lipids 2021, 2021, 9913969. [Google Scholar] [CrossRef]
- Alhabib, K.F.; Al-Rasadi, K.; Almigbal, T.H.; Batais, M.A.; Al-Zakwani, I.; Al-Allaf, F.A.; Al-Waili, K.; Zadjali, F.; Alghamdi, M.; Alnouri, F.; et al. Familial Hypercholesterolemia in the Arabian Gulf Region: Clinical results of the Gulf FH Registry. PLoS ONE 2021, 16, e0251560. [Google Scholar] [CrossRef]
- Huang, C.C.; Niu, D.M.; Charng, M.J. Genetic Analysis in a Taiwanese Cohort of 750 Index Patients with Clinically Diagnosed Familial Hypercholesterolemia. J. Atheroscler. Thromb. 2021, 62773, Epub ahead of print. [Google Scholar]
- Mehta, R.; Martagon, A.J.; Ramirez, G.A.G.; Antonio-Villa, N.E.; Vargas-Vázquez, A.; Elias-Lopez, D.; Gonzalez-Retana, G.; Rodríguez-Encinas, B.; Ceballos-Macías, J.J.; Romero-Zazueta, A.; et al. Familial hypercholesterolemia in Mexico: Initial insights from the national registry. J. Clin. Lipidol. 2020, 15, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Averna, M.; Cefalu’, A.B.; Casula, M.; Noto, D.; Arca, M.; Bertolini, S.; Calandra, S.; Catapano, A.L.; Tarugi, P.; Pellegatta, F.; et al. Familial hypercholesterolemia: The Italian Atherosclerosis Society Network (LIPIGEN). Atheroscler. Suppl. 2017, 29, 11–16. [Google Scholar] [CrossRef]
- Mata, N.; Alonso, R.; Badimón, L.; Padró, T.; Fuentes, F.; Muñiz, O.; Perez-Jiménez, F.; López-Miranda, J.; Díaz, J.L.; Vidal, J.I.; et al. Clinical characteristics and evaluation of LDL-cholesterol treatment of the Spanish Familial Hypercholesterolemia Longitudinal Cohort Study (SAFEHEART). Lipids Health Dis. 2011, 10, 94. [Google Scholar] [CrossRef] [Green Version]
- Dyrbuś, K.; Gąsior, M.; Desperak, P.; Osadnik, T.; Banach, M. The prevalence and management of familial hypercholesterolemia in patients with acute coronary syndrome in Poland: Results from the TERCET Registry. Eur. Heart J. 2019, 40, P820. [Google Scholar] [CrossRef]
- Kutkiene, S.; Petrulioniene, Z.; Laucevicius, A.; Cerkauskiene, R.; Staigyte, J.; Saulyte, A.; Petrulionyte, E.; Gargalskaite, U.; Skiauteryte, E.; Matuzeviciene, G.; et al. Lipid profile evaluation and severe hypercholesterolaemia screening in the middle-aged population according to nationwide primary prevention programme in Lithuania. Atherosclerosis 2018, 277, 267–272. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Children (n = 16) | Adults (n = 87) | p | Total | |
|---|---|---|---|---|
| Clinical characteristics | ||||
| Age at baseline visit (years) | 9 ± 3 | 41 ± 16 | <0.01 | 39 ± 18 |
| Weight (kg) | 37 ± 17 | 82 ± 17 | <0.01 | 73 ± 25 |
| Height (m) | 146 (124–159) | 170 (163–176) | <0.01 | 165 (160–175) |
| Body mass index (kg/m2) | 25.3 ± 1.8 | 28.4 ± 5.3 | 0.42 | 28.2 ± 5.2 |
| Systolic blood pressure (mmHg) | 112 (104–116) | 135 (120–150) | 0.001 | 128 (117–147) |
| Diastolic blood pressure (mmHg) | 68 (65–74) | 82 (73–92) | 0.004 | 77 (71–90) |
| Heart rate (bpm) | 70 (60–71) | 75 (63–80) | 0.16 | 71 (60–80) |
| Laboratory test results | ||||
| Highest TC ever (mg/dL) | 343 ± 61 | 333 ± 91 | 0.74 | 335 ± 85 |
| Highest LDL ever (mg/dL) | 266 ± 51 | 235 ± 69 | 0.18 | 241 ± 66 |
| TC (mg/dL) | 313 (279–354) | 259 (213–334) | 0.04 | 266 (220–336) |
| LDL (mg/dL) | 247 (206–260) | 192 (144–251) | 0.02 | 195 (151–257) |
| HDL (mg/dL) | 53 (46–67) | 48 (40–58) | 0.009 | 48 (40–59) |
| TG (mg/dL) | 84 (67–101) | 112 (83–175) | 0.15 | 107 (79–162) |
| Glucose (mg/dL) | 83 (82–88) | 93 (86–97) | 0.04 | 90.5 (82–96) |
| AspAT (U/L) | 24 (22–29) | 24 (19–29) | 0.69 | 24 (19–29) |
| AlAT (U/L) | 19 (15–27) | 23 (17–33) | 0.21 | 22 (15–33) |
| Creatinine kinase (U/L) | 114 (58–149 | 76 (63–133) | 0.54 | 86 (62–140) |
| TSH (µLU/mL) | 1.72 (1.43–2.1) | 1.77 (1.38–2.57) | 0.89 | 1.73 (1.43–2.46) |
| Pharmacotherapy | ||||
| Statins (%) | 56 | 70 | ||
| Ezetymibe (%) | 0 | 29 | ||
| PCSK9 inhibitors (%) | 0 | 11.5 | ||
| Fibrates (%) | 0 | 13.7 | ||
| n-3 fatty acids (%) | 0 | 13.7 | ||
| Paired Differences | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Median | Interquartile Range | Min | Max | X2 | df | p | |
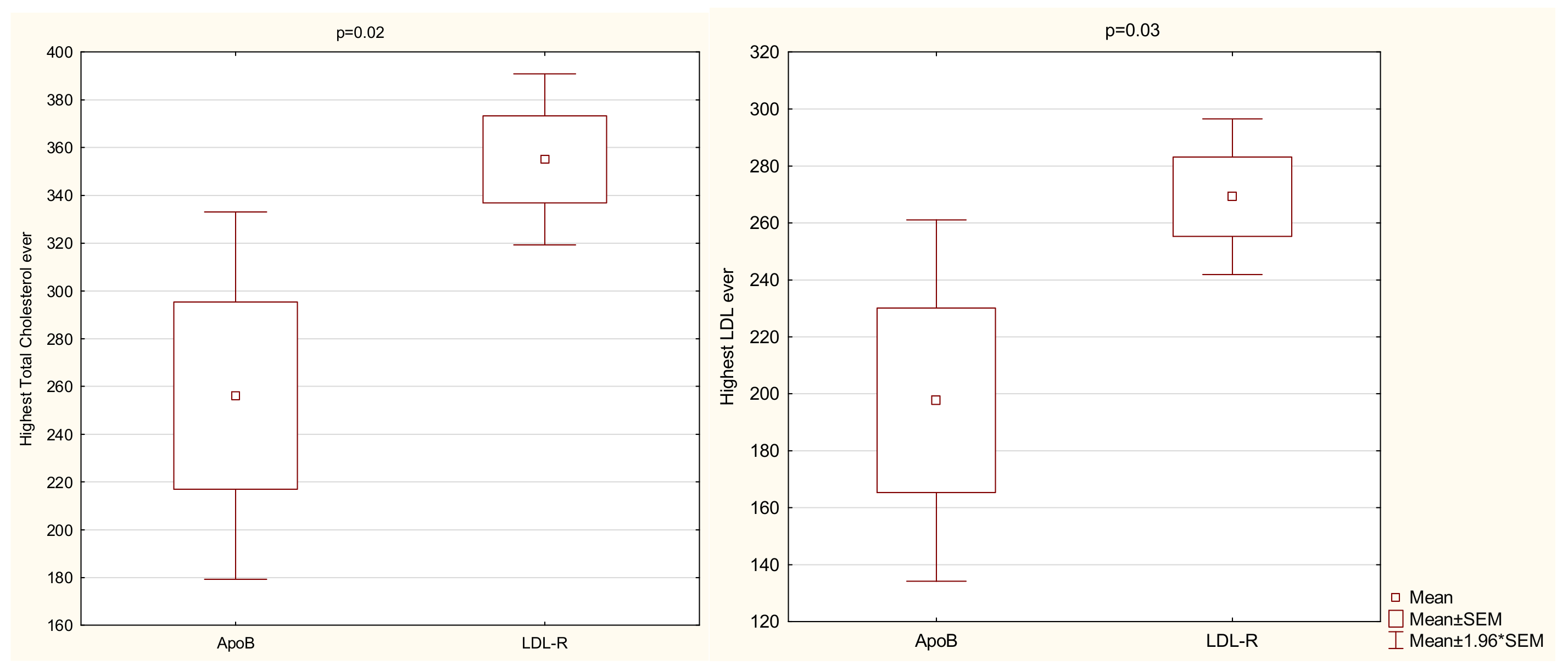
| Highest TC ever | 335 | 85 | 329 | 282–390 | 142 | 600 | 0.7 | 3 | 0.13 |
| Highest LDL-C ever | 241 | 67 | 243 | 196–293 | 80 | 400 | 3.5 | 2 | 0.19 |
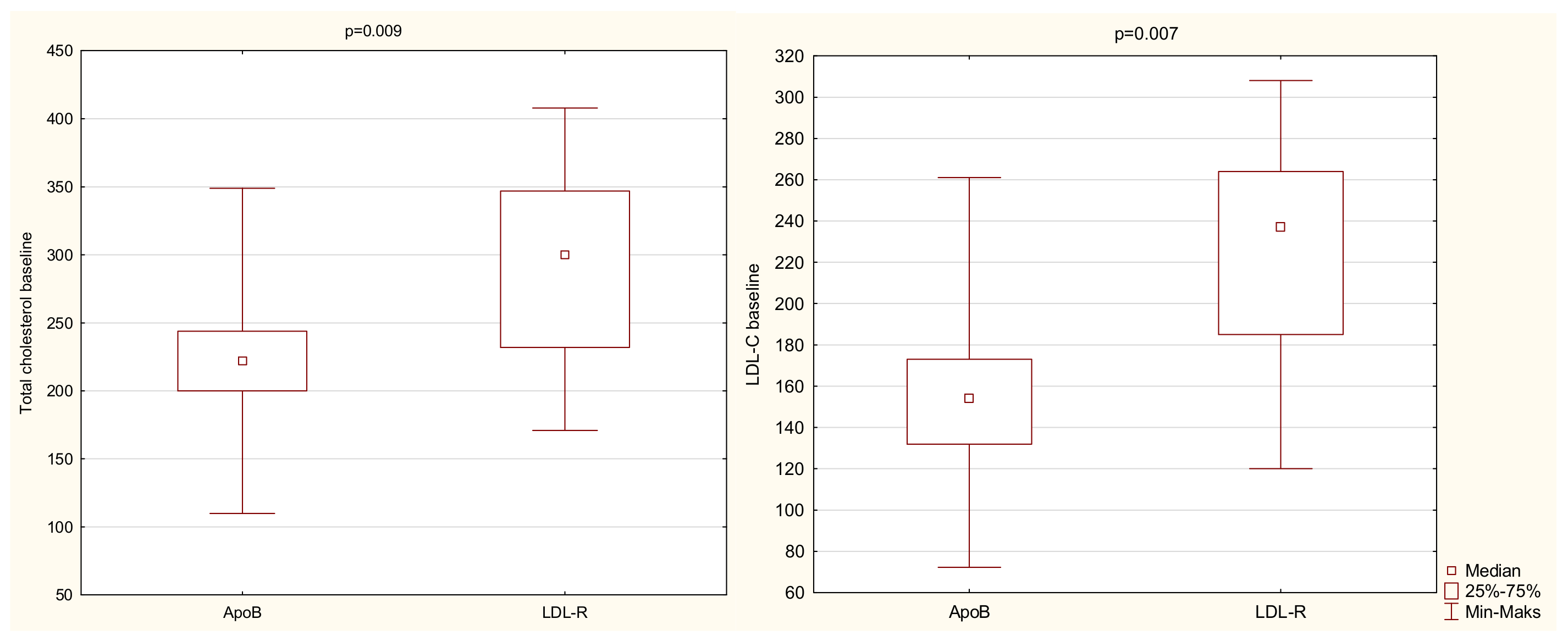
| TC at baseline | 274 | 71 | 267 | 220–336 | 110 | 419 | 8.5 | 2 | 0.02 |
| LDL-C at baseline | 200 | 64 | 195 | 151–257 | 55 | 331 | 3.9 | 2 | 0.02 |
| HDL-C at baseline | 51 | 15 | 48 | 40–59 | 27 | 102 | 2.5 | 2 | 0.81 |
| TG at baseline | 136 | 112 | 107 | 79–162 | 33 | 842 | 6.3 | 2 | 0.09 |
| Pairwise Comparison—Gene mutation | Mean Ranks | p of KW | |
|---|---|---|---|
| LDL-C at baseline | Lack—LDLR | 21.0/20.3 | 1.0 |
| LDLR—ApoB | 20.3/9.7 | 0.02 | |
| Lack—ApoB | 21.0/9.7 | 0.44 | |
| TC at baseline | Lack—LDLR | 26.0/19.8 | 1.0 |
| LDLR—ApoB | 19.8/9.7 | 0.03 | |
| Lack—ApoB | 26.0/9.7 | 0.11 | |
| TG at baseline | Lack—LDLR | 28.5/16.1 | 0.19 |
| LDLR—ApoB | 16.1/12.7 | 1.0 | |
| Lack—ApoB | 28./12.7 | 0.08 |
| TC | LDL-C | |
|---|---|---|
| Triple lipid lowering therapy | ||
| Mean decrease (%) | 58 | 71 |
| Mean absolute reduction | 200 ± 121 mg/dL (5.17 ± 3.13 mmol/L) | 190 ± 92 mg/dL (4.91 ± 2.38 mmol/L) |
| Dual therapy | ||
| Mean decrease (%) | 41 | 38 |
| Mean absolute reduction | 174 ± 59 mg/dL (4.5 ± 1.53 mmol/L) | 108 ± 75 mg/dL (2.79 ± 1.94 mmol/L) |
| Statins | ||
| Mean decrease (%) | 38 | 45 |
| Mean absolute reduction | 139 ± 65 mg/dL (3.59 ± 1.68 mmol/L) | 119 ± 57 mg/dL (3.08 ± 1.47 mmol/L) |
| Ezetimibe | ||
| Mean decrease (%) | 4 | 5 |
| Mean absolute reduction | 14 mg/dL (0.36 mmol/L) | 14 mg/dL (0.36 mmol/L) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewek, J.; Konopka, A.; Starostecka, E.; Penson, P.E.; Maciejewski, M.; Banach, M. Clinical Features of Familial Hypercholesterolemia in Children and Adults in EAS-FHSC Regional Center for Rare Diseases in Poland. J. Clin. Med. 2021, 10, 4302. https://doi.org/10.3390/jcm10194302
Lewek J, Konopka A, Starostecka E, Penson PE, Maciejewski M, Banach M. Clinical Features of Familial Hypercholesterolemia in Children and Adults in EAS-FHSC Regional Center for Rare Diseases in Poland. Journal of Clinical Medicine. 2021; 10(19):4302. https://doi.org/10.3390/jcm10194302
Chicago/Turabian StyleLewek, Joanna, Agnieszka Konopka, Ewa Starostecka, Peter E. Penson, Marek Maciejewski, and Maciej Banach. 2021. "Clinical Features of Familial Hypercholesterolemia in Children and Adults in EAS-FHSC Regional Center for Rare Diseases in Poland" Journal of Clinical Medicine 10, no. 19: 4302. https://doi.org/10.3390/jcm10194302
APA StyleLewek, J., Konopka, A., Starostecka, E., Penson, P. E., Maciejewski, M., & Banach, M. (2021). Clinical Features of Familial Hypercholesterolemia in Children and Adults in EAS-FHSC Regional Center for Rare Diseases in Poland. Journal of Clinical Medicine, 10(19), 4302. https://doi.org/10.3390/jcm10194302